Back to Journals » International Journal of Nanomedicine » Volume 15
PLGA Nanoparticle Platform for Trans-Ocular Barrier to Enhance Drug Delivery: A Comparative Study Based on the Application of Oligosaccharides in the Outer Membrane of Carriers
Authors Jiang G ![]() , Jia H, Qiu J, Mo Z, Wen Y, Zhang Y, Wen Y, Xie Q, Ban J
, Jia H, Qiu J, Mo Z, Wen Y, Zhang Y, Wen Y, Xie Q, Ban J ![]() , Lu Z, Chen Y, Wu H, Ni Q, Chen F, Lu J
, Lu Z, Chen Y, Wu H, Ni Q, Chen F, Lu J ![]() , Wang Z, Li H
, Wang Z, Li H ![]() , Chen J
, Chen J
Received 18 July 2020
Accepted for publication 24 September 2020
Published 24 November 2020 Volume 2020:15 Pages 9373—9387
DOI https://doi.org/10.2147/IJN.S272750
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Ge Jiang,1,* Huanhuan Jia,2,* Jindi Qiu,1,* Zhenjie Mo,1 Yifeng Wen,1 Yan Zhang,1 Yuqin Wen,1 Qingchun Xie,1,3– 5 Junfeng Ban,1,3– 5,* Zhufen Lu,1,3– 5 Yanzhong Chen,1,3– 5 Hao Wu,6 Qingchun Ni,7 Fohua Chen,1 Jiashu Lu,1 Zhijiong Wang,1 Haoting Li,1 Junming Chen1
1Guangdong Pharmaceutical University, Guangzhou, People‘s Republic of China; 2Key Laboratory of Guangdong Laboratory Animals, Guangdong Laboratory Animals Monitoring Institute, Guangzhou, People‘s Republic of China; 3Guangdong Provincial Key Laboratory of Advanced Drug Delivery Systems, Guangdong Pharmaceutical University, Guangzhou, People‘s Republic of China; 4Guangdong Provincial Engineering Center of Topical Precision Drug Delivery System, Center for Drug Research and Development, Guangdong Pharmaceutical University, Guangzhou, People‘s Republic of China; 5R&D Innovation Team for Controlled-Release Microparticle Drug Delivery System, Guangdong Pharmaceutical University, Guangzhou, People‘s Republic of China; 6Community Health Service Center of South China Agricultural University, Guangzhou, People‘s Republic of China; 7Guangzhou General Pharmaceutical Research Institute Co., Ltd, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhufen Lu; Yanzhong Chen
Guangdong Provincial Key Laboratory of Advanced Drug Delivery Systems, Guangdong Pharmaceutical University, Guangzhou 510006, People‘s Republic of China
Tel +86 20 39352506
Email [email protected] [email protected]
Purpose: The trans-ocular barrier is a key factor limiting the therapeutic efficacy of triamcinolone acetonide. We developed a poly(lactic-co-glycolic acid) nanoparticles (PLGA NPs) surface modified respectively with 2-hydroxypropyl-β-cyclodextrin (2-HP-β-CD), chitosan oligosaccharide and trehalose. Determination of the drug/nanoparticles interactions, characterization of the nanoparticles, in vivo ocular compatibility tests, comparisons of their corneal permeability and their pharmacokinetics in aqueous humor were carried out.
Methods: All PLGA NPs were prepared by the single emulsion and evaporation method and the drug-nanoparticle interaction was studied. The physiochemical features and in vitro corneal permeability of NPs were characterized while the aqueous humor pharmacokinetics was performed to evaluate in vivo corneal permeability of NPs. Ocular compatibility of NPs was investigated through Draize and histopathological test.
Results: The PLGA NPs with lactide/glycolide ratio of 50:50 and small particle size (molecular weight 10 kDa) achieved optimal drug release and corneal permeability. Surface modification with different oligosaccharides resulted in uniform particle sizes and similar drug-nanoparticle interactions, although 2-HP-β-CD/PLGA NPs showed the highest entrapment efficiency. In vitro evaluation and aqueous humor pharmacokinetics further revealed that 2-HP-β-CD/PLGA NPs had greater trans-ocular permeation and retention compared to chitosan oligosaccharide/PLGA and trehalose/PLGA NPs. No ocular irritation in vivo was detected after applying modified/unmodified PLGA NPs to rabbit’s eyes.
Conclusion: 2-HP-β-CD/PLGA NPs are a promising nanoplatform for localized ocular drug delivery through topical administration.
Keywords: ocular drug delivery, PLGA nanoparticle, ocular barrier, oligosaccharide, local bioavailability, triamcinolone acetonide, eye drop administration
Introduction
The constancy of the microenvironment of the intraocular tissues and organs is protected from foreign particles by static and dynamic ocular barriers, like the tear film and cornea. The major dilemma encountered while developing new formulations of ocular drug with poor solubility is low bioavailability.1 Other factors such as nasolacrimal drainage can necessitate recurrent drug administration. Therefore, lipophilicity, the degree of plasma protein binding and active membrane transport are prerequisites for effective ocular drugs.2,3 Poly(lactide-co-glycolide) nanoparticles (PLGA NPs) as a carrier may effectively overcome ocular barriers owing to unique effects in terms of specific surface area, surface energy and surface atoms, and has become an effective strategy to solve the limitation of the ocular barrier.4–6 The targeting efficiency of PLGA NPs depends on their cellular uptake, which in turn varies according to the epithelial cell types and the surface potential of the PLGA NPs itself. For instance, PLGA NPs are endocytosed and internalized by vascular smooth muscle cells through clathrin, whereas neither clathrin nor caveolin mediate endocytosis of PLGA NPs in rabbit's conjunctival epithelium cells.7,8 Therefore, drug delivery through PLGA NPs to increase the amount of drugs transported across barriers can be selectively targeted by altering their particle size, surface potential and by introducing surface modification. Desai et al showed the effect of PLGA NPs size, concentration, and temperature on endocytosis of NPs through epithelial cells, and found that endocytosis is a process of activating endocytosis and the epithelial cell types determined the absorption of NPs.9 Furthermore, the hydrophilicity, colloidal stability and mucoadhesiveness of the NPs surface also affect their cellular uptake and drug transport volume. The research of Alonso et al showed that surface PEGylation can significantly improve hydrophilicity, stability and adhesion on the surface of intestinal cells of the NPs in vivo, as well as the bioavailability of the encapsulated macromolecular drugs.10,11 In the past two decades, ophthalmic research has moved toward the formulation of novel and safe drug delivery systems to counteract eye barriers and maintain drug levels in target tissues.12 Anterior segment drug delivery dosage forms are produced by modifying traditional topical formulations such as solutions, suspensions, emulsions, gels, and ointments. Novel advanced ocular delivery systems have been extensively investigated, such as in-situ forming gel, nanocarrier systems, inserts, and vesicular systems.12,13 The improvement of surface hydrophilicity, targeting and transport rate of NPs via incorporation of hydrophilic and mucoadhesive biopolymers can increase their ability to penetrate the mucus layer and selectively adhere to the epithelial cells, thereby prolonging their retention, facilitating their endocytosis and increasing drug accumulation to the therapeutically significant dose.14,15 In addition, a particle size range of 40–120 nm enables epithelial absorption through cellular uptake and paracellular pathway simultaneously.16 There was a growing need to develop new approaches that control particle size, enhance drug dissolution rate, and accordingly improve its bioavailability. Oligosaccharides like 2-hydroxypropyl-β-cyclodextrin (2-HP-β-CD) and chitosan oligosaccharide are promising biomaterials for constructing trans-ocular nano drug carriers.17,18 Encapsulating a drug in biodegradable, biocompatible, natural or synthetic polymeric nanoparticles is one such approach. In the present study, we designed a triamcinolone acetonide-loaded PLGA nanoparticle platform for topical corticosteroids application, explored the transmembrane transport pathways and the distribution rules of the drug in the eyes, and analyzed the effect of different oligosaccharides on the physiochemical features and corneal permeability of the NPs in order to select the optimum formulation for ocular drug delivery.
