Back to Journals » International Medical Case Reports Journal » Volume 19
Phenotypic Spectrum and Chromosomal Discordance in Alobar Holoprosencephaly: A Comparative Case Series from a Tertiary Referral Center
Authors Caropeboka MFA, Nisa AS ![]() , Pramatirta AY, Pribadi A
, Pramatirta AY, Pribadi A ![]() , Mose JC
, Mose JC
Received 1 November 2025
Accepted for publication 30 January 2026
Published 11 February 2026 Volume 2026:19 569641
DOI https://doi.org/10.2147/IMCRJ.S569641
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Muhamad Faizal Arif Caropeboka, Aisyah Shofiatun Nisa, Akhmad Yogi Pramatirta, Adhi Pribadi, Johanes Cornelius Mose
Department of Obstetrics and Gynecology, Hasan Sadikin General Hospital-Padjadjaran University, Bandung, Indonesia
Correspondence: Muhamad Faizal Arif Caropeboka, Department of Obstetrics and Gynecology, Hasan Sadikin General Hospital-Padjadjaran University, Pasteur 38, Bandung, 40161, Indonesia, Tel +62 812-2233-3431, Email [email protected]
Introduction: Holoprosencephaly (HPE) is a rare congenital malformation of the forebrain caused by incomplete midline cleavage, often accompanied by craniofacial abnormalities. The condition arises from multifactorial etiologies, including genetic, environmental, and maternal factors. Early prenatal diagnosis is essential for parental counseling, management decisions, and detection of associated anomalies.
Case Report: We report two cases of alobar HPE diagnosed in the third trimester by ultrasonography. The first case involved a 41-year-old primigravida at 32 weeks of gestation, with ultrasound findings of fused thalami, single ventricle, microcephaly, and hypotelorism. The fetus was delivered with severe midline craniofacial abnormalities and a normal female karyotype (46,XX). The second case involved a 41-year-old multiparous woman at 32 weeks of gestation. Ultrasound revealed fused thalami, absent falx cerebri and corpus callosum, and a proboscis. The infant presented with synophthalmia and proboscis, and karyotyping confirmed trisomy 13. Both pregnancies were terminated after counseling, and postnatal findings confirmed the prenatal diagnosis.
Conclusion: This case series demonstrates the phenotypic and chromosomal variability within alobar HPE and underscores the diagnostic value of detailed ultrasonography combined with genetic analysis. Late detection in the third trimester limited reproductive options and highlights the importance of improved anomaly screening pathways. Early diagnosis remains crucial for comprehensive parental counselling and perinatal planning.
Keywords: holoprosencephaly, alobar holoprosencephaly, prenatal ultrasonography, craniofacial abnormalities
Introduction
Holoprosencephaly (HPE) is the most common malformation of the forebrain and results from incomplete separation of the prosencephalon, often accompanied by craniofacial anomalies.1 The severity of facial malformations typically parallels the degree of brain involvement, with features ranging from hypotelorism to cyclopia and proboscis.1–3 The condition occurs in roughly 1 in 250 fetuses and 1 in 16,000 live births across diverse populations.2 Several factors contribute to the disease such as genetic, environment, and mechanical factors. Around 75% of holoprosencephaly cases had normal chromosomes with no identified mutations which indicates the contribution of other factors towards the disease.2,4–6
The etiology of HPE include both genetic and non-genetic factors. Genetic causes may be syndromic or non-syndromic, while non-genetic contributors include maternal conditions such as pregestational diabetes, folic acid deficiency, exposure to teratogenic substances, and other environmental factors.1 HPE is typically classified into three main forms: alobar, semilobar, and lobar. Alobar HPE is the most common and severe, characterized by failure of hemispheric separation, resulting in a single lobe with a centrally located ventricle. Semilobar HPE shows partial cleavage of the forebrain, whereas lobar HPE demonstrates nearly complete separation. Rare variants include the middle interhemispheric variant (syntelencephaly), with abnormal midline fusion of the posterior frontal and parietal lobes, and the septo-preoptic variant, the rarest form, with midline fusion restricted to the septal or preoptic regions of the telencephalon.4,5
Genetic and chromosomal abnormalities are frequently implicated. Aneuploidy is present in up to 50–60% of cases of alobar or semilobar HPE, with trisomy 13 being the most commonly reported abnormality.6,7 However, a substantial proportion of cases demonstrate normal karyotypes, suggesting additional genetic, environmental, or multifactorial contributors.6,7
Prenatal diagnosis relies on ultrasonography, typically in the first trimester, although late-gestation cases continue to be encountered, particularly in resource-limited referral settings Early recognition may facilitate parental counseling, consideration of pregnancy termination, and neonatal management planning. Here we report a case series of alobar HPE diagnosed in the third trimester, each exhibiting discrepancy craniofacial phenotypes and chromosomal findings. These cases highlight the phenotypic variability of alobar HPE and underscore the continued diagnostic value of ultrasonography in late gestation.
