Back to Archived Journals » Research and Reports in Biochemistry » Volume 6
Phenylalanyl-tRNA synthetase
Authors Chakraborty S, Banerjee R
Received 30 July 2015
Accepted for publication 7 November 2015
Published 8 June 2016 Volume 2016:6 Pages 25—38
DOI https://doi.org/10.2147/RRBC.S83482
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Nikolay Dokholyan
Shruti Chakraborty, Rajat Banerjee
Department of Biotechnology, University of Calcutta, Kolkata, West Bengal, India
Abstract: Phenylalalnyl-tRNA synthetase (PheRS), a member of class II aminoacyl-tRNA synthetases, catalyzes the synthesis of phenylalanyl-tRNAPhe (Phe-tRNAPhe). Hence, like other aminoacyl-tRNA synthetases, PheRS also plays a crucial role in the cellular translation process. Structural characterization demonstrates remarkable architectural diversity ranging from monomer to hetero-oligomer. Heterotetrameric PheRS contains an editing domain to proofread misincorporation of non-cognate amino acids. However, editing activity is absent in monomeric PheRS. PheRS has also shown some noncanonical functions, such as DNA binding properties, suggesting its involvement in complex regulatory pathways. Engineered mutants with relaxed substrate specificities of PheRS can be a promising tool for chemical and synthetic biology. Because of substantial structural variations among species due to evolutionary divergence, PheRS may be validated as a novel drug target. Human PheRS gene mutations have recently been implicated in several neurological disorders prompting structure-function studies to elucidate the molecular role of PheRS in such pathologies. In this review on PheRS, we will briefly cover all of the aforementioned aspects and our current understanding about the enzyme.
Keywords: PheRS, structural organization, editing, disease, unnatural amino acid, drug targeting
Introduction
Aminoacyl-tRNA synthetases (aaRSs) form a set of housekeeping enzymes that play a crucial role in cellular translation. aaRSs chemically ligate tRNAs with their cognate amino acids producing the aminoacyl-tRNAs (aa-tRNAs) that subsequently bind to the elongation factor, facilitating delivery to the ribosome for mRNA decoding.1 The charging of tRNA is a two-step process. In the first step, aaRS activates the cognate amino acid (in a tRNA-dependent or a tRNA-independent manner) in the presence of ATP to form aaRS-aminoacyladenylate complex (aaRS-AMP). In the second step, this activated aminoacyl moiety is transferred to the 2′ OH or 3′ OH group of the terminal adenine of the acceptor end of cognate tRNA.2–4

Twenty-two different amino acids are ligated to their corresponding cognate tRNAs by 22 different aaRSs. However, based on their sequence motifs and catalytic domain topologies, aaRSs can be divided into two classes, class I and class II (Table 1).5–8
 | Table 1 Classification of aaRSs |
Every aaRS contains a catalytic domain and an anticodon-binding domain (ABD). In addition to these two domains several aaRSs also possess an editing domain to maintain the translational fidelity. During evolution some aaRSs have also acquired additional domains that are mostly involved in noncanonical functions, like transcriptional regulation, signal transduction, angiogenesis, inflammation, etc.9
In mammalian systems, two separate sets of genes encode two distinct sets of aaRSs, cytosolic, and mitochondrial. Mitochondrial aaRSs (mtaaRSs) are also encoded in the nucleus, translated in the cytosol, and transported to the mitochondrial lumen. In humans, out of 37 aaRSs, 17 aaRSs are exclusively functional in the cytosol, including bifunctional glutamyl-prolyl-tRNA synthetase (GluProRS; responsible for charging both Glu and Pro), 18 are exclusively active in the mitochondria and 2 are dually localized (GlyRS and LysRS).10
In plant systems, a total of 45 aaRSs are encoded for three translationally active chambers – cytosol, mitochondria, and chloroplast. Among these 45 aaRSs, 24 are organellar aaRSs, of which 22 are found to be dually targeted – 17 are found both in the mitochondria and the plastid, 5 are shared between the mitochondria, and the cytosol and 2 are localized exclusively in the chloroplast.11,12
Protozoans also have three translationally active compartments; namely the cytosol, the mitochondria and the relic plastid or the apicoplast. aaRSs are encoded by nuclear genes and dually targeted to both the cytosol and the apicoplast. However, the mitochondrial fractions of aaRSs are found to be absent in protozoa, suggesting mitochondrial import of aa-tRNAs.13–15 Among the 35 aaRSs found in plasmodia, 16 are restricted to the cytoplasm, 15 for apicoplast, and 4 single copy aaRSs are shared between the cytoplasm and the apicoplast. Recently, it is reported that malarial parasite Plasmodium falciparum encodes a copy of phenylalalnyl-tRNA synthetase (PheRS) that is exclusively located in the mitochondria, apart from two other copies, one each for the cytosol and the apicoplast.16
aaRSs have been evaluated as a potential drug target for infectious diseases. Considering that drug resistance happens to be the key nemesis of current antibiotic therapeutics, aaRSs are very attractive as novel drug targets to the researchers, both academia and industries.17,18 Evolutionary divergence of prokaryotic and eukaryotic aaRSs allow orthogonal drug targeting; justifying pathogenic aaRSs as attractive choice for drug development.
In recent years, several disease-causing mutations have been described for both cytosolic aaRSs (ctaaRSs) and mtaaRSs encoding genes and mostly found to be associated with disorders of the nervous system.19 Whereas the mutations found in the ctaaRSs encoding genes are associated with Charcot–Marie–Tooth disease and related neuropathies, mutations in the mtaaRSs encoding genes are found to be associated with a wider range of diseases.10
In this review, we will discuss PheRS, which is responsible for producing Phe-tRNAPhe. PheRS has been identified as a member of subclass IIc based on the structural makeup of the catalytic domain and three signature motifs (1, 2, and 3) of α subunit. PheRS is the only exception among the class II aaRSs that attaches the amino acid moiety to the 2′ OH group of the terminal ribose of the acceptor end of the cognate tRNA.20 Here we provide an update on recent studies of PheRS including its role in the pathophysiology, as a protein engineering tool, as a drug target, with a focus on its structural, and functional diversity across the kingdom.