Materials and Methods
Materials
Triamcinolone acetonide was purchased from the He Hui Pharmaceutical Group Co. Ltd. (Tianjin, China), PLGA from Dai Gang Bioengineering Co. Ltd. (Jinan, China), and polyvinyl alcohol (PVA) from Cola International Trade Co. Ltd. (Shanghai, China). The 2-hydroxypropyl-β-cyclodextrin (2-HP-β-CD) was a kind gift from Qian Hui Biotechnology Co. Ltd. (Zibo, China). Trehalose was obtained from Wei Jia Technology Co. Ltd. (Guangzhou, China), and chitosan oligosaccharide from AK Biotech Co. Ltd. (Shandong, China). All other reagents and solvents were of analytical reagent grade.
Preparation of Nanoparticles
Triamcinolone acetonide-loaded PLGA NPs were prepared by the single emulsion and evaporation method as previously described.19 Briefly, 100 mg PLGA and 10 mg triamcinolone acetonide were dissolved in 5 mL acetone/ethanol (4:1) mixture to form the organic phase. Later, the organic phase was slowly injected into 2% w/v aqueous solution of PVA (40 mL) using a syringe under sonication (ultrasonic probe, JY88IIN, Ningbo, China). The resulting emulsion was stirred overnight (400 rpm) at room temperature to evaporate the organic phase. The different oligosaccharide-modified NPs were prepared by first dissolving 1.5% (w/v) 2-HP-β-CD, 0.2% (w/v) trehalose or 0.02% (w/v) chitosan oligosaccharide in aqueous PVA, and subsequent steps were followed as described. The carrier materials were hydrated and swollen during preparation, and their pH and osmotic pressure were adjusted to obtain uniform, non-aggregating NPs.20
Determination of Drug-Nanoparticles Interactions
Differential Scanning Calorimetry (DSC)
DSC is used to evaluate crystalline structure of the interacting molecules in terms of enthalpy changes. Briefly, 8–12 mg samples were sealed in aluminum pans and the scans (DSC4000, Elma Perkin technology company, USA) were recorded from 30 °C to 300 °C at the heating rate of 10 °C·min−1 in a nitrogen atmosphere. Empty pans were used as reference.
X-Ray Diffraction (XRD)
X-ray powder diffraction analyzes crystalline structure through the diffraction effect of X-ray, and the peak positions are used to determine the presence of specific compounds. XRD of different samples were measured using D8-Advance (Bruker company, Germany) at 40 mA and 40 kV, and scanned from 8° to 80° (2θ) at room temperature.
Fourier Transform Infrared Spectroscopy (FT-IR)
Infrared spectroscopy identifies organic and inorganic compounds and some inorganic substances based on the intensity and shape of infrared absorption peaks. The samples were mixed with potassium bromide and pressed into tablets. FT-IR (Spectrum100, Elma Perkin technology company, USA) scans were taken at ambient temperature from 400 to 4000 cm−1, with blank potassium bromide as reference.
Characterization of Nanoparticles
Entrapment Efficiency and Drug Loading
Entrapment efficiency (EE%) and drug loading (DL%) were used to evaluate the drug encapsulation of different nanoparticle formulations and determined by the ultrafiltration method.21 Briefly, freshly prepared nanoparticles suspension (0.5 mL) was pipetted into an ultrafiltration filter tube (MW: 10 kDa) and centrifuged at 9000 rpm for 30 mins. The filtrate was collected and injected into a HPLC column to determine the amount of the non-encapsulated free drug. The nanoparticles were then lyophilized and weighed. The HPLC parameters for analyzing different concentrations of triamcinolone acetonide were based on a method previously described.22 Triamcinolone acetonide contents were determined for all formulations, with an anticipated range of 200.13 ± 0.15 μg·mL−1. EE% and DL% of the nanoparticles were calculated using Equations (1) and (2):23


Particle Size Distribution and Zeta Potential Analysis
Particle size and zeta potential are important physicochemical parameters determining the homogeneity and stability of nanoparticles. The average particle size, polydispersity index (PDI), and zeta potential of the different NPs were determined by dynamic light scattering (Delsa Nano C® particle size and ξ-potential analyzer, Beckman Coulter Inc., USA) at 25 ± 1 °C. All the samples were diluted with deionized water, and the experiment was performed thrice.
Morphological Observation Using Transmission Electron Microscopy (TEM)
The surface morphology of NPs was analyzed by transmission electron microscopy (JEM-1400, JEOL, JAPAN). A drop of the NPs dispersion was placed on a carbon-coated copper grid and air dried, followed by staining with 2% (w/v) phosphotungstic acid solution. The copper grid was dried at room temperature and observed.