Case Report
This case report presents 2 cases of alobar holoprosencephaly that were detected by ultrasound examination and later found to have discrepancy facial abnormalities. Karyotyping examination was also performed on the 2 cases, each showing discrepancy results. The discrepancy of case 1 and 2 was reported in Table 1.
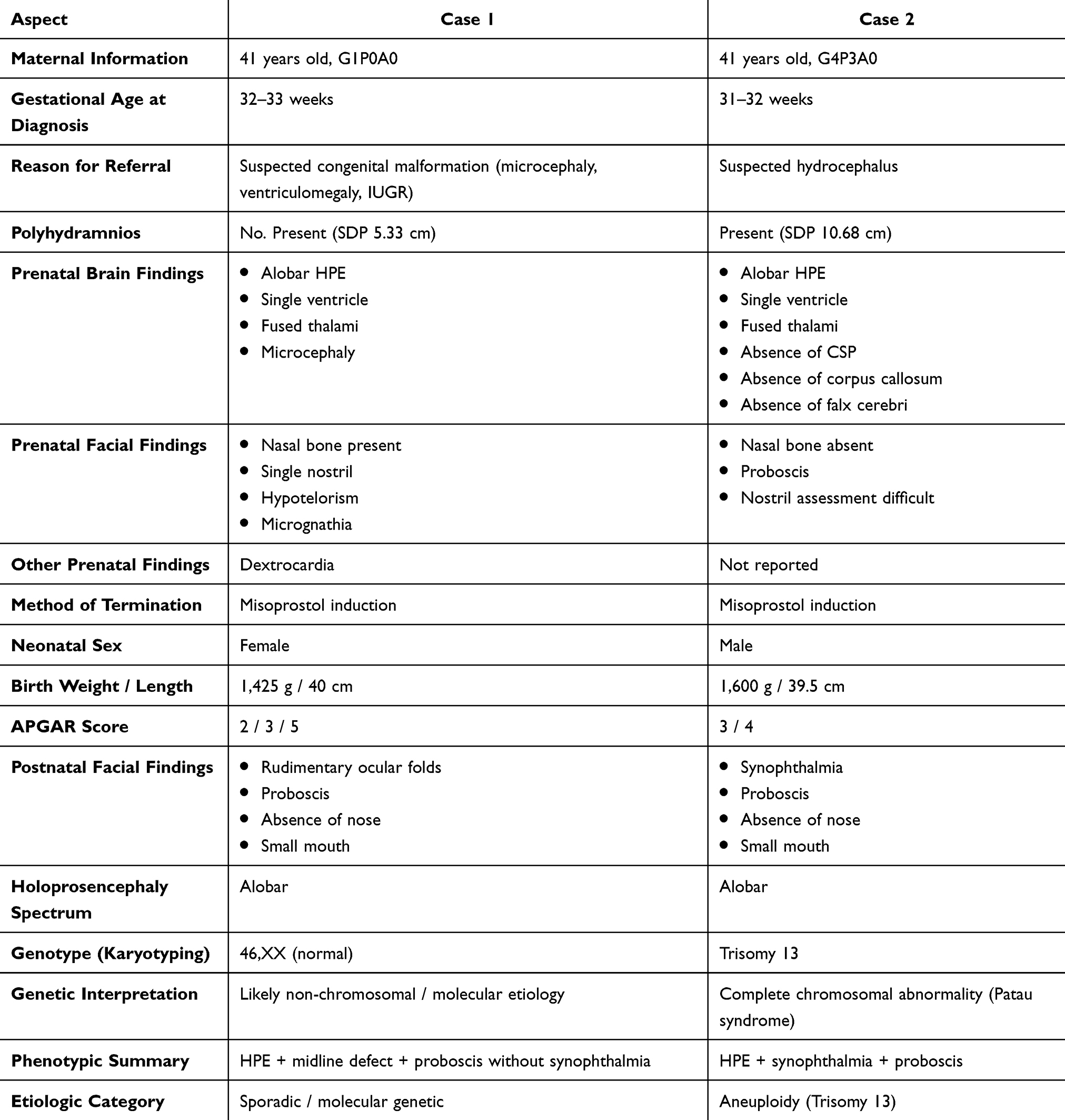 |
Table 1 The Discrepancy of Case 1 and Case 2 |
Case 1
A 41-year-old woman was referred from a district hospital with a diagnosis of G1P0A0, 32–33 weeks of gestation, with suspected congenital abnormalities including microcephaly, ventriculomegaly, and intrauterine growth restriction (IUGR). The patient had no complaints related to her pregnancy, and there were no signs of labor at the time of referral. Referral was made due to abnormalities detected on an ultrasound examination performed a week prior.
Ultrasound revealed a fetus smaller than expected for gestational age, fused thalami, a single ventricle, and microcephaly (Figure 1). Facial examination showed a present nasal bone, single nostril, no cleft, micrognathia, and features suggestive of hypotelorism. The heart was located in the right thoracic cavity. Measurement of the single deepest pocket (SDP) was 5.33 cm. Based on these findings, the diagnosis was a G1P0A0 pregnancy at 32 weeks 5 days with polyhydramnios, alobar holoprosencephaly, and dextrocardia.