Structural diversity of PheRS
Structurally, PheRS is the most diverse enzyme of the aaRSs family. They show great variations in subunit structure/composition across the three kingdoms of life and exist in three forms – heterotetrameric (aβ2) in eubacacteria, heterotetrameric (α2β2) in archaea/eukaryotic cytoplasm, and monomeric in organelles.21 While heterotetrameric (α2β2) form is the most complex among the aaRSs family, monomeric organellar PheRS is the smallest that exhibits aminoacylation activity (Figure 1).7 Multiple sequence alignment shows that archaeal/eukaryotic cytosolic PheRS (ctPheRS) have originated from a single ancestor, while eubacterial PheRS is thought to be the predecessor of monomeric organellar PheRS.22
 | Figure 1 Schematic representation of localization and function of HctPheRS and HmtPheRS.Abbreviations: FARS, HctPheRS encoding gene; FARS2, HmtPheRS encoding gene; HctPheRS, human cytosolic phenylalanyl-tRNA synthetase; HmtPheRS, human mitochondrial phenylalanyl-tRNA synthetase; Phe, phenylalanine; Tyr, tyrosine. |
Heterotetrameric PheRS (α2β2): structural insight
The heterotetrameric organization (α2β2) is evolutionarily conserved among eubacterial and archaeal/eukaryotic ctPheRS. Whereas α subunit is responsible for catalytic activity, the major function of β subunit is tRNA recognition, binding, and editing of misacylated tRNAPhe.23,24 The eubacterial PheRS has 11 distinct structural domains: A0–A2 in the α subunit and B1–B8 in the β subunit (Table 2).25
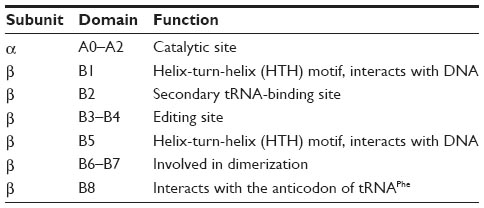 | Table 2 Structural organisation of eubacterial PheRS |
Both a and β subunits of archaeal/eukaryotic ctPheRS exhibit significant length difference from their bacterial counterparts: 1) the α subunit of archaeal/eukaryotic ctPheRS is ~150 residues longer at the N terminus than the bacterial one, 2) β subunit is ~200 amino acids shorter than its bacterial counterpart due to the absence of B2 and B8 domains in the former (Table 3).26 Apart from difference in length, there is also marked divergence in domain organization between the two kingdoms.27,28
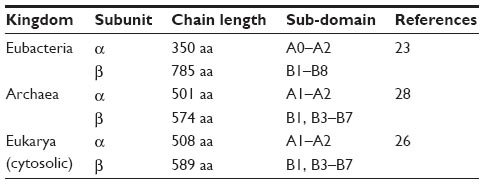 | Table 3 Domain organisation divergence between the kingdoms |
Conformational switch: “non-active” to “active” state
α or β subunit on its own is catalytically inactive.29,30 Affinity labeling to localize the substrate binding site in Escherichia coli PheRS (ecPheRS) along with crystallographic data of Thermus thermophilus PheRS (ttPheRS) complexed with phenylalanine (Phe) and phenylalaninyl-adenylate (PheOH-AMP), the synthetic analog of phenylalanyl-adenylate (Phe-AMP), corroborate that the α subunit of PheRS on its own is unable to carry out the first step of the aminoacylation reaction.31,32 Sequence analysis shows that the amino acid binding loop (residues α257–α263 of ttPheRS; TyrPheProPheValGluPro) of α subunit together with the metal-binding loop (residues β450–β465 of ttPheRS; ArgLeuAspLeuArgLeuGluGluAspLeuValGluGluValAspArg) of β subunit are evolutionarily retained among different species and form metal-mediated conserved α/β subunit interface. Conservation of these residues among different PheRS across species implies the importance of the metal-mediated α/β subunit interface in enzymatic activity, especially in the first step of the aminoacylation reaction.32 Crystal structure of ttPheRS/tRNAPhe complex suggests that the C-terminal domain of β subunit (B8 domain) plays a significant role in recognizing anticodon triplet of tRNAPhe.24 Comparative studies of the ttPheRS/PheOH-AMP and ttPheRS/tRNAPhe binary complexes and ttPheRS/PheOH-AMP/tRNAPhe ternary complex reveal that the structural rearrangements of the enzyme induced by small substrates enable the correct positioning for charging of tRNAPhe. Conformational transition of ttPheRS from “closed” to “open” conformation is driven by the PheOH-AMP and the tRNAPhe induced concerted movement of the motif 2 and helical loops that promote the second step of aminoacylation.33
The tRNAPhe binding pattern is different in two kingdoms, as the B8 domain involved in anticodon-binding is missing in the archaeal and the eukaryotic ctPheRS.34,35 Crystal structure of the human ctPheRS (HctPheRS) in complex with Phe reveals a novel structural module, containing three domains with helix-turn-helix (HTH) motif (DBD-1, DBD-2, and DBD-3) at the N-terminus of the α subunit, whose functional role in recognition and binding of tRNAPhe has been probed by truncation mutagenesis studies. Two mobile loops (motif 2 and helical loops of α subunit) play a key role in enzymatic function of HctPheRS, similar to ttPheRS (Figure 2). Conformation of the motif 2 loop is found to be similar for both the enzymes. Similarly, the helical loop in HctPheRS containing a highly conserved sequence [xProxxHisProAlaArgAsp(Met/x)(Trp/Gln/His)AspThrPhe] is important for the proper positioning of CCA end of tRNAPhe.27 The tRNAPhe makes contact with all four subunits of the enzyme. Two different α subunits of PheRS are involved in the acceptor end and the anticodon stem binding of a tRNAPhe.27,32 Crystal structures clearly depict that the heterotetrameric PheRS is able to bind two molecules of tRNAPhe simultaneously.36
 | Figure 2 Crystal structures of HctPheRS and ttPheRS. |
Organelle-specific PheRS: a chimera
Eukaryotic cells contain many sub-cellular compartments designed for performing specialized functions, eg, mitochondria, chloroplast, endoplasmic reticulum, golgi apparatus etc. Among them, the mitochondria and the chloroplast have their own genomes and encode a few proteins. The proteins responsible for organelle translation including aaRSs are encoded in the nucleus, synthesized in the cytosol and transported to the organelle.11,37 Due to the endosymbiotic origin of the mitochondria and the chloroplast, there is a notion that the prokaryotic aaRSs are the ancestor of the organelle-specific aaRSs.38,39 Whereas a wealth of literature exists on mtaaRSs, information about chloroplast-specific aaRSs is very limited.
The organelle-specific PheRS lacks the conventional heterotetrameric (α2β2) organization. It is monomer with a minimal set of structural domains. It is basically a chimera of the catalytic domain of the α subunit and B8 domain of the β subunit of bacterial PheRS. Sequence alignment of the mitochondrial PheRS (mtPheRS) family with prokaryotic PheRS shows that there is an insertion (35–70 amino acid residues) following motif 2. However, the function of this insertion is unknown.40,41
Like other mtaaRSs, mtPheRS is also encoded in the nucleus. It is translated in the cytoplasm as a pre-protein containing mitochondrial targeted sequence. The pre-protein is imported in the mitochondria through translocase of the inner membrane–translocase of the outer membrane complexes.42,43 Once inside the mitochondria, the mitochondrial targeted sequence is processed by the mitochondrial processing peptidase and mitochondrial intermediate peptidase.44,45 Postprocessing, mtPheRS must refold correctly within the organelle matrix to ensure proper enzymatic function. Defects in refolding can lead to mitochondrial dysfunction, either by triggering an unfolded protein response or by lowering the levels of stable folded active protein.46
HmtPheRS: conformational plasticity for enzymatic function
Human mtPheRS (HmtPheRS) is the smallest known enzyme among the aaRSs family and by far the most extensively characterized both structurally and functionally among the mtPheRS family. Crystal structures of HmtPheRS, complexed with Phe-AMP (Protein Data Bank accession number 3CMQ, at 2.2 Å resolution) and in complex with both tRNAPhe and Phe (Protein Data Bank accession number 3TUP, at a resolution of 3.0 Å) have been solved (Figure 3).47,48 The mature HmtPheRS consists of four major parts: the N-terminal region (residues 1–47), the catalytic domain (residues 48–289), the linker region (residues 290–322), and the C-terminal domain (residues 323–415). Crystal structure of HmtPheRS complexed with Phe-AMP reveals a “closed” conformation where the ABD located near the C-terminus overlaps with the acceptor stem of tRNAPhe, if the substrate is fitted in a manner similar to that observed in the bacterial PheRS-tRNAPhe complex. Hence, ABD must undergo a ~160° hinge-type rotation for correct positioning of tRNAPhe.47 HmtPheRS therefore exists in two conformations; the “closed” or non-active conformation where it cannot bind tRNAPhe and the “open” or active conformation where it can bind tRNAPhe. This conformational plasticity of HmtPheRS may be attributed to the presence of an extended flexible linker region.48,49
 | Figure 3 Crystal structures of HmtPheRS. |
Plasmodiam falciparum mtPheRS: an exception