In vitro Drug Release
The amount of triamcinolone acetonide released from the NPs was determined by dialysis.24 Briefly, 2 mL NPs suspension was placed into a dialysis bag (SWB-2000, Tianjin Automatic Science Instrument Co. Ltd, China) of molecular weight cut-off 8 kDa, which was then immersed in 25 mL simulated tear fluid. The samples were incubated in a shaker bath at 34 ± 0.5 °C, and 2 mL aliquots of the buffer were collected at 1, 2, 4, 6, 8, 12 and 24 h, and immediately replaced with the same volume of fresh buffer. The amounts of triamcinolone acetonide released from all formulations were investigated by HPLC after membrane filtration (φ = 0.22 μm). Three independent experiments were performed simultaneously. The cumulative release rate (Q) of the drug was calculated as follows:25

 : drug concentration at time point (μg·mL−1);
: drug concentration at time point (μg·mL−1); 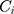 : drug concentration at the sampling points (μg·mL−1);
: drug concentration at the sampling points (μg·mL−1);  : total amount of drug in nanoparticles (μg);
: total amount of drug in nanoparticles (μg); 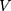 : sampling volume (2 mL).
: sampling volume (2 mL).
In vitro Evaluation of Corneal Permeation
The corneal permeation ability of the NPs was determined ex vivo in rabbit corneas. The corneas were removed and mounted on Franz diffusion cells between the donor and receptor chamber, with the epithelial surface facing the former. The receptor chamber was filled with 5 mL artificial aqueous humor, and 0.5 mL of each sample was placed in the donor chamber. The chambers were incubated at 34 ± 0.5 °C in a thermostatic water bath with continuous stirring at 200 rpm for 24 h. Two milliliters aliquots were taken from the receptor chamber at different time points, and immediately replaced with an equal volume of fresh medium. The withdrawn samples were analyzed by HPLC in triplicate after membrane filtration (φ = 0.22 μm). The data processing was calculated from the linear portion of the permeation curve according to the following equation.26,27
The cumulative amount (Q, μg·cm−2) of triamcinolone acetonide in the receiving chamber was calculated using Equation (4) based on a standard calibration curve:
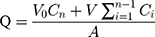
 : drug concentration at time point (μg·mL−1);
: drug concentration at time point (μg·mL−1);  : drug concentration at the sampling points (μg·mL−1);
: drug concentration at the sampling points (μg·mL−1);  : the volume of the medium in the receiving chamber (5 mL);
: the volume of the medium in the receiving chamber (5 mL);  : effective penetration area (0.5024 cm2);
: effective penetration area (0.5024 cm2);  : sampling volume (2 mL).
: sampling volume (2 mL).
The apparent permeability coefficient (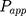 , cm·s−1) was calculated according to Equation (5):
, cm·s−1) was calculated according to Equation (5):

 : initial drug concentration; A: effective penetration area (0.5024 cm2);
: initial drug concentration; A: effective penetration area (0.5024 cm2);  : the slope of cumulative permeation versus time curves.
: the slope of cumulative permeation versus time curves.
The steady flow (Jss, μg·s−1·cm−2) was calculated using Equation (6):

Pharmacokinetics of Nanoparticles in Aqueous Humor
The pharmacokinetics of nanoparticles in aqueous humor was determined by microdialysis.28 Healthy New Zealand albino rabbits were anesthetized by injecting urethane (1 g·kg−1) into the marginal ear vein, and their body temperature was kept constant using a homeothermic blanket. After dilating the pupils with tropicamide phenylephrine eye drops (5 mg·mL−1), a linear probe (CMA 30; Sweden; Cuprophane membrane; 10 mm length; molecular weight cut-off, 6 kDa) was inserted into the aqueous humor using a 25-gauge needle just above the cornea scleral limbus, and traversed through the center of the anterior chamber. The needle was then removed, and the active dialysis membrane was adjusted within the anterior chamber. The probe was secured in the correct position with surgical tapes to prevent any disturbance during sampling. After setting the pump flow rate at 10 μL·min−1, the probe was perfused with normal saline till liquid was ejected from the outlet. The flow rate was then reduced to 0.5 μL·min−1, and the animals were stabilized for at least 1 h before adding 180 μL of each sample (22.5 μg·kg−1) onto the corneal surface. Dialyzed samples were collected every 30 min over a period of 6 h, and analyzed by HPLC. The study was approved by the Institutional Animal Care and Use committee of Guangdong Pharmaceutical University, and performed as per the guidelines of the National Institutes of Health guide for the care and use of laboratory animals.
In vivo Ocular Compatibility Test
The biocompatibility of the different PLGA NPs was determined by the irritation test. Briefly, 50 μL of a single dose formulation was administrated to the left eyes of the rabbit model, and the same volume of normal saline was applied into the right eyes (n = 3) for a period of 2 weeks. The irritation of the eyes was scored according to the Draize scale.29 The animals were sacrificed after the final observation, and the eyeballs were removed and stained with H&E for histopathological evaluation.
Pharmacokinetic Calculation and Data Analysis
The pharmacokinetic parameters were calculated using the DAS 3.0 software package (Chinese Pharmacological Society, Shanghai, China), and all values were expressed as mean ± SD. The non-compartmental pharmacokinetic model was used to calculate maximum concentration (Cmax), time to reach maximum concentration (Tmax), area under curve from 0 to t (AUC0–t), mean residence time (MRT) and variance of residence time (VRT).
Results and Discussion
Preparation and Characterization of PLGA NPs
The lactide/glycolide weight ratios (LA/GA ratios) and molecular weights (MWs) of PLGA may affect the particle size, biodegradation and even corneal permeation of the NPs. Therefore, we formulated PLGA NPs with LA/GA ratios of 50:50, 75:25 and 90:10, and MWs 10 kDa, 20 kDa and 30 kDa, and analyzed the physicochemical properties of each. As shown in Figure 1, the D90 or cumulative particle size distribution of 90% NPs (LA/GA 50:50) increased with their MWs, whereas the cumulative drug release rate (%) at 24 h showed an inverse correlation with the MW of PLGA (P < 0.05). A higher MW increased the inherent viscosity of the polymer network. Therefore, the net sheer stress upon PLGA polymers is reduced during preparation of NPs, resulting in larger particle size of NPs.23,30,31 With a constant LA/GA ratio, a higher MW of PLGA impedes formation of sufficient water-filled pores on NPs surface compared to a PLGA with lower MW, which limits drug diffusion through the NPs matrix and therefore protracts drug release.32,33 However, the release kinetics of all PLGA NPs with different MWs followed the Higuchi model, indicating that the MWs did not significantly affect release kinetics.