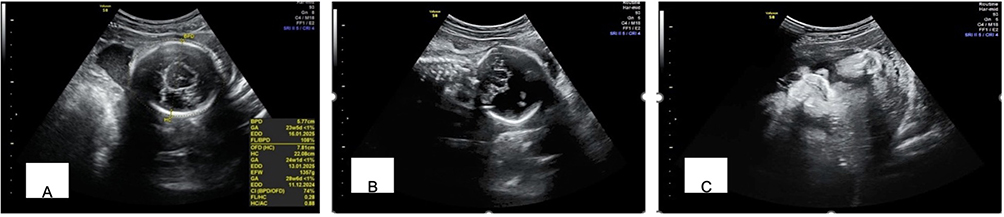 |
Figure 1 Ultrasonography examination of case 1. (A) Alobar holoprosencephaly, transthalamic section; (B) Alobar holoprosencephaly, coronal section; (C) Appearance of a single nostril. |
Pregnancy termination was performed, and a female infant was delivered with an APGAR score of 2/3/5. The baby weighed 1,425 grams and measured 40 cm in length. On facial examination, the newborn presents with a severe congenital anomaly. The eyes are not normally developed, with only rudimentary eyelid-like folds visible and no distinct globes. In the midline, there is a bulbous protrusion resembling a proboscis that replaces the normal nasal structure, indicating complete absence of a functional nose. The mouth appears small with thin lips. This overall appearance is consistent with a severe midline craniofacial malformation, most likely within the spectrum of alobar holoprosencephaly (Figure 2). Karyotyping revealed a normal female karyotype (46,XX) with no mosaicism (Figure 3). Genetic mutations, however, could not be excluded as a potential cause of the observed abnormalities.
