Back to Journals » The Application of Clinical Genetics » Volume 13
PHEX Gene Mutation in a Patient with X-Linked Hypophosphatemic Rickets in a Developing Country
Authors Forero-Delgadillo JM ![]() , Cleves D
, Cleves D ![]() , Ochoa V
, Ochoa V ![]() , Londoño-Correa H, Restrepo JM, Nastasi-Catanese JA
, Londoño-Correa H, Restrepo JM, Nastasi-Catanese JA ![]() , Pachajoa H
, Pachajoa H ![]()
Received 25 September 2019
Accepted for publication 23 December 2019
Published 13 February 2020 Volume 2020:13 Pages 57—62
DOI https://doi.org/10.2147/TACG.S232448
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Jessica María Forero-Delgadillo,1,2 Daniela Cleves,2,3 Vanessa Ochoa,1,2 Hernando Londoño-Correa,4 Jaime Manuel Restrepo,4 José Antonio Nastasi-Catanese,4,5 Harry Pachajoa2,5
1Pediatric Nephrology Fellow, Universidad Icesi-Fundación Valle de Lili, Cali, Colombia; 2Facultad de Ciencias de la Salud, Universidad Icesi, Cali, Colombia; 3Pediatrics Resident, Universidad Icesi-Fundación Valle de Lili, Cali, Colombia; 4Pediatric Nephrology Department, Fundación Valle del Lili, Cali, Colombia; 5Clinical Genetics Department, Fundación Valle del Lili, Cali, Colombia
Correspondence: Harry Pachajoa
Genetics Division, Fundación Valle del Lili, Carrera 98 # 18-49, Cali, Valle del Cauca, Colombia
Tel +57 2 3319090
Email [email protected]
Introduction: X-linked hypophosphatemic rickets is part of a larger group of hereditary diseases characterized by renal phosphate loss, which causes growth disorders, rickets, and osteomalacia. These conditions are characterized by disorders in phosphate equilibrium, which is essential for bone formation.
Case Report: A female patient presented with bone deformities of the inferior extremities, prominent joints, and loss of teeth. She received initial management with oral calcium and orthotics in inferior extremities, with poor clinical outcome. PHEX gene sequencing revealed a pathogenic variant c.1601C>T (p.Pro534Leu).
Discussion: XLHR is caused by mutations in the PHEX gene; to date, more than 460 mutations have been associated with the disease. Clinically, it is characterized by bowing of the lower extremities, decreased growth, musculoskeletal complaints, dental abscesses, and other clinical signs and symptoms of rickets.
Keywords: XLHR, Phex gene, Colombia, pediatric, rickets
Introduction
X-linked hypophosphatemic rickets (XLHR, MIM 307,800), also known as vitamin D resistant rickets, familial hypophosphatemic rickets, or phosphate diabetes, is part of a larger group of hereditary diseases characterized by renal phosphate loss causing growth disorders, rickets, and osteomalacia.1,2 These conditions are characterized by disorders in phosphate equilibrium, which is essential for bone formation.1,2 As in XLHR, autosomal dominant hypophosphatemic rickets (MIM 193,100), tumor-induced osteomalacia; hyperphosphatemic familial tumoral calcinosis (MIM 617,993), and oncogenic osteomalacia and hereditary hypophosphatemic rickets with hypercalciuria (MIM 241,530) are part of a group of conditions known as hypophosphatemic syndrome.1,3,4 It is estimated that XLHR has a prevalence of one in every 20,000 newborns and follows a dominant X-linked inheritance.5–8
XLHR is caused by mutations in the PHEX gene; to date, more than 460 mutations associated with this condition have been reported in the literature.5,9,10 The gene is located in Xp22.1.5–8 Clinically, the disease is characterized by bowing of the lower extremities, decreases in the growth rate after the child starts to walk, musculoskeletal complaints, stress fractures, dental abscesses, and clinical signs characteristic of rickets.3,4,11
We report the case of a pediatric Colombian patient with clinical, radiological, and molecular diagnoses of XLHR. Written parental consent was obtained in order to include case details and accompanying images.