 |
Figure 1 The effects of PLGA MWs on the NPs (LA/GA 50:50) particle size and in vitro drug release. |
The parameters of PLGA NPs (MW 10 kDa) like cumulative drug release rate (%) and cumulative corneal permeation per unit area (μg·cm−2) varied with the LA/GA ratios (Figure 2). The cumulative drug release rate (%) over 12 h decreased continuously from 55.73% to 48.98% (P < 0.05) when the LA/GA ratio was increased from 50:50 to 90:10, and a moderate decrease in the cumulative corneal permeation per unit area (μg·cm−2) was also observed over the same duration. PLGA is a copolymer of a hydrophobic (lactide) and a hydrophilic (glycolide) monomer in the optimal ratio that allows drug release from PLGA NPs.34,35 Increased proportion of lactide enhances the hydrophobicity of the NPs, which slows degradation of NPs in water and reinforces the drug-NPs affinity.35 Accordingly, NPs with low LA/GA ratio release a higher amount of the drug compared to NPs with high LA/GA ratio in order to achieve a relatively higher corneal permeation. Regardless of the LA/GA ratios and MW, however, the PDI values of the PLGA NPs were within 0.2 and did not differ significantly. But the zeta potential distribution of PLGA NPs with LA/GA ratio 90:10 was the largest, indicating a risk of nano-system instability.
 |
Figure 2 The effects of PLGA LA/GA ratios on in vitro drug release and corneal permeability of the NPs (MW 10,000). |
In conclusion, the PLGA NPs with LA/GA ratio 50:50 and MW 10 kDa were the smallest and showed fastest drug release and corneal permeation, and were therefore selected for subsequent analyses.
Evaluation of Drug-Polymer Interactions
The physical state of drug-loaded PLGA NPs was determined by DSC analysis of pure drug/nanoparticles physical mixtures and drug-loaded nanoparticles. As shown in Figure 3, a single sharp endothermic melting peak of triamcinolone acetonide was observed at 292.57 °C for both the pure drug and nanoparticles physical mixtures. However, the peak disappeared in the DSC spectra of all drug-loaded nanoparticles, indicating the absence of crystalline drug in the NPs or drug solvation in the amorphous carrier.36 We surmised therefore that triamcinolone acetonide existed in an amorphous or disorderly crystalline phase in dispersion or a solid solution state in the polymeric NPs.
 |
Figure 3 Differential scanning calorimetry thermograms of (A) PLGA NPs, (B) 2-HP-β-CD/PLGA NPs, (C) trehalose/PLGA NPs and (D) chitosan oligosaccharide/PLGA NPs. |
XRD analysis was performed to determine the crystalline structure of triamcinolone acetonide in the NPs. As shown in Figure 4, triamcinolone acetonide displayed numerous distinct peaks at 8–30°, indicating a crystalline nature.37 The physical mixtures also exhibited relatively weak peaks at the same positions, which could be attributed to the low drug content and the amorphous nature of PVA, resulting in poor crystallization. In contrast, no distinct triamcinolone acetonide peaks were seen in the different NPs, further confirming the amorphous state of the drug as observed in the DSC analysis.
 |
Figure 4 X-ray diffraction patterns of (A) PLGA NPs, (B) 2-HP-β-CD/PLGA NPs, (C) trehalose/PLGA NPs, and (D) chitosan oligosaccharide/PLGA NPs. |
The FT-IR spectra of the different formulations are shown in Figure 5. The triamcinolone acetonide spectrum showed four characteristic peaks in the range of 1708–1600 cm−1, indicating its C=C and C=O stretching vibrations.36,38 The physical mixture also had the same characteristic peaks in the same wavelength range. However, these peaks were absent in the different NPs, which instead showed a major peak at 1740 cm−1. This could be attributed to the formation of a hydrogen bond between the C=O group of triamcinolone acetonide with the polymer matrix.6,36 Combined with the XRD and DSC spectra, these results further indicate that triamcinolone acetonide is present in the nanoparticles in an amorphous form.
 |
Figure 5 Infrared spectrograms of (A) PLGA NPs, (B) 2-HP-β-CD/PLGA NPs, (C) trehalose/PLGA NPs, and (D) chitosan oligosaccharide/PLGA NPs. |
Characterization of Physical Features of Oligosaccharides/PLGA NPs
The particle size distribution and zeta potential of the NPs is shown in Figure 6. The diameters of the PLGA, 2-HP-β-CD/PLGA, trehalose/PLGA and chitosan oligosaccharide/PLGA NPs were (145.3 ± 1.36), (132.7 ± 3.34), (117.7 ± 3.10) and (137.1 ± 2.19) nm respectively. The size distribution was homogenous in all samples, with PDI values of all within 0.2. Previous studies show that drug carriers larger than 10 μm irritate the ocular surface and can even damage healthy tissue structure.39 Thus, all NPs were smaller than 200 nm and suitable for topical administration. Moreover, the small size of PLGA NPs and their surface-modified forms may offer several advantages like low toxicity, improved solubility, high surface area, dissolution rate and enhanced biodistribution.40–42
 |
Figure 6 Particle size distribution and zeta potential of (A) PLGA NPs, (B) 2-HP-β-CD/PLGA NPs, (C) trehalose/PLGA NPs, (D) chitosan oligosaccharide/PLGA NPs. |
The zeta potential of the PLGA, 2-HP-β-CD/PLGA, trehalose/PLGA and chitosan oligosaccharide/PLGA NPs were (−22.46 ± 4.76), (−19.09 ± 1.99), (−16.24 ± 4.65) and (−7.12 ± 0.75) mV respectively. Since the surface charge is an indicator of NP stability, the results indicate that PLGA NPs were the most stable, followed by trehalose/PLGA NPs, 2-HP-β-CD/PLGA NPs and chitosan oligosaccharide/PLGA NPs.39
The shape and surface morphology of the NPs were assessed by TEM. The particle size of the NPs as observed by TEM were different from that measured by DLS, which is likely due to the drying step in the former that results in NPs aggregation. As shown in Figure 7, all NPs were spherical and of uniform size.
 |
Figure 7 TEM images of (A) PLGA NPs, (B) 2-HP-β-CD/PLGA NPs, (C) trehalose/PLGA NPs, and (D) chitosan oligosaccharide/PLGA NPs. |
The EE (%) of the PLGA, 2-HP-β-CD/PLGA, trehalose/PLGA and chitosan oligosaccharide/PLGA NPs were (39.1 ± 1.41%), (63.2 ± 3.51%), (47.6 ± 3.01%) and (40.7 ± 1.92%) respectively. The highest EE (%) of 2-HP-β-CD/PLGA NPs suggested a synergy between 2-HP-β-CD and the PLGA matrix that increased drug loading capacity.43 However, the DL (%) of all PLGA NPs were similar, and that of 2-HP-β-CD/PLGA and trehalose/PLGA NPs were in fact lower compared to the PLGA NPs. This could be due to greater particle weight of 2-HP-β-CD and trehalose-modified PLGA NPs, which decreased the calculated DL (%) as per Equation (2).