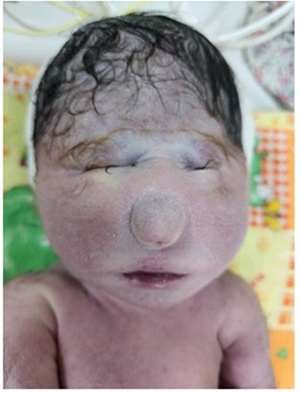 |
Figure 2 Facial appearance of case 1 with rudimentary eyelid-like folds, proboscis, and thin lips. |
 |
Figure 3 Chromosome analysis of case 1 resulted 46XX. |
Case 2
A 41-year-old woman, G4P3A0, at 31–32 weeks of gestation, with suspected hydrocephalus, was referred from Cahaya Kawaluyaan Hospital to the Fetomaternal Division of RSHS. The patient had no complaints and showed no signs of labor. The day prior to admission, abnormalities of the fetal head were detected on ultrasound, prompting referral.
Ultrasound examination revealed a fetus with fused thalami, positive vernix, absence of the cavum septum pellucidum, absence of the falx cerebri, absence of the corpus callosum, and a single ventricle. Facial evaluation showed absence of the nasal bone, difficult-to-assess nostrils, presence of a proboscis, and no cleft. The single deepest pocket (SDP) was measured at 10.68 cm (Figure 4).
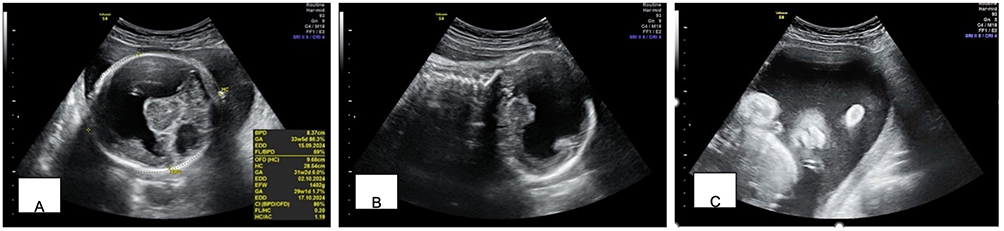 |
Figure 4 Ultrasonography examination of case 2. (A) Alobar holoprosencephaly, transthalamic section; (B) Alobar holoprosencephaly, coronal section; (C) Appearance of a single nostril. |
The final diagnosis was G4P3A0 pregnancy at 32 weeks of gestation with polyhydramnios and alobar holoprosencephaly. Pregnancy termination was planned using misoprostol induction. A male infant was delivered, weighing 1,600 grams and measuring 39.5 cm in length, with an APGAR score of 3/4. All the prenatal ultrasound findings were confirmed postnatally.
On facial examination, the newborn presents with a severe congenital anomaly characterized by synophtalmia, in which a single orbital cavity is located at the midline of the face. Just above this orbital cavity, there is a tubular protrusion resembling a trunk, known as a proboscis, replacing the normal nasal structure. The nose is absent, while the mouth appears small with a narrow opening. This overall appearance is consistent with a severe midline malformation, most commonly associated with alobar holoprosencephaly (Figure 5). Karyotyping revealed a male karyotype with trisomy 13, with no additional chromosomal abnormalities or mosaicism (Figure 6).