Case Report
A 9-year-old patient presented with symptoms that began during the first year of life. The symptoms consisted of bone deformities in the lower limbs, coupled with prominent joints and loss of teeth (Figure 1A and B). She received management using calcium, and orthosis in the lower limbs, without improvement of the symptoms. She reported having a maternal, male cousin with a history of deformities in the lower extremities with no genetic or metabolic studies, as he lived in a rural, disperse area and had no access to specialty medicine. No other family members reported bone deformities, growth alterations or genetic studies done.
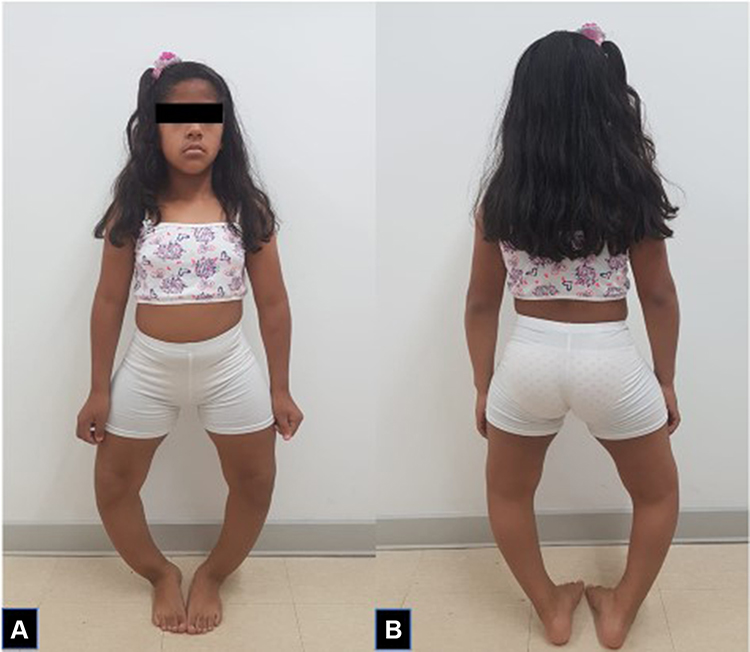 |
Figure 1 Anterior (A) and posterior (B) view of the patient at 15 years of age. |
Elevated alkaline phosphatase was documented, at 656 μ/L (maximum reference value of 269 μ/L), normal serum calcium of 8.9 mg/dL (reference range 8.4–10.2 mg/dL), and decreased serum phosphorus of 2.7 mg/dL (reference range 2.9–5.1 mg/dL). Additionally, decreased levels of 25-OH-vitamin D: 12.77 ng/mL (reference range: 30–70 ng/mL) and slightly elevated parathyroid hormone (PTH) levels of 67.24 pg/mL (reference range 12–65 pg/mL) were found. Renal function (creatinine 0.3 mg/dL, BUN 17), other electrolytes (sodium 138 mg/dL, potassium 4.4 mg/dL, and chloride 114 mg/dL), and the renal ultrasound were within normal limits. The urine sample did not show proteinuria or hematuria, and 24 hrs urine calcium was normal. The urine phosphorus/creatinine relation in urine was greater than 1 (reference range 0.05–0.25), and the maximum tubular excretion for phosphate relative to the glomerular filtration rate was 2.3 mg/dL (normal range 2.5–4.5 mg/dL).
X-rays of the long bones, taken at 4 years of age, showed widening of the proximal metaphyses of femurs and poor definition of bone contours (Figure 2A–C). Deformities in flexion of the distal diaphyses were observed, and there were similar findings in radii, ulnas, and tibias, as well as dorsal column deterioration (Figure 3A and B). All findings were compatible with a metabolic disorder such as rickets or osteomalacia.