Evaluation of in vitro Characteristics
Topical eye drops usually have low therapeutic efficacy due to poor ocular bioavailability.44 Therefore, it is essential to formulate NPs that can sustain a therapeutically effective concentration of the drug over a certain time window. The encapsulated triamcinolone acetonide showed an initial burst release from all oligosaccharides modified PLGA NPs, and the release of trehalose/PLGA and chitosan oligosaccharides/PLGA NPs decreased thereafter. During the first 4 hours, surface-embedded or loosely bound hydrophobic drug was released rapidly from the polymer phase into the simulated tear fluid. The steady diffusion of the buffer into the polymer matrix and subsequent swelling then released the PLGA-entrapped drug. The initial burst release can help achieve the therapeutic dose of the drug in minimal time, and the later slow release maintained effective drug concentration.
As shown in Figure 8A, 89.66% of triamcinolone acetonide was released from 2-HP-β-CD/PLGA NPs into the simulated tear fluid within 12 h compared to only 55.63%, 47.35% and 48.64% from the PLGA, trehalose/PLGA and chitosan oligosaccharide/PLGA NPs respectively. This could be due to the presence of a hydrophobic cavity in 2-HP-β-CD which can accommodate and increase aqueous solubility of triamcinolone acetonide. Furthermore, the amorphous, non-crystalline nature of 2-HP-β-CD further increased its solubility. In addition, the presence of cyclodextrin in the NPs can enhance drug permeability into the cornea by fusing with the cholesterol and phospholipids present in the epithelial cell membrane.45 Consistent with the in vitro observations, the cumulative amount of drug that permeated into the cornea from 2-HP-β-CD/PLGA NPs reached 28.71 ± 3.24 μg·cm−2 at 8 h, which was 5.71-, 3.55- and 3.25-fold higher compared to that from PLGA, trehalose/PLGA and chitosan oligosaccharide/PLGA NPs respectively (P < 0.05) (Figure 8B).
 |
Figure 8 (A) The cumulative percentage of triamcinolone acetonide released in artificial tear fluid. (B) Permeability of triamcinolone acetonide across excised rabbit corneas (■: PLGA NPs; ▼: 2-HP-β-CD/PLGA NPs; ▲: trehalose/PLGA NPs; ●: chitosan oligosaccharide/PLGA NPs). |
The effect of triamcinolone acetonide dose on the corneal permeation of different formulations were analyzed using a series of kinetic models (Table 1). The Ritger-Peppas kinetic model was best-fitted with the PLGA, trehalose/PLGA and chitosan oligosaccharide/PLGA NPs based on the correlation coefficient (R2), whereas 2-HP-β-CD/PLGA NPs was best fitted with a first-order kinetic model that is directly proportional to the drug concentration.46 The Ritger-Peppas kinetic model with n > 0.85 is possibly controlled by the skeleton dissolution release mechanism.25,47 Based on the results so far, we concluded that surface modification with 2-HP-β-CD can significantly enhance the ability of NPs to penetrate the cornea and increase drug concentration in the aqueous humor.
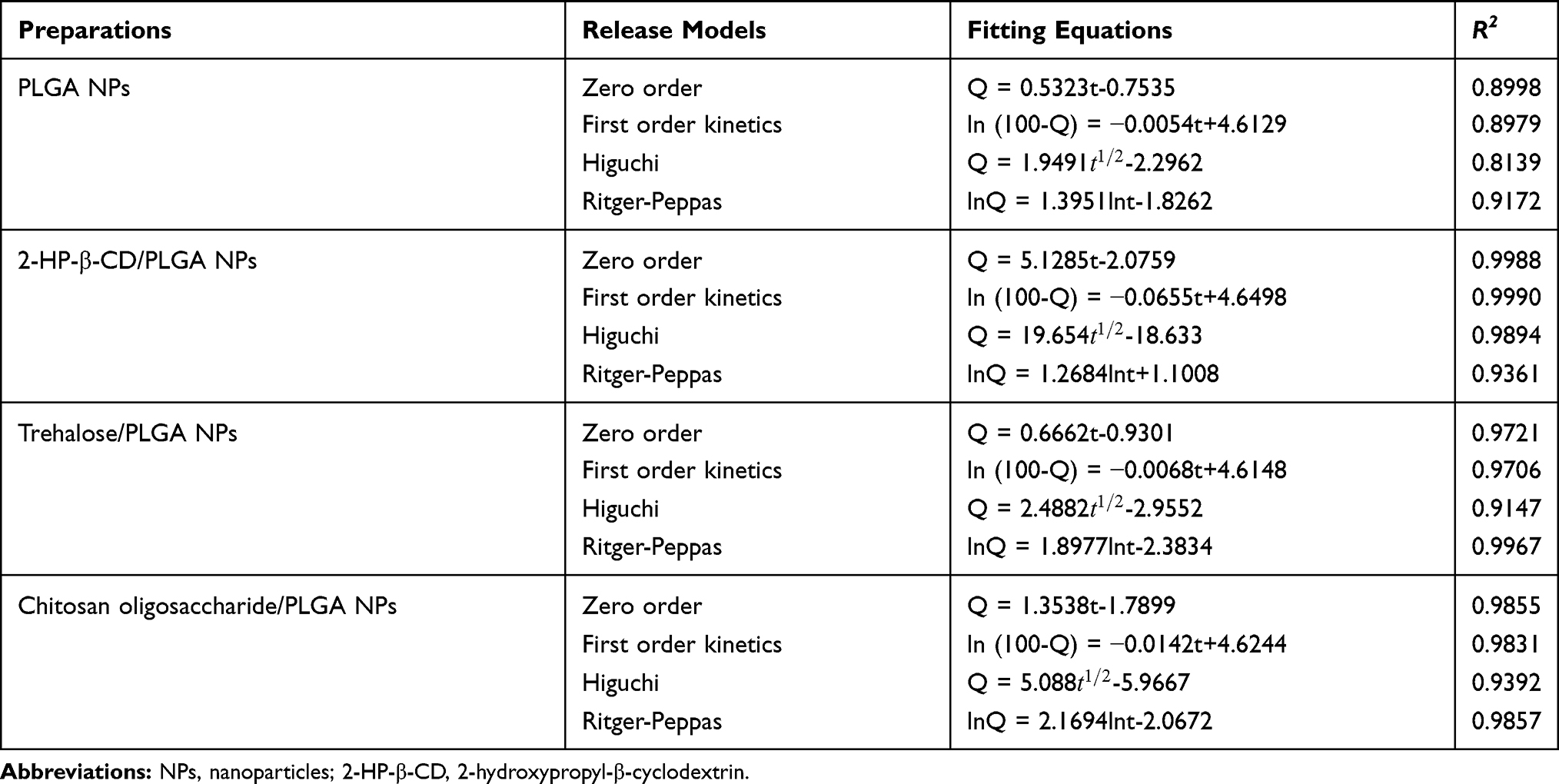 |
Table 1 Release Models of Triamcinolone Acetonide from Determination Preparations Across Corneas in vitro |
As shown in Table 2, 2-HP-β-CD/PLGA NPs had the greatest  thus confirming its optimum corneal permeability. In addition, the
thus confirming its optimum corneal permeability. In addition, the  for 2-HP-β-CD/PLGA NPs, chitosan oligosaccharide/PLGA NPs and trehalose/PLGA NPs were respectively 4.76, 1.38 and 1.33 folds higher compared to that of PLGA NPs (P < 0.05). The
for 2-HP-β-CD/PLGA NPs, chitosan oligosaccharide/PLGA NPs and trehalose/PLGA NPs were respectively 4.76, 1.38 and 1.33 folds higher compared to that of PLGA NPs (P < 0.05). The  value of 2-HP-β-CD/PLGA NPs indicated that they can form a drug reservoir by adhering to the corneal epithelium and allowing sustained drug release. Therefore, administering triamcinolone acetonide via 2-HP-β-CD/PLGA NPs can prolong its corneal retention and permeation.