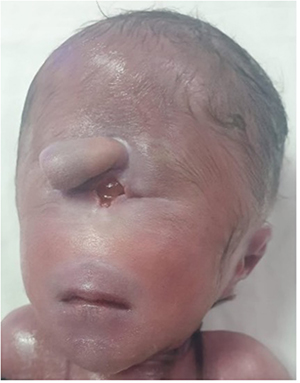 |
Figure 5 Facial appearance of case 2 with synophthalmia, proboscis, and thin lips. |
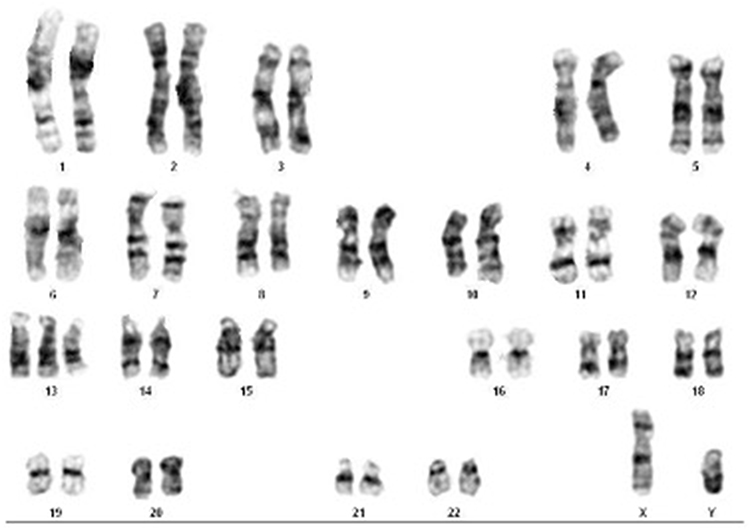 |
Figure 6 Chromosome analysis of case 2 resulted with trisomy 13. |
Discussion
In this case series, we report two cases diagnosed prenatally with alobar holoprosencephaly who demonstrated differing craniofacial phenotypes and chromosomal results. These cases illustrate the phenotypic variability of holoprosencephaly and demonstrate the importance of integrating ultrasonography with genetic investigations to support prenatal counseling. The risk factors include transplacental infections, first-trimester bleeding, a history of miscarriage, and maternal diabetes mellitus. Early ultrasonographic diagnosis facilitates timely counseling regarding pregnancy termination, potentially preventing the psychological trauma of delivering a severely malformed infant, while also allowing evaluation for other anomalies. Chromosomal abnormalities are frequently implicated, as illustrated by one of the present cases.7–9
The key diagnostic features of alobar holoprosencephaly, including fused thalami, absence of the falx cerebri, and a single monoventricle, were clearly identified on ultrasonography in both cases. These findings consistent with existing literature, which describes ultrasound as a reliable modality for the recognition of holoprosencephaly as early as 11–12 weeks of gestation.10,11 However, similar to many reported series in developing regions, both diagnoses in our report were made in the third trimester, limiting reproductive decision-making options, including medical termination, and affecting maternal psychological preparation. Delayed diagnosis in low-resource settings may reflect restricted access to anomaly screening and late referral pathways, a pattern also noted in previous studies.
The most notable discrepancy between the two cases was the variation in craniofacial phenotype. The first case showed severe ocular hypoplasia without a functional nasal structure, while the second presented with synophthalmia and a midline proboscis. These craniofacial patterns are consistent with the well-established concept that facial malformations in holoprosencephaly correlate with the degree of forebrain non-separation.12 Our findings reinforce the clinical value of assessing facial structures, particularly in the coronal plane, as the facial phenotype often alerts clinicians to the diagnosis even before intracranial anomalies are fully characterized.
Our findings also reinforce DeMyer’s classic axiom that “the face predicts the brain.” Case 2 exhibited the most severe facial dysmorphism—synophthalmia and a proboscis—both highly characteristic of alobar HPE. In contrast, Case 1 showed a milder yet still significant craniofacial phenotype, including a single nostril and hypotelorism. Notably, the case with less severe facial malformation (Case 1) had a normal 46,XX karyotype, whereas the case with the most extreme midline non-separation (Case 2) was associated with Trisomy 13. This gradient supports the well-documented correlation between the severity of craniofacial dysmorphism and the likelihood of chromosomal aneuploidy, underscoring the diagnostic value of careful facial evaluation in predicting underlying genetic etiology.