A recent study has reported the presence of mtPheRS in malaria parasite, P. falciparum. Bioinformatic study reveals that the gene for mtPheRS is exclusive to malaria parasite among the apicomplexa phyla. Apicoplast PheRS (aPheRS) is a ~67 kDa protein with a signal and transit peptide for apicoplast targeting. mtPheRS is the smallest of the three PheRSs present in Plasmodia with a molecular mass of ~53 kDa. Both the organelle specific PheRSs contain a conserved aminoacylation domain at the N-terminus and an ABD at the C-terminus, however, the editing domain is absent. mtPheRS of P. falciparum also contains an insertion of ~45 residues in the aminoacylation domain, as reported for other mtPheRSs. P. falciparum contains two copies of tRNAPhe, one is encoded in the nucleus and the other in the apicoplast. On the contrary, Plasmodia mitochondrial genome lacks any copy of tRNA genes. Thus, it is presumed that like some other organisms, such as Leishmania, Trypanosoma, and Toxoplasma sp., mitochondrial tRNA import is also active in P. falciparum. Indeed, it has been shown that tRNAPhe is imported into mitochondria from the cytoplasm. Although, the charging status of imported tRNAPhe is uncertain, mtPheRS is able to charge cytoplasmic tRNAPhe in vitro.16
Editing activity: a canonical function of PheRS
Accurate translation of the genetic code is a high fidelity process. aaRSs play a crucial role in the quality control of mRNA decoding and maintaining the overall error rate of translation ~10–4.1 Two major checkpoints for maintaining the translation fidelity are : 1) aminoacylation of tRNA with the cognate amino acid by aaRS and 2) selection of aa-tRNA by the ribosome.50 aa-tRNA is delivered to ribosome by the elongation factor (EF) (EF thermo unstable [EF-Tu] in bacteria and EF-1α in archaea and eukaryotes).3,51 In case of class II aaRSs, once the misacylated tRNA escapes the editing step, EF-Tu binds it and delivers it to the ribosome and thus the noncognate amino acid is incorporated into the growing polypeptide chain. EF-Tu competes with the editing domain for aa-tRNA binding. Therefore, in class II aaRSs, editing activity is the sole quality control checkpoint that determines translational fidelity.52 A “double-sieve” model for aaRSs has been proposed to explain the high fidelity of aminoacylation process. According to this hypothesis, the first sieve is the amino acid recognition by the active site of the aaRSs, which acts as a coarse sieve and excludes most of the non-cognate amino acids that are larger than the cognate one. The editing site serves as a second fine sieve, which selectively hydrolyzes structurally similar, smaller near-cognate amino acids based on size and chemical character.53 While the first sieve is functional in all aaRSs, only approximately half of the aaRSs are found to harbor the second sieve.54 The editing site hydrolyzes the near-cognate amino acid either by hydrolyzing misactivated aminoacyl-adenylate – known as pre-transfer editing or the mischarged aa-tRNA – known as post-transfer editing.55 Inaccuracies in aminoacylation activity resulting from charging non-cognate tRNAs are less frequent, as selection of cognate tRNA is guided by the presence of tRNA identity elements.56
Structural analyses have revealed that the editing domains of class I aaRSs share structural homology. A unique insertion domain, called connective peptide 1 (CP1) domain is responsible for the editing activities in IleRS, ValRS, LeuRS via both pre- and post-transfer editing.4 In addition to CP1, LeuRS is found to harbor a second insertion domain, called CP2, which is involved in both aminoacylation and editing activities.57 The editing mechanism of MetRS of class I, however, differs from the other class I aaRS members. MetRS proofreads the non-cognate homocysteine via intramolecular cyclization by the aminoacylation active site.58 On the contrary, editing domains of class II aaRSs are structurally diverse. For example, ThrRS has an N-terminal N2 domain dedicated for editing activity,59 ProRS contains an insertion domain involved in editing,60 whereas, unlike other class II aaRSs, SerRS does not contain any distinct editing domain.61 The heterotetrameric PheRS from two kingdoms (eubacterial and archaeal/eukaryotic) possess an editing domain, located at the B3/B4 domain of β subunit, sharing no structural resemblance with any known editing domains.62 The monomeric organelle-specific counterpart, however, lacks any editing domain.63–65 While Phe is the cognate amino acid for PheRS, it can mischarge tRNAPhe with isosteric tyrosine (Tyr) and a non-cognate product, tyrosyl-tRNAPhe (Tyr-tRNAPhe), is synthesized at low frequency.64
Critical residues for editing activity of PheRS
There is an ambiguity over the predominant mode of PheRS editing activity. Both pre-transfer and post-transfer editing have been suggested to be operative in yeast PheRS.64,66 However, pre-transfer editing triggered by native tRNAPhe has been shown to predominate in yeast ctPheRS.64 In contrast, eubacterial PheRS hydrolyzes near-cognate Tyr at the post-transfer stage. Conservation mapping has suggested that the editing site is ~40 Å away from the synthetic site in PheRS62,67 and it has been suggested that cis editing is complemented by trans editing for efficient hydrolysis of the mischarged Tyr-tRNAPhe that dissociates from the enzyme. αAla294 in motif 3 of ecPheRS, present in amino acid binding pocket of the enzyme, has been found to be the responsible residue for amino acid specificity. The mutant αAla294Gly has been shown to have relaxed substrate specificity for para-substituted Phe analogs.68 Not only αAla294Gly mutant of ecPheRS, but wild-type ttPheRS is also able to activate p-chloro-l-Phe (p-Cl-Phe).67 The editing pocket recognizes near-cognate Tyr, not by size, but based on specific interactions with side chain hydroxyl groups.54 In eubacterial PheRS editing site, amino acid discrimination is achieved by two highly conserved motifs, “GlyValMetGlyGlyxxSer/Thr” and “GlnProxHisxPheAsp” and highly conserved amino acid residues, βArg244, βPhe263, βGly315, βGlu334, and βAla356. Mutagenesis studies supplemented with biochemical assays establish that all these residues are important for editing.62,67
Phylogenetic analysis of the editing domain shows very low sequence similarity between the two groups, eubacterial and archaeal/eukaryotic. However, there is some resemblance in the architecture of the editing site between these two groups. In case of P. horikoshii PheRS (phPheRS), βAsp234 residue, corresponding to βGlu334 of ttPheRS, has been shown to be important for preferentially selecting Phe over Tyr. Replacement of this residue with Ala allows Phe-tRNAPhe hydrolysis accompanied by a decrease in Tyr-tRNAPhe hydrolysis activity. Whereas, βHis261 (ttPheRS) residue involved in hydrolysis of Tyr, is found to be replaced by βAsn217 in phPheRS.35 In HctPheRS, βGlu254, and βAsn238 corresponding to βGlu334 and βHis261 in ttPheRS respectively, play a crucial role in recognition and editing of the Tyr moiety.27 The corresponding residues, which are crucial for editing in E. coli are βGlu334 and βHis26562 and in yeast are Aspβ243 and Hisβ158.65 The B2 domain has also been shown to play a key role in post-transfer editing. Mutagenesis studies have suggested that B2 domain may play a role in translocating aa-tRNA from synthetic site to editing site.25
Organelle-specific PheRS lacks editing activity
Organelle-specific counterparts of PheRS do not contain any domain homologous to B3/B4 domain involved in editing and are devoid of proofreading activity.65 In mammalian mitochondria only 13 proteins are encoded, whereas in plants more than 150 proteins are encoded within the organelles. These editing deficient organellar PheRSs may lead to mistranslation, giving rise to the accumulation of non-native proteins and compromise organellar homeostasis. Some post-translational check-point must operate to maintain the protein populations in functional forms within the organelles. It has been reported that a conserved protein degradation machinery operates as a major contributor of the protein quality control systems in the organelle. The key role of the proteases that are the prime components of the protein degradation machinery is rapid and selective proteolysis of misfolded, non-native proteins to ensure organelles’ viability.69,70
Error-prone editing of PheRS under oxidative stress