 |
Figure 2 X-rays showing deterioration of bone structures of lower limbs, as well as the progression of the deformity ((A). 4 years of age (B) 11 years of age (C) 16 years of age). |
 |
Figure 3 X-rays showing deterioration of dorsal spine ((A) 4 years old (B) 11 years old). |
The clinical result raised the suspicion of XLHR, so sequencing of the gene PHEX was performed. Whole exome sequencing (WES) approach was performed. Approximately 214,000 exons from all encoded sequences were enriched for consensus using genomic DNA fragments of >340,000 probes against the human genome. The sequencing was performed on the Illumina HiSeq 4000 platform until approximately 100–130X read depth was attained. Only variants in the coding region and flanking intronic regions with a minor allele frequency <1% were evaluated. Selected MAFs were compared using the following datasets: 1000 Genomes Project Consortium, dbSNP, Exome Variant Server, and Exome Aggregation Consortium (ExAc). A pathogenic variant, c.1601C>T (p.Pro534Leu) heterozygote, was found. The finding was confirmed by Sanger sequencing and was compatible with the diagnosis of XLHR.
This mutation causes a substitution of semi-conserved amino acid, which can affect the secondary structure of PHEX protein. The position at which this substitution occurs is preserved in all species, and in silico analysis predicted that this variant is likely to be detrimental to the structure and function of the protein, with bioinformatics analyses predicting the changes to be either harmful or pathogenic.12 This mutation has been previously been described in the literature and has two entries in the ClinVar database as a pathogenic variant, confirming the diagnosis of XLHR.12–16
Discussion
The PHEX gene (MIM 307,800) has been implicated in XLHR, and more than 460 associated mutations associated with this condition have been reported in the literature.17 The variant found in the patient was described for the first time in 1997 by a multicenter group that performed genetic sequencing of patients with XHLR using an analog mouse gene and included centers in Germany, the United Kingdom, and the United States.18 It has also been the subject of many studies, mainly case reports that analyzed the genetic trees of families including individuals with a diagnosis of XLHR. One of these studies, published in 2016, analyzed data from 18 families in China with XLHR-affected family members and found that three members of the family with decreased growth rates and limb deformities had the c.1601C>T mutation.7,13–20 Other studies of Korean children with hypophosphatemic rickets have reported the same variant found in our patient.21 In Colombia, there is a subdiagnosis of this pathology and we found no reports in literature with genetic confirmation.
The PHEX gene encodes a metalloprotease, a member of the M13 family of zinc-dependent proteases of the cell membrane type II, and is expressed in the bone, teeth, lung, ovary, testes, and parathyroid gland.9,22,23 The PHEX gene has been reported to be involved in the regulation of fibroblast growth factor-23 (FGF-23) by degrading it into inactive fragments.9 Inactivation of the PHEX gene prevents the normal degradation of FGF-23, leading to a decrease in the reabsorption of renal phosphorus by suppression of the expression of the sodium–phosphorus co-transporters IIA and IIC in the apical surface of the proximal tubule of the nephron, causing an increase in the urinary excretion of phosphorus.9,22,23 FGF-23 also inhibits the action of 1-α-hydroxylase, which decreases 1α, 25 (OH) 2D3 and likewise the absorption of phosphate in the intestine and bone.9,22,23 There are other more complex models that explain the pathophysiology of the disease, including the participation not only of PHEX, FGF-23 but also of other molecules such as MEPE (an extracellular matrix phosphoglycoprotein).9,22,23
The kidney is the most important organ in the regulation of the balance of phosphorus. The rate of reabsorption of phosphorus depends on the physiological needs of the body, and 70% to 80% of reabsorption is performed in the proximal tubules, 5% to 10% in the distal tubules, and 2% to 3% in the collecting tubules.5,6 Sodium phosphate co-transporters are responsible for ensuring the reabsorption of phosphorus in the proximal tubules.5,6 There are three types of sodium–phosphate co-transporters: I, II, and III, of which IIA is responsible for 70% of phosphorus reabsorption in the proximal tubules.5,6 In patients with hypophosphatemic rickets, these mechanisms are altered, and phosphorus is not adequately reabsorbed.5,6