value of 2-HP-β-CD/PLGA NPs indicated that they can form a drug reservoir by adhering to the corneal epithelium and allowing sustained drug release. Therefore, administering triamcinolone acetonide via 2-HP-β-CD/PLGA NPs can prolong its corneal retention and permeation.
 |
Table 2 Permeation Parameters of the Different Preparations Through the Excised Corneas |
Pharmacokinetic Study of Aqueous Humor
Microdialysis is a well-established, minimally invasive technique for in vivo pharmacokinetics assays in different tissues, including the eye. The ocular pharmacokinetics of triamcinolone acetonide was accordingly analyzed in anesthetized rabbits. The time-concentration profiles of the drug in aqueous humor after topical application of the different formulations are shown in Figure 9, and the corresponding kinetic parameters are summarized in Table 3. Continuous dilution by tears can disrupt the nanocarriers in contact with ocular membranes, impeding more drugs from penetrating the cornea and releasing in the aqueous humor to implement their therapeutic effects.44,48 Taken together, 2-HP-β-CD/PLGA is a highly promising nanocarrier for targeted ocular drug delivery and sustained drug release.
 |
Table 3 Pharmacokinetics Parameters of Triamcinolone Acetonide in Aqueous Humor After Topical Instillation |
 |
Figure 9 Pharmacokinetic profiles of the NPs in aqueous humor. (■: PLGA NPs; ▼: 2-HP-β-CD/PLGA NPs; ▲: trehalose/PLGA NPs; ●: chitosan oligosaccharide/PLGA NPs). |
In vivo Ocular Compatibility
The Draize test showed that neither single nor multiple-dosing administration caused any abnormities in the cornea, iris or conjunctiva of the rabbit eyes. Moreover, the corneal sections of eyeballs post-treated with different oligosaccharide-modified NPs did not show any significant differences compared to PLGA NPs (control) (Figure 10), thus confirming that all PLGA NPs were non-irritant and completely biocompatible.
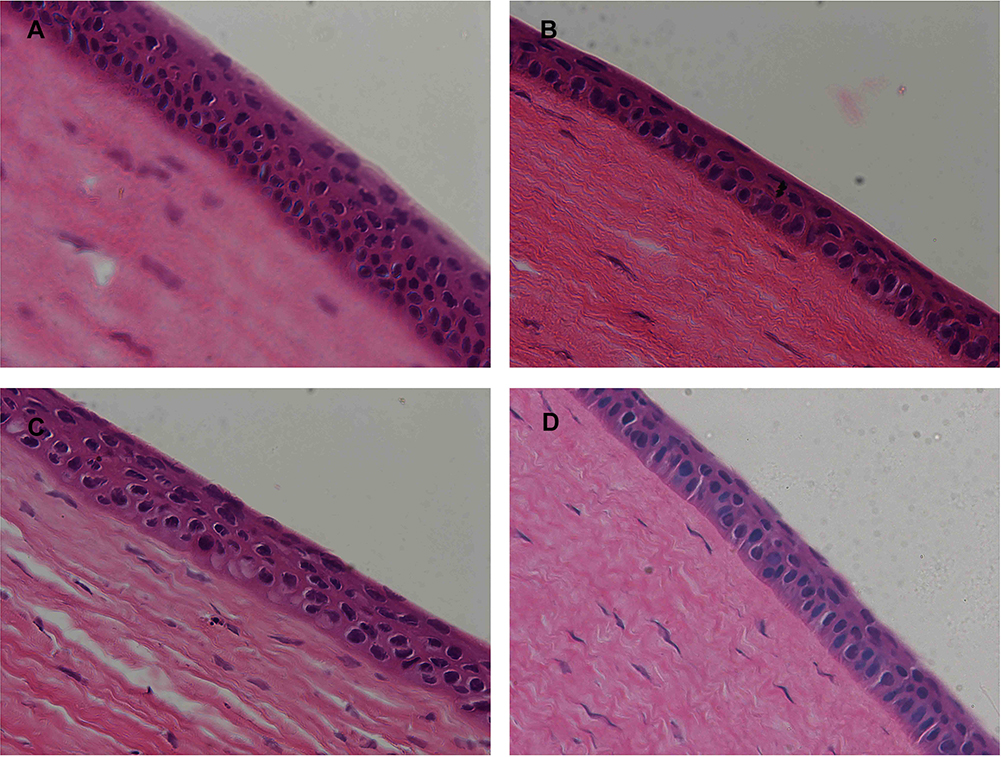 |
Figure 10 Histopathological images of rabbit corneas treated with (A) PLGA NPs, (B) 2-HP-β-CD/PLGA NPs, (C) trehalose/PLGA NPs, and (D) chitosan oligosaccharide/PLGA NPs. |
Conclusion
Mucoadhesiveness in ocular drug delivery is a robust approach that prolongs drug retention and increases the membrane permeability and intracellular uptake of drugs. Polymers like polylactic-co-glycolic acid (PLGA) and 2-HP-β-CD are often used to overcome the ocular barriers, and they also help achieve mucoadhesive ocular controlled drug delivery. The present study aimed to enhance ocular bioavailability of triamcinolone acetonide by preparing and evaluating 2-HP-β-CD/PLGA NPs. 2-HP-β-CD/PLGA NPs displayed rapid drug release and optimum corneal permeability, and the higher AUC and Cmax of 2-HP-β-CD/PLGA NPs compared to PLGA, trehalose/PLGA and chitosan oligosaccharide/PLGA NPs have indicated that 2-HP-β-CD/PLGA NPs can significantly enhance the bioavailability of triamcinolone acetonide.