Reactive oxygen species (ROS) are known to modify the naturally occurring amino acids within the cell and the incorporation of these damaged amino acids into proteins may have pathological consequences in age related disorders, such as Alzheimer’s, Parkinson’s, atherosclerosis, and cataractogenesis.71–73 Exposure of Phe to ROS generates multiple isomers of Tyr: m-Tyr, o-Tyr, and the standard p-Tyr.74 On the other hand, the exposure of Tyr to ROS or catalytic conversion of Tyr by tyrosinase enzyme generates 3,4-dihydroxy-l-Phe, known as levodopa or l-DOPA.75 l-DOPA is the immediate biosynthetic precursor of the neurotransmitter dopamine and is used to treat Parkinson’s disease and dopamine-responsive dystonia.76,77 Incorporation of oxidized amino acids in proteins may lead to protein aggregation. Proteins bearing oxidized residues evade recognition by protein degradation machinery leading to accumulation of noncanonical proteins inside the cell.78 It has been shown that both HctPheRS and HmtPheRS catalyze attachment of m-Tyr to tRNAPhe. The affinity of HctPheRS toward m-Tyr is one magnitude lower than that of HmtPheRS and it discriminates more efficiently between the cognate Phe and non-cognate m-Tyr.74 Both the enzymes, however, cannot discriminate l-DOPA as efficiently as Tyr and thereby mischarge tRNAPhe. Unlike HmtPheRS, which lacks any editing activity, HctPheRS is able to proofread these ROS modified analogs of Phe, but in much lower level than the standard substrate Tyr. Kinetic experimental data show that specificity constants for l-DOPA over Phe for both HctPheRS and HmtPheRS are much higher than the estimated translational error rate (<10–4).79,80 The overall error rate of aa-tRNA synthesis is determined by two factors; aaRSs’ intrinsic proofreading activity and the ratio of cognate/non-cognate amino acid pool inside the cell.81 l-DOPA is the most widely used medicine for Parkinson’s disease. Increased availability of l-DOPA may lead to mis-incorporation of l-DOPA into proteins. l-DOPA bound proteins are majorly found in age-related disorders, such as atherosclerosis and cataract. l-DOPA can be further modified to dopaquinone, which crosslinks proteins forming covalent links with other biomolecules.75 These modified proteins form stable aggregates that are resistant to proteolysis82,83 and extremely cytotoxic causing extensive damage to other biological molecules.84 Thus, protein-bound l-DOPA may play a critical role in age-related disease progression by contributing to lysosomal membrane permeabilization-initiated apoptosis.85 Indeed, it has been found that the levels of protein-incorporated l-DOPA have increased in cells of l-DOPA treated patients.86 Moor et al have demonstrated that PheRS has the potential to misacylate tRNAPhe with l-DOPA and incorporate it into proteins instead of Phe.79 Increased levels of l-DOPA bound proteins in the cells of l-DOPA treated patients may be attributed to the inability of proofreading activity of HmtPheRS.80 Thus, long-term usage of l-DOPA may contribute to the toxic effects associated with l-DOPA treatment, further complicating the disease scenario.84,86
Noncanonical function of PheRS
It is postulated that aaRSs have acquired a number of noncanonical functions during evolution, while preserving their canonical aminoacylation activity. Functional expansion may occur either by the introduction of new structural domains to the existing enzyme or through transformation of idle domains into ones with new functions without disrupting the enzymatic activity. ttPheRS has been demonstrated to bind double-stranded DNA. A detailed work has shown that the B5 domain of β subunit containing HTH motif is involved in DNA binding. Specifically, amino acid residues Glu438-Gly439-Pro440-Thr441 and Pro447 are found to be responsible for DNA binding as demonstrated by mutagenesis studies. Although, the exact function is not understood, PheRS may contribute to some other biological functions, apart from protein translation, via DNA binding.87,88 ttPheRS also shares significant structural homology (a HTH motif and a Src-homology 3 fold) with E. coli biotin synthetase/repressor (BirA) protein, which acts as a transcriptional regulator.89,90 However, no studies have been carried out till now to elucidate the functional significance of these conserved motifs in PheRS.
PheRS as a tool in protein engineering
Selective incorporation of unnatural amino acids into proteins has become a useful tool for systematic engineering of proteins. This can be achieved either by site-specific method or by residue-specific method.91,92 The site-specific method exploits an orthogonal tRNA-aaRS pair that operates independently of the endogenous pair of synthetase-tRNA of host cell for introducing a particular unnatural amino acid at a specific site of a protein by decoding a reassigned nonsense codon in the gene of interest.93 The amber codon (UAG) is most frequently exploited among the three stop codons (amber–UAG, ochre–UAA, and opal–UGA), because it is least utilized in E. coli and most suppressor tRNA can efficiently recognize it.94 Residue-specific method is comparatively less complicated in a way that it requires only engineering of aaRSs to expand the specificity of their amino acid recognition site. In this way, several analogs of cognate amino acid can be incorporated into a protein. Both the methods have been manipulated to introduce various analogs of Phe into protein. For in vitro site-specific incorporation of unnatural amino acid, E. coli cell-free translational system has been used containing a gene with a nonsense codon mutation and preacylated suppressor tRNA. Preacylation of suppressor tRNA with p-iodo-l-Phe (p-I-Phe) is carried out with αAla294Gly mutant of ecPheRS that has relaxed substrate specificity.95 In vivo incorporation of p-bromo-l-Phe (p-Br-Phe) has been carried out using yeast suppressor tRNA (tRNAPheCUA) and αThr415Gly mutant of yeast PheRS in E. coli. αAla294 mutant of ecPheRS has already been shown to have relaxed substrate specificity and is able to incorporate p-Cl-Phe into recombinant protein.68 A computer simulation study corroborates the finding that αThr251 and αAla294 of E. coli (corresponding to αVal261 and αAla314 in ttPheRS) are crucial for amino acid specificity and replacement of both the amino acids with Gly enables efficient incorporation of p-acetyl-l-Phe in vivo replacing Phe.96 Sequence alignment shows αThr251 in ecPheRS is equivalent to αThr415 in yeast PheRS. A high-throughput screening method has been developed to identify yeast PheRS variant from a library with high substrate-specificity for a Phe analog. The approach is to obtain yeast PheRS variants from the yeast PheRS library, which leads to loss of fluorescence of green fluorescence protein upon efficient incorporation of 2-l-naphthylalanine, a Phe analog, into multiple sites of green fluorescence protein by yeast PheRS, exploiting the degeneracy of the Phe codons. A yeast PheRS variant (yPheRS_naph: containing Asn412Gly, Thr415Gly, Ser418Cys, and Ser437Phe mutations) has been identified, which shows 60-fold increased selectivity toward 2-l-naphthylalanine vs tryptophan compared to αThr415Gly variant of yeast PheRS.97
In contrast to site-specific method that requires multiple gene manipulation, residue-specific method is a much simpler way for unnatural amino acid incorporation into protein. This can be achieved by enlarging the amino acid binding pocket of aaRSs by point mutations, which have relaxed substrate specificity toward structurally similar amino acid analogs.98 This strategy has been used in eukaryotic organism yeast99 as well as in mammalian cells.100 This method also has been utilized to incorporate Phe analogs, replacing Phe, in domesticated silkworm, Bombyx mori. Phe is an essential amino acid for B. mori101 and is incorporated in the major protein component of B. mori silk in a periodic manner,102 making it an appropriate target for unnatural amino acid incorporation. Silk proteins produced by B. mori have a wide range of industrial applications103–105 and this strategy will improve its application further. αAla450Gly mutant of B. mori PheRS (BmPheRS) has been generated and incorporation of Phe analogs, p-Cl- and p-Br-Phe, into proteins by this mutant are checked both in vitro and in vivo (in BmN, a B. mori cell line). Although, in vitro assay shows aminoacylation of B. mori tRNAPhe with both p-Cl- and p-Br-Phe, incorporation of only p-Cl-Phe is observed in vivo.106,107 Another point mutation, replacing αThr407 (corresponding to αThr415 residue of yeast PheRS) with Ala or Gly, has been introduced into BmPheRS to relax the amino acid specificity further and to increase the incorporation efficiency of unnatural amino acid into protein. Aminoacylation capabilities of αAla450Gly, αThr407Ala and αThr407Gly BmPheRS mutants are checked both in vitro and in vivo in BmN using six para-substituted Phe analogs. The data show that αThr407Ala mutant is able to incorporate p-Br-Phe in proteins in vivo. Further, p-Br-Phe, p-cyano-Phe, p-azido-Phe, and p-I-Phe can be incorporated in proteins in vivo by lowering the concentration of competing amino acid Phe in culture media.108 This strategy has been extended to generate transgenic B. mori expressing the αAla450Gly mutant in silk glands. p-Cl-, p-Br-, and p-azido-substituted analogs are efficiently incorporated into silk fibroin by adding them to the diet and decreasing the Phe content.109