In addition to renal mechanisms for the reabsorption of phosphorus, intestinal absorption, mainly in the jejunum, is an important factor for the maintenance of phosphorus homeostasis.5,6 Intestinal absorption must be similar to the renal excretion in order to maintain a neutral balance and depends upon the action of hormones including the parathyroid hormone, FGF-23, and the active form of vitamin D, the 1α, 25-dihydroxyvitamin D3 (1α, 25 (OH) 2D3).5,6
The most common clinical manifestations are alterations in growth, leading to short stature, usually associated with the presence of bone deformities and bowing of the lower limbs, and symptoms of resistance to vitamin D.3,4,11 Bone pain, enthesopathy, and infrequent, mild muscle complaints have also been described as part of the clinical spectrum.3,4,7,11 As growth continues, deformities appear in the lower limbs such as genu valgus or genu varus, secondary to involvement, in the epiphyseal regions of the distal part of the femur and the proximal part of the tibia.3,4,7,11 Other findings include pectus carinatum; dental defects such as abscesses without caries; clinical signs characteristic of rickets such as rachitic rosary, craniotabes, Harrison’s groove (a horizontal channel in the lower end of the chest caused by the diaphragm that pushes the ribs inwards at its attachment); and in adults, the development of osteomalacia.3,4,7,11
Patients present with hypophosphatemia, elevated alkaline phosphatase, and normal serum calcium levels, whereas PTH may be normal or increased. Molecular analysis is used to confirm the diagnosis of hypophosphatemic rickets, especially if there are high levels of FGF-23.23,24
Treatment is based on phosphate supplements in the form of oral salts and metabolites, calcitriol or 1-hydroxylated vitamin D analogs.3–5,24 Several different adverse effects, such as gastrointestinal side effects and the risk of hypercalciuria with nephrocalcinosis, have been reported using this regime.3–5,24 Its effectiveness may be incomplete because of a transient increase in serum phosphorus and bone deformities, with residual short stature.3–5,24 At the beginning of 2018, the use of Burosumab® was approved by the FDA for the treatment of XLHR.25 Burosumab® is a recombinant human IgG1 monoclonal antibody that is designed to bind to FGF-23 and reduce its activity.26,27 Some studies have shown positive clinical impact with the use of Burosumab® at a dose of 1 mg/kg every 2 weeks, finding improvement in serum phosphorus levels and tubular reabsorption of phosphorus, as well as a decrease in bone pain and the severity of bone deformities.26,27 Burosumab® has been included as part of the XLHR management protocols.26,27
In Colombia, there is an underdiagnosis of this pathology and we lack molecular characterization of this population. This is in part due to the health system. Many patients cannot access specialty care medicine as they live in rural and disperse areas. We emphasize the importance of molecular and genetic confirmation of these pathologies in developing countries in order to offer adequate and early treatment that will improve patients’ quality of life and decrease the economic burden on the health system.
Conclusions
XLHR leads to compromised function of bone metabolism in most patients. Early identification and multidisciplinary diagnostic studies are vital for the establishment of appropriate management and to improve the quality of life of affected patients.
Ethics Approval
This case report was approved by Fundación Valle de Lili's Ethical Committee on Biomedical Research after parents signed written consent form which included publication of the case details and images.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Blackard WG, Robinson RRWJ. Familial hypophosphatemia. Report of a case, with observations regarding pathogenesis. N Engl J Med. 1962;266:899–905.
2. De Luca HF. Current concepts: vitamin D. N Engl J Med. 1969;281(20):1103–1104. doi:10.1056/NEJM196911132812006
3. Pavone V, Testa G, Gioitta S, Evola I, Francesco Roberto Avondo S, Sessa G. Hypophosphatemic rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol Orthop Traumatol. 2015;25(2):221–226. doi:10.1007/s00590-014-1496-y
4. Bergwitz C, Miyamoto K. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch. 2019;471(1):149–163. doi:10.1007/s00424-018-2184-2
5. Reference GH. Hereditary hypophosphatemic rickets. Genetics Home Reference; 2018. Available from: https://ghr.nlm.nih.gov/condition/hereditary-hypophosphatemic-rickets.
6. Velasquez-Jones L, Medeiros-Domingo M. Raquitismos hipofosfatémicos hereditarios. Bol Méd Hosp Infant México. 2013;70(6):421–431.