Funding
This work was supported by the Guangdong “Climbing” Program for Undergraduates (2021); The Natural Science Foundation of Guangdong Province (Grant Number 2019A1515011018); The planned Science and Technology Project of Guangdong Province (2017B020231001); Guangdong Graduate Education Innovation Program: Project Funded by Guangdong Province Joint Training Graduate Demonstration Base (Guangzhou General Pharmaceutical Research Institute Co., Ltd.) Guangdong Pharmaceutical University “Innovation and Enhancing Project”; National Natural Science Foundation of China (81941010); National Innovation and Entrepreneurship Training Program for College Students (Grant Number 201910573004).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ge Jiang, Huanhuan Jia, Jindi Qiu and Junfeng Ban contributed equally to this study and should be regarded as joint first authors.
References
1. Garg P, Venuganti VVK, Roy A, Roy G. Novel drug delivery methods for the treatment of keratitis: moving away from surgical intervention. Expert Opin Drug Deliv. 2019;16(12):1381–1391. doi:10.1080/17425247.2019.1690451
2. Meng T, Kulkarni V, Simmers R, Brar V, Xu QG. Therapeutic implications of nanomedicine for ocular drug delivery. Drug Discov Today. 2019;24(8):1524–1538. doi:10.1016/j.drudis.2019.05.006
3. Singh RRT, Tekko I, McAvoy K, McMillan H, Jones D, Donnelly RF. Minimally invasive microneedles for ocular drug delivery. Expert Opin Drug Deliv. 2017;14(4):525–537. doi:10.1080/17425247.2016.1218460
4. Aksungur P, Demirbilek M, Denkbas EB, Vandervoort J, Ludwig A, Unlu N. Development and characterization of Cyclosporine A loaded nanoparticles for ocular drug delivery: cellular toxicity, uptake, and kinetic studies. J Control Release. 2011;151(3):286–294. doi:10.1016/j.jconrel.2011.01.010
5. Vasconcelos A, Vega E, Perez Y, Gomara MJ, Garcia ML, Haro I. Conjugation of cell-penetrating peptides with poly(lactic-co-glycolic acid)-polyethylene glycol nanoparticles improves ocular drug delivery. Int J Nanomedicine. 2015;10:609–631. doi:10.2147/IJN.S71198
6. Li F, Wen Y, Zhang Y, et al. Characterisation of 2-HP-β-cyclodextrin-PLGA nanoparticle complexes for potential use as ocular drug delivery vehicles. Artif Cells Nanomed Biotechnol. 2019;47(1):4097–4108. doi:10.1080/21691401.2019.1683567
7. Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly(D,L-lactide-co-glycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003;20(2):212–220. doi:10.1023/a:1022219003551
8. Qaddoumi MG, Ueda H, Yang J, Davda J, Labhasetwar V, Lee VH. The characteristics and mechanisms of uptake of PLGA nanoparticles in rabbit conjunctival epithelial cell layers. Pharm Res. 2004;21(4):641–648. doi:10.1023/b:pham.0000022411.47059.76
9. Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13(12):1838–1845. doi:10.1023/a:1016085108889
10. Tobio M, Sanchez A, Vila A, et al. The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloids Surf B Biointerfaces. 2000;18(3–4):315–323. doi:10.1016/s0927-7765(99)00157-5
11. Vila A, Gill H, McCallion O, Alonso MJ. Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J Control Release. 2004;98(2):231–244. doi:10.1016/j.jconrel.2004.04.026
12. Cabrera FJ, Wang DC, Reddy K, Acharya G, Shin CS. Challenges and opportunities for drug delivery to the posterior of the eye. Drug Discov Today. 2019;24(8):1679–1684. doi:10.1016/j.drudis.2019.05.035
13. Arafa MG, Girgis GNS, El-Dahan MS. Chitosan-coated PLGA nanoparticles for enhanced ocular anti-inflammatory efficacy of atorvastatin calcium. Int J Nanomedicine. 2020;15:1335–1347. doi:10.2147/IJN.S237314
14. Smart JD. The basics and underlying mechanisms of mucoadhesion. Adv Drug Deliv Rev. 2005;57(11):1556–1568. doi:10.1016/j.addr.2005.07.001
15. Junga T, Kamm W, Breitenbacha A, Kaiserlingc E, Xiaoc JX, Kissel T. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur J Pharm Biopharm. 2000;50(1):147–160. doi:10.1016/S0939-6411(00)00084-9
16. Mathiowitz E, Jacob JS, Jong YS, et al. Biologically erodable microsphere as potential oral drug delivery system. Nature. 1997;386(6623):410–414. doi:10.1038/386410a0
17. Sayed S, Elsayed I, Ismail MM. Optimization of beta-cyclodextrin consolidated micellar dispersion for promoting the transcorneal permeation of a practically insoluble drug. Int J Pharm. 2018;549(1–2):249–260. doi:10.1016/j.ijpharm.2018.08.001
18. Li J, Tan G, Cheng B, Liu D, Pan W. Transport mechanism of chitosan-N-acetylcysteine, chitosan oligosaccharides or carboxymethyl chitosan decorated coumarin-6 loaded nanostructured lipid carriers across the rabbit ocular. Eur J Pharm Biopharm. 2017;120:89–97. doi:10.1016/j.ejpb.2017.08.013
19. Ding D, Zhu Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater Sci Eng C Mater Biol Appl. 2018;92:1041–1060. doi:10.1016/j.msec.2017.12.036
20. Chen SH, Jia HH, Cui XH, et al. Characterization of stimuli-responsive and cross-linked nanohydrogels for applications in ophthalmiatrics therapy. Appl Nanosci. 2020. doi:10.1007/s13204-020-01450-7
21. Liu Y, Wang Y, Yang J, Zhang H, Gan L. Cationized hyaluronic acid coated spanlastics for cyclosporine A ocular delivery: prolonged ocular retention, enhanced corneal permeation and improved tear production. Int J Pharm. 2019;565:133–142. doi:10.1016/j.ijpharm.2019.05.018
22. Tipnis NP, Shen J, Jackson D, Leblanc D, Burgess DJ. Flow-through cell-based in vitro release method for triamcinolone acetonide poly (lactic-co-glycolic) acid microspheres. Int J Pharm. 2020;579:119130. doi:10.1016/j.ijpharm.2020.119130
23. Song X, Zhao Y, Wu W, et al. PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: systematic study of particle size and drug entrapment efficiency. Int J Pharm. 2008;350(1–2):320–329. doi:10.1016/j.ijpharm.2007.08.034