PheRS as a therapeutic target
The dramatic increase of multidrug resistance in pathogens against existing drugs drives researchers to search for novel targets that can be exploited to develop novel therapeutics.17,110 Several reasons have made aaRS an attractive target for the development of antimicrobial agents, i) they are indispensable for cellular function, ii) abundant genetic and protein structural information available, iii) due to their conserved nature, unlikely to undergo mutations and develop resistance upon being targeted, iv) sequence and structural divergence among the kingdoms facilitate easy targeting of pathogenic aaRSs with high selectivity and hence reduced toxicity.111,112 Although, several antimicrobial agents against aaRSs have been reported and patented so far, mupirocin (Bactroban) is the only commercially available antibacterial agent, used against methicillin-resistant Staphylococcus aureus. It selectively inactivates eubacterial IleRS.113 The structural differences between eukaryotic and prokaryotic PheRS lead to search for novel PheRS inhibitors with broad spectrum efficacy and high selectivity. A series of compounds have been isolated, namely, spirocyclic furans and pyrrolidines, ethanolamines, and benzyl phenyl ethers, but with nominal efficacy.114–117
A novel class of PheRS inhibitor, phenyl-thiazolylurea-sulfonamides, has been reported to be a potent antibiotic against both gram-positive and gram-negative bacteria, with IC50 in the nanomolar range. These compounds competitively inhibit the aminoacylation of tRNAPhe with respect to the cognate substrate Phe. The advantage of these compounds is that they do not show any cross-reactivity to the mammalian enzymes, both cytoplasmic and mitochondrial.118 However, the compounds also have some drawbacks, eg, low aqueous solubility, high level of plasma protein binding, and Phe complementation reduces the efficacy of the compounds significantly. To overcome these weaknesses, a high throughput screening has been performed to isolate novel chemical scaffolds with improved efficacy. Finally, three novel chemical scaffolds (pyridinyl anilines, trifluoromethyl pyrazoles, and thiazole amides) have been identified and crystal structures of Pseudomonas aeruginosa PheRS have been determined in complex with phenyl-thiazolylurea-sulfonamides as well as these chemical scaffolds to identify the binding mode of the inhibitor. In addition to Phe binding pocket, an auxiliary hydrophobic pocket, just below the substrate binding pocket has been identified, where the inhibitors can bind but Phe cannot. Subsequent studies have identified three phenyl-thiazolylurea-sulfonamide-resistant mutations in E. coli, which suggest that the auxiliary pocket should be avoided for drug designing against PheRS in the future.119
PheRS in pathophysiology
Human disorders associated with PheRS mutations
Human cells harbor two distinct sets of aaRSs encoded by two specific sets of genes; cytoplasmic and mitochondrial.120 Recent studies have revealed the association of aaRSs mutations with human pathologies.10,19,121 Many disease-associated mutations have been reported for ctaaRSs, but none involving HctPheRS to date. On the contrary, so far five disease-causing mutations have been identified within FARS2 gene encoding HmtPheRS (Figure 4 and Table 4).122–125 FARS2 gene is located in chromosome 6; containing 7 exons, of which 2–7 are protein coding.122
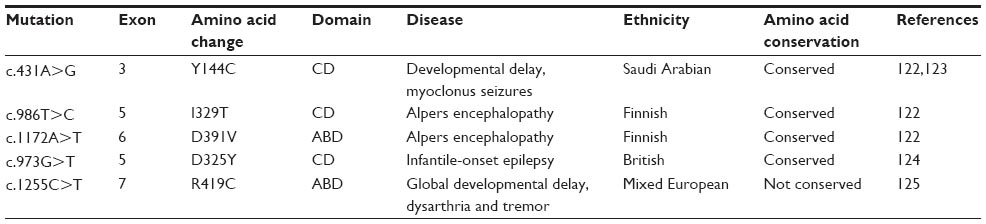 | Table 4 Disease-related mutations of HmtPheRS |
 | Figure 4 Sites of genetic mutations in HmtPheRS. |
The effects of the disease-associated mutations on aminoacylation activity and enzyme stability have been analyzed in HmtPheRS. Structural modeling has shown that the mutation I329T is located near the ATP binding site of the catalytic domain, impairing the ATP binding capacity of the mutant protein. The D391V mutation has been found to be located at the interface of the catalytic domain and ABD, impairing Phe binding affinity. Y144C mutation is also located at the interface and found to impair tRNAPhe binding.122 D325Y mutation is located near the ATP binding site of catalytic domain and found to impair ATP binding and consequently aminoacylation activity of the enzyme.124 A previous report shows that mutating amino acid residues (S57C and N280S), located far from the catalytic or tRNA binding site, do not alter aminoacylation activity but greatly affect refolding ability and protein stability. This suggests that improper folding and reduced stability may also contribute to diseases through mutations; even if they do not directly compromise enzymatic activity.126
Autoantibody against PheRS: antisynthetase syndrome
Antisynthetase syndrome is a rare, chronic autoimmune disease characterized by myositis, arthralgia, Raynaud phenomenon, skin changes called mechanic’s hands and interstitial lung disease. The hallmark of the disease is the presence of serum autoantibodies against the aaRSs. To date, a total of eight antisynthetase autoantibodies have been reported, including autoantibody against PheRS (Table 5).127–133 A combination of immunoprecipitation and mass spectrometric data have identified a novel pattern with two bands corresponding to 60 and 70 kDa, matching to PheRS α and β chain, respectively.127
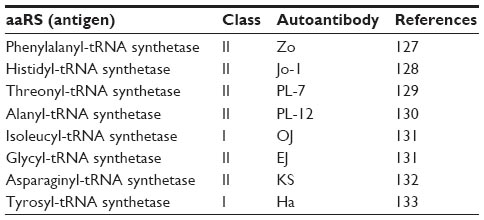 | Table 5 Antisynthetase antibodies |
Outlook
The crystal structures of PheRS, both with and without ligand, have been solved from all three kingdoms of life. This is an unprecedented advantage for several applications as observed in some recent studies. All these following observations have proved that PheRSs indeed snugly fit into future emerging field of research, particularly in the area of synthetic biology, proteomics, genetics, a model system to understand the mechanism for aaRS related diseases, to name a few. All of these applications can be attributed to our knowledge of PheRS structure. The approach of unnatural amino acid incorporation during protein synthesis has emerged as a probe for studying protein structure and function as well as enabled designing new proteins with novel applications. Recently, one group has demonstrated that yeast PheRS can be manipulated to incorporate Phe analog NaI at several specific sites of a protein very efficiently.97 Caenorhabditis elegans PheRS has been engineered to achieve cell-selective bioorthogonal noncanonical amino acid tagging (cell-selective BONCAT) of low-abundance proteins. Coupling this method with stable-isotope labeling of amino acids in cell culture enables identification of expression patterns of proteins in targeted subsets of cells. This study provides first-hand information that proteomic analysis of rare cells is possible that otherwise pose difficulty for enriching, identifying and quantifying proteins in complex biological systems.134 Two recently published reports state that PheRS can be used as a counter selection marker in genome recombineering, genome mutagenesis, and plasmid curing experiments. Mayazaki develops two highly efficient variants (Thr251Ala/Ala294Gly and Thr251Ser/Ala294Gly) of E. coli PheRS that show improved incorporation efficiency of a toxic analog 4-chloro-phenylalanine over the single mutant (Ala294Gly) leading to the high lethality in common bacterial growth media. These two variants can be used as counter selection marker with high selection efficiency.135 Another group has developed a genetic model of T. thermophilus that can potentially be used for genome engineering of this microorganism. Interestingly, this model has been generated by chromosomal manipulation of PheRS gene that is sensitive to 4-chloro-phenylalanine. This conditionally lethal mutant has been used as a counter selection marker for introducing genomic point mutations or deletions in T. thermophilus.136 To date, the mechanism of aaRS mutation related genetic disorders is poorly understood. Lu et al have recently developed a double-sieving-defective model of PheRS in Drosophila melanogaster and demonstrated its usefulness in understanding the underlying mechanism of aaRS related genetic disorders.137
Acknowledgments
SC is supported by a fellowship from the University Grant Commission (UGC), Government of India, and is currently working as a senior research fellow. This work was supported by funds from the Department of Science and Technology (India) to RB (SR/SO/BB- 0018/2012).
Disclosure
The authors report no conflicts of interest in this work.