7. Li SS, Gu J-M, Yu W-J, He J-W, Fu W-Z, Zhang Z-L. Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic rickets. Int J Mol Med. 2016;38(6):1703–1714. doi:10.3892/ijmm.2016.2796
8. Ma S, Vega-Warner V, Gillies C, et al. Whole exome sequencing reveals novel PHEX splice site mutations in patients with hypophosphatemic rickets. PLoS One. 2015;10(6):e0130729. doi:10.1371/journal.pone.0130729
9. PHEX Gene. Genetics home reference; 2010. Available from: ghr.nlm.nih.gov/gene/PHEX.
10. HGMD Professional. Available from: https://portal.biobase-international.com.
11. Ruppe MD. X-Linked Hypophosphatemia. GeneReviews; 2012. Available from: http://www.ncbi.nlm.nih.gov/books/NBK83985/.
12. NM_000444.4(PHEX):c.1601C>T(p.Pro534Leu). ClinVar; 2018. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/280076/.
13. Filsetti D, Ostermann G, Von Bredow M, et al. Non-random distribution of mutations in the PHEX gene, and under-detected missense mutations at non-conserved residues. Eur J Hum Genet. 1999;7(5):615–619. doi:10.1038/sj.ejhg.5200341
14. Holm IA, Nelson AE, Robinson BG, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001;86(8):3889–3899. doi:10.1210/jcem.86.8.7761
15. Yavropoulou MP, Kotsa K, Psarrakou A, et al. Cinacalcet in hyperparathyroidism secondary to X-linked hypophosphatemic rickets: case report and brief literature review. Hormones. 2010;9(3):274–278. doi:10.14310/horm.2002.1149
16. Mumm S, Huskey M, Cajic A, et al. PHEX 3ʹ-UTR c.*231A>G near the polyadenylation signal is a relatively common, mild, American mutation that masquerades as sporadic or X-linked recessive hypophosphatemic rickets. J Bone Min Res. 2015;30(1):137–143. doi:10.1002/jbmr.2307
17. Francis F, Strom TM, Hennig S, et al. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res. 1997;7(6):573–585. doi:10.1101/gr.7.6.573
18. Zhang S, Zhang Q, Cheng L, et al. Analysis of PHEX gene mutations in three pedigrees affected with hypophosphatemic rickets. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018;35(5):644–647. doi:10.3760/cma.j.issn.1003-9406.2018.05.005
19. Song HR, Park JW, Cho DY, Jae HY, Yoon HR, Jung SC. PHEX gene mutations and genotype-phenotype analysis of Korean patients with hypophosphatemic rickets. J Korean Med Sci. 2007;22(6):981–986. doi:10.3346/jkms.2007.22.6.981
20. Dixon PH, Christie PT, Wooding C, et al. Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab. 1998;83(10):3615–3623. doi:10.1210/jcem.83.10.5180
21. Cho HY, Lee BH, Kang JH, Ha IS, Cheong H, Choi Y. A clinical and molecular genetic study of hypophosphatemic rickets in children. Pediatr Res. 2005;58(2):329–333. doi:10.1203/01.PDR.0000169983.40758.7B
22. Kinoshita Y, Saito T, Shimizu Y, et al. Mutational analysis of patients with FGF23-related hypophosphatemic rickets. Eur J Endocrinol. 2012;167(2):165–172. doi:10.1530/EJE-12-0071
23. Saito T, Nishii Y, Yasuda T, et al. Familial hypophosphatemic rickets caused by a large deletion in PHEX gene. Eur J Endocrinol. 2009;161(4):647–651. doi:10.1530/EJE-09-0261
24. Baroncelli G, Toschi B, Bertelloni S. Hypophosphatemic rickets. Curr Opin Endocrinol Diabetes Obes. 2012;19(6):460–467. doi:10.1097/MED.0b013e328358be97
25. Eisenman T. Press Announcements - FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia. FDA; 2018. Available from: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm604810.htm.
26. Carpenter TO, Whyte MP, Imel EA, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018;378(21):1987–1998. doi:10.1056/NEJMoa1714641
27. Kutilek S. Burosumab: a new drug to treat hypophosphatemic rickets. Sudan J Paediatr. 2017;17(2):71–73. doi:10.24911/SJP.2017.2.11
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.