24. Yu M, Yuan W, Li D, Schwendeman A, Schwendeman SP. Predicting drug release kinetics from nanocarriers inside dialysis bags. J Control Release. 2019;315:23–30. doi:10.1016/j.jconrel.2019.09.016
25. Wen Y, Ban J, Mo Z, et al. A potential nanoparticle-loaded in situ gel for enhanced and sustained ophthalmic delivery of dexamethasone. Nanotechnology. 2018;29(42):425101. doi:10.1088/1361-6528/aad7da
26. Hahne M, Zorn-Kruppa M, Guzman G, et al. Prevalidation of a human cornea construct as an alternative to animal corneas for in vitro drug absorption studies. J Pharm Sci Us. 2012;101(8):2976–2988. doi:10.1002/jps.23190
27. Juretic M, Cetina-Cizmek B, Filipovic-Grcic J, Hafner A, Lovric J, Pepic I. Biopharmaceutical evaluation of surface active ophthalmic excipients using in vitro and ex vivo corneal models. Eur J Pharm Sci. 2018;120:133–141. doi:10.1016/j.ejps.2018.04.032
28. Wang JL, Zhao F, Liu R, et al. Novel cationic lipid nanoparticles as an ophthalmic delivery system for multicomponent drugs: development, characterization, in vitro permeation, in vivo pharmacokinetic, and molecular dynamics studies. Int J Nanomed. 2017;12:8115–8127. doi:10.2147/Ijn.S139436
29. Zhang WJ, Li XD, Ye TT, et al. Nanostructured lipid carrier surface modified with Eudragit RS 100 and its potential ophthalmic functions. Int J Nanomed. 2014;9:4305–4315. doi:10.2147/Ijn.S63414
30. Kohno M, Andhariya JV, Wan B, et al. The effect of PLGA molecular weight differences on risperidone release from microspheres. Int J Pharmaceut. 2020;582:119339. doi:10.1016/j.ijpharm.2020.119339
31. Song XR, Zhao Y, Hou SX, et al. Dual agents loaded PLGA nanoparticles: systematic study of particle size and drug entrapment efficiency. Eur J Pharm Biopharm. 2008;69(2):445–453. doi:10.1016/j.ejpb.2008.01.013
32. Mylonaki I, Allemann E, Delie F, Jordan O. Imaging the porous structure in the core of degrading PLGA microparticles: the effect of molecular weight. J Control Release. 2018;286:231–239. doi:10.1016/j.jconrel.2018.07.044
33. Fredenberg S, Wahlgren M, Reslow M, Axelsson A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems-a review. Int J Pharm. 2011;415(1–2):34–52. doi:10.1016/j.ijpharm.2011.05.049
34. Mir M, Ahmed N, Rehman AU. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf B Biointerfaces. 2017;159:217–231. doi:10.1016/j.colsurfb.2017.07.038
35. Mittal G, Sahana DK, Bhardwaj V, Kumar MNVR. Estradiol loaded PLGA nanoparticles for oral administration: effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J Control Release. 2007;119(1):77–85. doi:10.1016/j.jconrel.2007.01.016
36. Abou-ElNour M, Ishak RAH, Tiboni M, et al. Triamcinolone acetonide-loaded PLA/PEG-PDL microparticles for effective intra-articular delivery: synthesis, optimization, in vitro and in vivo evaluation. J Control Release. 2019;309:125–144. doi:10.1016/j.jconrel.2019.07.030
37. Nirbhavane P, Sharma G, Singh B, et al. Triamcinolone acetonide loaded-cationic nano-lipoidal formulation for uveitis: evidences of improved biopharmaceutical performance and anti-inflammatory activity. Colloids Surf B Biointerfaces. 2020;190:110902. doi:10.1016/j.colsurfb.2020.110902
38. Meng Y, Sun S, Li J, et al. Sustained release of triamcinolone acetonide from an episcleral plaque of multilayered poly-epsilon-caprolactone matrix. Acta Biomater. 2014;10(1):126–133. doi:10.1016/j.actbio.2013.09.022
39. Gonzalez-Pizarro R, Carvajal-Vidal P, Halbault Bellowa L, Calpena AC, Espina M, Garcia ML. In-situ forming gels containing fluorometholone-loaded polymeric nanoparticles for ocular inflammatory conditions. Colloids Surf B Biointerfaces. 2019;175:365–374. doi:10.1016/j.colsurfb.2018.11.065
40. Ridolfo R, Tavakoli S, Junnuthula V, Williams DS, Urtti A, van Hest JCM. Exploring the impact of morphology on the properties of biodegradable nanoparticles and their diffusion in complex biological medium. Biomacromolecules. Epub 2020 Jun 8. doi:10.1021/acs.biomac.0c00726.
41. Zhang W, Liu CP, Chen SQ, et al. Poloxamer modified florfenicol instant microparticles for improved oral bioavailability. Colloids Surf B Biointerfaces. 2020;193:111078. doi:10.1016/j.colsurfb.2020.111078
42. Agrahari V, Burnouf PA, Burnouf T, Agrahari V. Nanoformulation properties, characterization, and behavior in complex biological matrices: challenges and opportunities for brain-targeted drug updates delivery applications and enhanced translational potential. Adv Drug Deliver Rev. 2019;148:146–180. doi:10.1016/j.addr.2019.02.008
43. Mura P. Advantages of the combined use of cyclodextrins and nanocarriers in drug delivery: A review. Int J Pharmaceut. 2020;579:119181. doi:10.1016/j.ijpharm.2020.119181
44. Janagam DR, Wu LF, Lowe TL. Nanoparticles for drug delivery to the anterior segment of the eye. Adv Drug Deliver Rev. 2017;122:31–64. doi:10.1016/j.addr.2017.04.001
45. Hammoud Z, Khreich N, Auezova L, Fourmentin S, Elaissari A, Greige-Gerges H. Cyclodextrin-membrane interaction in drug delivery and membrane structure maintenance. Int J Pharmaceut. 2019;564:59–76. doi:10.1016/j.ijpharm.2019.03.063
46. Costa P, Manuel J, Lobo S. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. doi:10.1016/S0928-0987(01)00095-1
47. Siepmann J, Siepmann F. Mathematical modeling of drug delivery. Int J Pharm. 2008;364(2):328–343. doi:10.1016/j.ijpharm.2008.09.004
48. Jumelle C, Gholizadeh S, Annabi N, Dana R. Advances and limitations of drug delivery systems formulated as eye drops. J Control Release. 2020;321:1–22. doi:10.1016/j.jconrel.2020.01.057
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.