References
Ibba M, Söll D. Quality control mechanisms during translation. Science. 1999;286(5446):1893–1897. | |
Carter CW Jr. Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases. Annu Rev Biochem. 1993;62:715–748. | |
Ibba M, Söll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. | |
Mascarenhas AP, An S, Rosen AE, Martinis SA, Musier-Forsyth K. Fidelity mechanisms of the aminoacyl-tRNA synthetases. In: Köhrer C, RajBhandary UL, editors. Protein Engineering. Berlin: Springer-Verlag; 2009:155–203. | |
Burbaum JJ, Schimmel P. Structural relationships and the classification of aminoacyl-tRNA synthetases. J Biol Chem. 1991;266(26):16965–16968. | |
Cusack S. Sequence, structure and evolutionary relationships between class 2 aminoacyl-tRNA synthetases: an update. Biochimie. 1993; 75(12):1077–1081. | |
Klipcan L, Finarov I, Moor N, Safro MG. Structural aspects of phenylalanylation and quality control in three major forms of phenylalanyl-tRNA synthetase. J Amino Acids. 2010;2010:983503. | |
Bullwinkle TJ, Ibba M. Emergence and evolution. Top Curr Chem. 2014;344:43–87. | |
Guo M, Yang XL, Schimmel P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat Rev Mol Cell Biol. 2010;11(9):668–674. | |
Peng Y, Paul LF. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med. 2013;5(3):332–343. | |
Duchêne AM, Giritch A, Hoffmann B, et al. Dual targeting is the rule for organellar aminoacyl-tRNA synthetases in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2005;102(45):16484–16489. | |
Berglund AK, Pujol C, Duchene AM, Glaser E. Defining the determinants for dual targeting of amino acyl-tRNA synthetases to mitochondria and chloroplasts. J Mol Biol. 2009;393(4):803–814. | |
Jackson KE, Pham JS, Kwek M, et al. Dual targeting of aminoacyl-tRNA synthetases to the apicoplast and cytosol in Plasmodium falciparum. Int J Parasitol. 2012;42(2):177–186. | |
Pino P, Aeby E, Foth BJ, et al. Mitochondrial translation in absence of local tRNA aminoacylation and methionyl tRNA Met formylation in Apicomplexa. Mol Microbiol. 2010;76(3):706–718. | |
Jackson KE, Habib S, Frugier M, et al. Protein translation in Plasmodium parasites. Trends Parasitol. 2011;27(10):467–476. | |
Sharma A, Sharma A. Plasmodium falciparum mitochondria import tRNAs along with an active phenylalanyl-tRNA synthetase. Biochem J. 2015;465(3):459–469. | |
Schimmel P, Tao J, Hill J. Aminoacyl tRNA synthetases as targets for new anti-infectives. FASEB J. 1998;12(15):1599–1609. | |
Kim S, Lee SW, Choi EC, Choi SY. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl Microbiol Biotechnol. 2003;61(4):278–288. | |
Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet. 2008;9:87–107. | |
Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347(6289):203–206. | |
Safro M, Moor N, Lavrik O. Phenylalanyl-tRNA synthetase. In: Ibba M, Francklyn CS, Cusack S, editors. The Aminoacyl-tRNA Synthetases, George Town: Landes Bioscience; 2004:250–265. | |
Woese CR, Olsen GJ, Ibba M, Söll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64(1):202–236. | |
Mosyak L, Reshetnikova L, Goldgur Y, Delarue M, Safro MG. Structure of phenylalanyl-tRNA synthetase from Thermus thermophilus. Nat Struct Biol. 1995;2(7):537–547. | |
Goldgur Y, Mosyak L, Reshetnikova L, et al. The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe. Structure. 1997;5(1):59–68. | |
Roy H, Ibba M. Phenylalanyl-tRNA synthetase contains a dispensable RNA-binding domain that contributes to the editing of noncognate aminoacyl-tRNA. Biochemistry. 2006;45(30):9156–9162. | |
Moor N, Linshiz G, Safro M. Cloning and expression of human phenylalanyl-tRNA synthetase in Escherichia coli: comparative study of purified recombinant enzymes. Protein Expr Purif. 2002;24(2):260–267. | |
Finarov I, Moor N, Kessler N, Klipcan L, Safro MG. Structure of human cytosolic phenylalanyl-tRNA synthetase: evidence for kingdom-specific design of the active sites and tRNA binding patterns. Structure. 2010; 18(3):343–353. | |
Shiraki K, Tsuji M, Hashimoto Y, et al. Genetic, enzymatic, and structural analyses of phenylalanyl-tRNA synthetase from Thermococcus kodakaraensis KOD1. J Biochem. 2003;134(4):567–574. | |
Zykova NA, Nevinskiĭ GA, Lavrik OI. The separation of subunits of phenylalanyl-tRNA-synthetase from Escherichia coli MRE-600 by means of affinity chromatography in dissociation conditions. Mol Biol (Mosk). 1982;16(6):1165–1172. | |
Ducruix A, Hounwanou N, Reinbolt J, Boulanger Y, Blanquet S. Purification and reversible subunit dissociation of overproduced Escherichia coli phenylalanyl-tRNA synthetase. Biochim Biophys Acta. 1983;741(2):244–250. | |
Khodyreva SN, Moor NA, Ankilova VN, Lavrik OI. Phenylalanyl-tRNA synthetase from E. coli MRE-600: analysis of the active site distribution on the enzyme subunits by affinity labelling. Biochim Biophys Acta. 1985;830(2):206–212. | |
Reshetnikova L, Moor N, Lavrik O, Vassylyev DG. Crystal structures of phenylalanyl-tRNA synthetase complexed with phenylalanine and a phenylalanyl-adenylate analogue. J Mol Biol. 1999;287(3):555–568. | |
Moor N, Kotik-Kogan O, Tworowski D, Sukhanova M, Safro M. The crystal structure of the ternary complex of phenylalanyl-tRNA synthetase with tRNAPhe and a phenylalanyl-adenylate analogue reveals a conformational switch of the CCA end. Biochemistry. 2006; 45(35):10572–10583. | |
Rodova M, Ankilova V, Safro MG. Human phenylalanyl-tRNA synthetase: cloning, characterization of the deduced amino acid sequences in terms of the structural domains and coordinately regulated expression of the alpha and beta subunits in chronic myeloid leukemia cells. Biochem Biophys Res Commun. 1999;255(3):765–773. | |
Sasaki HM, Sekine S, Sengoku T, et al. Structural and mutational studies of the amino acid-editing domain from archaeal/eukaryal phenylalanyl-tRNA synthetase. Proc Natl Acad Sci U S A. 2006; 103(40):14744–14749. | |
Fasiolo F, Remy P, Pouyet J, Ebel JP. Yeast phenylalanyl-tRNA synthetase. Molecular weight and interaction with tRNAPhe and phenylalanine. Eur J Biochem. 1974;50(1):227–236. | |
Diodato D, Ghezzi D, Tiranti V. The mitochondrial aminoacyl tRNA synthetases: genes and syndromes. Int J Cell Biol. 2014;2014:787956. | |
Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2(6):reviews 1018. | |
Martin W, Stoebe B, Goremykin V, Hapsmann S, Hasegawa M, Kowallik KV. Gene transfer to the nucleus and the evolution of chloroplasts. Nature. 1998;393(6681):162–165. | |
Sanni A, Walter P, Boulanger Y, Ebel JP, Fasiolo F. Evolution ofaminoacyl-tRNA synthetase quaternary structure and activity: Saccharomyces cerevisiae mitochondrial phenylalanyl-tRNA synthetase. Proc Natl Acad Sci U S A.1991;88(19):8387–8391. | |
Bullard JM, Cai YC, Demeler B, Spremulli LL. Expression and characterization of a human mitochondrial phenylalanyl-tRNA synthetase. J Mol Biol. 1999;288(4):567–577. | |
Baker MJ, Frazier AE, Gulbis JM, Ryan MT. Mitochondrial protein-import machinery: correlating structure with function. Trends Cell Biol. 2007;17(9):456–464. | |
Endo T, Yamano K. Multiple pathways for mitochondrial protein traffic. Biol Chem. 2009;390(8):723–730. | |
Vögtle FN, Wortelkamp S, Zahedi RP, et al. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell. 2009;139(2):428–439. | |
Gakh O, Cavadini P, Isaya G. Mitochondrial processing peptidases. Biochim Biophys Acta. 2002;1592(1):63–77. | |
Longley MJ, Humble MM, Sharief FS, Copeland WC. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J Biol Chem. 2010;285(39):29690–29702. | |
Klipcan L, Levin I, Kessler N, Moor N, Finarov I, Safro M. The tRNA-induced conformational activation of human mitochondrial phenylalanyl-tRNA synthetase. Structure. 2008;16(7):1095–1104. | |
Klipcan L, Moor N, Finarov I, Kessler N, Sukhanova M, Safro MG. Crystal structure of human mitochondrial PheRS complexed with tRNA(Phe) in the active “open” state. J Mol Biol. 2012;415(3):527–537. | |
Yadavalli SS, Klipcan L, Zozulya A, et al. Large-scale movement of functional domains facilitates aminoacylation by human mitochondrial phenylalanyl-tRNA synthetase. FEBS Lett. 2009;583(19):3204–3208. | |
Cochella L, Green R. Fidelity in protein synthesis. Curr Biol. 2005; 15(14):R536–R540. | |
Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem. 2005;74:129–177. | |
Ling J, So BR, Yadavalli SS, et al. Resampling and editing of mischarged tRNA prior to translation elongation. Mol Cell. 2009;33(5):654–660. | |
Fersht AR. Editing mechanisms in protein synthesis. Rejection of valine by the isoleucyl-tRNA synthetase. Biochemistry. 1977; 16(5):1025–1030. | |
Ling J, Reynolds N, Ibba M. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol. 2009;63:61–78. | |
Ling J, Roy H, Ibba M. Mechanism of tRNA-dependent editing in translational quality control. Proc Natl Acad Sci U S A. 2007;104(1):72–77. | |
Giegé R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26(22):5017–5035. | |
Zhou XL, Zhu B, Wang ED. The CP2 domain of leucyl-tRNA synthetase is crucial for amino acid activation and post-transfer editing. J Biol Chem. 2008;283(52):36608–36616. | |
Kim HY, Ghosh G, Schulman LH, Brunie S, Jakubowski H. The relationship between synthetic and editing functions of the active site of an aminoacyl-tRNA synthetase. Proc Natl Acad Sci U S A. 1993; 90(24):11553–11557. | |
Dock-Bregeon A, Sankaranarayanan R, Romby P, et al. Transfer RNA-mediated editing in threonyl-tRNA synthetase. The class II solution to the double discrimination problem. Cell. 2000;103(6):877–884. | |
Wong FC, Beuning PJ, Nagan M, Shiba K, Musier-Forsyth K. Functional role of the prokaryotic proline-tRNA synthetase insertion domain in amino acid editing. Biochemistry. 2002;41(22):7108–7115. | |
Gruic-Sovulj I, Rokov-Plavec J, Weygand-Durasevic I. Hydrolysis of non-cognate aminoacyl-adenylates by a class II aminoacyl-tRNA synthetase lacking an editing domain. FEBS Lett. 2007; 581(26):5110–5114. | |
Roy H, Ling J, Irnov M, Ibba M. Post-transfer editing in vitro and in vivo by the beta subunit of phenylalanyl-tRNA synthetase. EMBO J. 2004;23(23):4639–4648. | |
Igloi GL, von der Haar F, Cramer F. Aminoacyl-tRNA synthetases from yeast: generality of chemical proofreading in the prevention of misaminoacylation of tRNA. Biochemistry. 1978;17(17):3459–3468. | |
Lin SX, Baltzinger M, Remy P. Fast kinetic study of yeast phenylalanyl-tRNA synthetase: role of tRNAPhe in the discrimination between tyrosine and phenylalanine. Biochemistry. 1984;23(18):4109–4116. | |
Roy H, Ling J, Alfonzo J, Ibba M. Loss of editing activity during the evolution of mitochondrial phenylalanyl-tRNA synthetase. J Biol Chem. 2005;280(46):38186–38192. | |
Lin SX, Baltzinger M, Remy P. Fast kinetic study of yeast phenylalanyl-tRNA synthetase: an efficient discrimination between tyrosine and phenylalanine at the level of the aminoacyladenylate-enzyme complex. Biochemistry. 1983;22(3):681–689. | |
Kotik-Kogan O, Moor N, Tworowski D, Safro M. Structural basis for discrimination of L-phenylalanine from L-tyrosine by phenylalanyl-tRNA synthetase. Structure. 2005;13(12):1799–1807. | |
Ibba M, Kast P, Hennecke H. Substrate specificity is determined by amino acid binding pocket size in Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry. 1994;33(23):7107–7112. | |
Augustin S, Nolden M, Müller S, Hardt O, Arnold I, Langer T. Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J Biol Chem. 2005;280(4):2691–2699. | |
Janska H, Kwasniak M, Szczepanowska J. Protein quality control in organelles -AAA/FtsH story. Biochim Biophys Acta. 2013;1833(2):381–387. | |
Stadtman ER. Protein oxidation and aging. Science. 1992;257(5074):1220–1224. | |
Dean RT, Fu S, Stocker R, Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. Biochem J. 1997;324(Pt 1):1–18. | |
Fu S, Davies MJ, Stocker R, Dean RT. Evidence for roles of radicals in protein oxidation in advanced human atherosclerotic plaque. Biochem J. 1998;333(Pt 3):519–525. | |
Klipcan L, Moor N, Kessler N, Safro MG. Eukaryotic cytosolic and mitochondrial phenylalanyl-tRNA synthetases catalyze the charging of tRNA with the meta-tyrosine. Proc Natl Acad Sci U S A. 2009;106(27):11045–11048. | |
Rodgers KJ, Dean RT. Metabolism of protein-bound DOPA in mammals. Int J Biochem Cell Biol. 2000;32(9):945–955. | |
Lawrence AD, Evans AH, Lees AJ. Compulsive use of dopamine replacement therapy in Parkinson’s disease: reward systems gone awry? Lancet Neurol. 2003;2(10):595–604. | |
Nutt JG, Nygaard TG. Response to levodopa treatment in dopa-responsive dystonia. Arch Neurol. 2001;58(6):905–910. | |
Malkus KA, Tsika E, Ischiropoulos H. Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: how neurons are lost in the Bermuda triangle. Mol Neurodegener. 2009;4:24. | |
Moor N, Klipcan L, Safro MG. Bacterial and eukaryotic phenylalanyl-tRNA synthetases catalyze misaminoacylation of tRNA(Phe) with 3,4-dihydroxy-L-phenylalanine. Chem Biol. 2011;18(10):1221–1229. | |
First EA. L-DOPA ropes in tRNA(Phe). Chem Biol. 2011;18(10):1201–1202. | |
Reynolds NM, Ling J, Roy H, et al. Cell-specific differences in the requirements for translation quality control. Proc Natl Acad Sci U S A. 2010;107(9):4063–4068. | |
Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83(3–4):301–310. | |
Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10(12):524–530. | |
Rodgers KJ, Shiozawa N. Misincorporation of amino acid analogues into proteins by biosynthesis. Int J Biochem Cell Biol. 2008; 40(8):1452–1466. | |
Dunlop RA, Brunk UT, Rodgers KJ. Proteins containing oxidized amino acids induce apoptosis in human monocytes. Biochem J. 2011;435(1):207–216. | |
Rodgers KJ, Hume PM, Morris JG, Dean RT. Evidence for L-dopa incorporation into cell proteins in patients treated with levodopa. J Neurochem. 2006;98(4):1061–1067. | |
Lechler A, Kreutzer R. The phenylalanyl-tRNA synthetase specifically binds DNA. J Mol Biol. 1998;278(5):897–901. | |
Dou X, Limmer S, Kreutzer R. DNA-binding of phenylalanyl-tRNA synthetase is accompanied by loop formation of the double-stranded DNA. J Mol Biol. 2001;305(3):451–458. | |
Wilson KP, Shewchuk LM, Brennan RG, Otsuka AJ, Matthews BW. Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc Natl Acad Sci U S A. 1992;89(19):9257–9261. | |
Safro M, Mosyak L. Structural similarities in the noncatalytic domains of phenylalanyl-tRNA and biotin synthetases. Protein Sci. 1995;4(11):2429–2432. | |
Voloshchuk N, Montclare JK. Incorporation of unnatural amino acids for synthetic biology. Mol Biosyst. 2010;6(1):65–80. | |
Link AJ, Tirrell DA. Reassignment of sense codons in vivo. Methods. 2005;36(3):291–298. | |
Chin JW. Expanding and reprogramming the genetic code of cells and animals. Annu Rev Biochem. 2014;83:379–408. | |
Kwon I, Wang P, Tirrell DA. Design of a bacterial host for site-specific incorporation of p-bromophenylalanine into recombinant proteins. J Am Chem Soc. 2006;128(36):11778–11783. | |
Kodama K, Fukuzawa S, Sakamoto K, et al. A new protein engineering approach combining chemistry and biology, part I; site-specific incorporation of 4-iodo-L-phenylalanine in vitro by using misacylated suppressor tRNAPhe. Chembiochem. 2006;7(10):1577–1581. | |
Datta D, Wang P, Carrico IS, Mayo SL, Tirrell DA. A designed phenylalanyl-tRNA synthetase variant allows efficient in vivo incorporation of aryl ketone functionality into proteins. J Am Chem Soc. 2002; 124(20):5652–5653. | |
Kwon I, Lim SI. Tailoring the substrate specificity of yeast phenylalanyl-tRNA synthetase toward a phenylalanine analog using multiple- site-specific incorporation. ACS Synth Biol. 2015;4(5):634–643. | |
Johnson JA, Lu YY, Van Deventer JA, Tirrell DA. Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Curr Opin Chem Biol. 2010;14(6):774–780. | |
Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An expanded eukaryotic genetic code. Science. 2003; 301(5635):964–967. | |
Liu W, Brock A, Chen S, Chen S, Schultz PG. Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat Methods. 2007;4(3):239–244. | |
Arai N, Ito T. Amino acid requirements of the silkworm, Bombyx mori L. J Sericult Sci Jpn. 1964;33:107–110. | |
Zhou CZ, Confalonieri F, Medina N, et al. Fine organization of Bombyx mori fibroin heavy chain gene. Nucleic Acids Res. 2000; 28(12):2413–2419. | |
Omenetto FG, Kaplan DL. New opportunities for an ancient material. Science. 2010;329(5991):528–531. | |
Kaplan D, Adams WW, Farmer B, Viney C. Silk polymers: materials science and biotechnology. ACS Symp Ser. 1994;544:291–358. | |
Leal-Egaña A, Scheibel T. Silk-based materials for biomedical applications. Biotechnol Appl Biochem. 2010;55(3):155–167. | |
Teramoto H, Kojima K. Cloning of Bombyx mori phenylalanyl-tRNA synthetase and the generation of its mutant with relaxed amino acid specificity. J Insect Biotechnol Sericol. 2010;79:53–65. | |
Teramoto H, Kojima K, Kajiwara H, Ishibashi J. Expansion of the amino acid repertoire in protein biosynthesis in silkworm cells. Chembiochem. 2012;13(1):61–65. | |
Teramoto H, and Kojima K. Residue-specific incorporation of phenylalanine analogues into protein biosynthesis in silkworm cultured cells. J. Insect Biotechnol Sericol. 2013;82(3):61–69. | |
Teramoto H, Kojima K. Production of Bombyx mori silk fibroin incorporated with unnatural amino acids. Biomacromolecules. 2014; 15(7):2682–2690. | |
McDevitt D, Rosenberg M. Exploiting genomics to discover new antibiotics. Trends Microbiol. 2001;9(12):611–617. | |
Vondenhoff GH, Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur J Med Chem. 2011;46(11):5227–5236. | |
Chopra I, Hesse L, O’Neill AJ. Exploiting current understanding of antibiotic action for discovery of new drugs. J Appl Microbiol. 2002;92(Suppl s1):4S–15S. | |
Hurdle JG, O’Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob Agents Chemother. 2005;49(12):4821–4833. | |
Yu XY, Finn J, Hill JM, et al. A series of spirocyclic analogues as potent inhibitors of bacterial phenylalanyl-tRNA synthetases. Bioorg Med Chem Lett. 2004;14(5):1339–1342. | |
Yu XY, Finn J, Hill JM, et al. A series of heterocyclic inhibitors of phenylalanyl-tRNA synthetases with antibacterial activity. Bioorg Med Chem Lett. 2004;14(5):1343–1346. | |
Jarvest RL, Erskine SG, Forrest AK, et al. Discovery and optimisation of potent, selective, ethanolamine inhibitors of bacterial phenylalanyl tRNA synthetase. Bioorg Med Chem Lett. 2005;15(9):2305–2309. | |
Montgomery JI, Toogood PL, Hutchings KM, et al. Discovery and SAR of benzyl phenyl ethers as inhibitors of bacterial phenylalanyl-tRNA synthetase. Bioorg Med Chem Lett. 2009;19(3):665–669. | |
Beyer D, Kroll HP, Endermann R, et al. New class of bacterial phenylalanyl-tRNA synthetase inhibitors with high potency and broad-spectrum activity. Antimicrob Agents Chemother. 2004;48(2):525–532. | |
Abibi A, Ferguson AD, Fleming PR, et al. The role of a novel auxiliary pocket in bacterial phenylalanyl-tRNA synthetase druggability. J Biol Chem. 2014;289(31):21651–21662. | |
Bonnefond L, Fender A, Rudinger-Thirion J, Giegé R, Florentz C, Sissler M. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry. 2005;44(12):4805–4816. | |
Park SG, Schimmel P, Kim S. Aminoacyl tRNA synthetases and their connections to disease. Proc Natl Acad Sci U S A. 2008; 105(32):11043–11049. | |
Elo JM, Yadavalli SS, Euro L, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet. 2012;21(20):4521–4529. | |
Shamseldin HE, Alshammari M, Al-Sheddi T, et al. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet. 2012;49(4):234–241. | |
Almalki A, Alston CL, Parker A, et al. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim Biophys Acta. 2014;1842(1):56–64. | |
Vernon HJ, McClellan R, Batista DA, Naidu S. Mutations in FARS2 and non-fatal mitochondrial dysfunction in two siblings. Am J Med Genet A. 2015;167A(5):1147–1151. | |
Banerjee R, Reynolds NM, Yadavalli SS, et al. Mitochondrial aminoacyl-tRNA synthetase single-nucleotide polymorphisms that lead to defects in refolding but not aminoacylation. J Mol Biol. 2011;410(2):280–293. | |
Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology. 2007;46(6):1005–1008. | |
Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum. 1980;23(8):881–888. | |
Mathews MB, Reichlin M, Hughes GR, Bernstein RM. Anti-threonyl-tRNA synthetase, a second myositis-related autoantibody. J Exp Med. 1984;160(2):420–434. | |
Bunn CC, Bernstein RM, Mathews MB. Autoantibodies against alanyl-tRNA synthetase and tRNAAla coexist and are associated with myositis. J Exp Med. 1986;163(5):1281–1291. | |
Targoff IN. Autoantibodies to aminoacyl-transfer RNA synthetases for isoleucine and glycine. Two additional synthetases are antigenic in myositis. J Immunol. 1990;144(5):1737–1743. | |
Hirakata M, Suwa A, Nagai S, et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol. 1999;162(4):2315–2320. | |
Hashish L, Trieu EP, Sadanandan P, Targoff IN. Identification of autoantibodies to tyrosyl-tRNA synthetase in dermatomyositis with features consistent with antisynthetase syndrome. Arthritis Rheum. 2005;52(Suppl 9):S312. | |
Yuet KP, Doma MK, Ngo JT, et al. Cell-specific proteomic analysis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2015; 112(9):2705–2710. | |
Miyazaki K. Molecular engineering of a PheS counterselection marker for improved operating efficiency in Escherichia coli. Biotechniques. 2015;58(2):86–88. | |
Carr JF, Danziger ME, Huang AL, Dahlberg AE, Gregory ST. Engineering the genome of Thermus thermophilus using a counter selectable marker. J Bacteriol. 2015;197(6):1135–1144. | |
Lu J, Bergert M, Walther A, Suter B. Double-sieving-defective aminoacyl-tRNA synthetase causes protein mistranslation and affects cellular physiology and development. Nat Commun. 2014;5:5650. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.