Genomic evaluation remains crucial given that approximately 50% of holoprosencephaly cases are associated with chromosomal abnormalities.4 In our series, karyotype analysis revealed trisomy 13 in one case, consistent with the most frequently reported aneuploidy associated with alobar holoprosencephaly, whereas the other case had a normal 46,XX karyotype.12 The latter suggests the possibility of pathogenic variants in holoprosencephaly-associated genes such as SHH, ZIC2, SIX3, or TGIF1, which were not ruled out due to unavailability of sequencing. This distinction is clinically relevant because syndromic cases tend to carry a higher recurrence risk and may influence counseling for future pregnancies.1,6,13
Both fetuses developed polyhydramnios, likely due to impaired swallowing mechanisms secondary to severe neurological dysfunction. Polyhydramnios has been described in up to one-third of alobar holoprosencephaly cases and may serve as a secondary sonographic indicator.8,9 Both newborns had markedly poor postnatal outcomes, consistent with the known prognosis of alobar holoprosencephaly, which carries the highest mortality among its subtypes.1,8,9 Complications such as endocrine disturbances, epilepsy, and profound motor and cognitive impairment have been well described among survivors, although most infants die in the neonatal period, as occurred in our cases.1,14,15
In summary, the findings of this case series are consistent with the existing literature and support the understanding that alobar holoprosencephaly represents a structural, genetic, and phenotypic spectrum rather than a single uniform entity. This case series adds to the current body of knowledge by demonstrating differing craniofacial and chromosomal outcomes within the same subtype of holoprosencephaly and emphasizes the importance of a comprehensive prenatal assessment that includes neurosonography, facial evaluation, and genetic testing. Additionally, the late gestational detection in both cases underscores the need for improved anomaly screening systems in order to optimize parental counseling, reproductive options, and multidisciplinary perinatal planning.
Conclusion
Holoprosencephaly is a rare and heterogeneous malformation of the developing forebrain. In this case series, alobar holoprosencephaly manifested with differing craniofacial phenotypes and contrasting chromosomal outcomes, underscoring the phenotypic and genetic variability of the condition. Prenatal ultrasonography proved central to diagnosis, while genetic evaluation further informed etiology and recurrence risk. These findings highlight the importance of comprehensive prenatal assessment and timely anomaly screening in supporting reproductive counselling and perinatal planning.
Ethical Approval
This study is exempted from an ethical approval as determined by the institutional and department review board.
Informed Consent Patient Statement
The authors confirm that written informed consent has been obtained from the involved patients (and/or their legal guardians). The patients (and/or their legal guardians) has been informed about the details of the case and has provided approval for the information to be published in this case report. A copy of the written consent is available for review by the Editor of this journal.
Author Contributions
MFAZ conceived the design in article and collected the data. MFAZ and ASN wrote the draft. AYP, AP, and JCM directed and supervise the review article. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The study did not receive external funding.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Malta M, AlMutiri R, Martin CS, Srour M. Holoprosencephaly: review of embryology, clinical phenotypes, etiology and management. Children. 2023;10(4):647. doi:10.3390/children10040647
2. Geng X, Oliver G. Pathogenesis of holoprosencephaly. J Clin Invest. 2009;119(6):1403–8. doi:10.1172/JCI38937
3. Petryk A, Graf D, Marcucio R. Holoprosencephaly: signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans. Wiley Interdiscip Rev Dev Biol. 2015;4(1):17–32. doi:10.1002/wdev.161
4. Suwardewa TGA, Mulyana RS, Setiawan WA. Alobar holoprosencephaly: a case report. Indones J Perinatol. 2022;3(1):4–7. doi:10.51559/inajperinatol.v3i1.23
5. Ramakrishnan S, Das JM. Holoprosencephaly. In: StatPearls. StatPearls Publishing; 2024.
6. Roessler E, Muenke M, editors. The molecular genetics of holoprosencephaly. In: American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Wiley Online Library; 2010.
7. Norton ME. Callen’s Ultrasonography in Obstetrics & Gynecology: 1SAE-E-Book. Elsevier Health Sciences; 2016.
8. Raman R, Mukunda Jagadesh G. Antenatal diagnosis of alobar holoprosencephaly. Case Rep Radiol. 2014;2014(1):724671. doi:10.1155/2014/724671
9. Obanife O, Adekanye AG, Ekpo RG, et al. Management of alobar holoprosencephaly associated with fronto-nasal encephalocoele and type I (closed-lips) schizencephaly at the university of calabar teaching Hospital: a case report and literature review. Interdiscip Neurosurg. 2022;27:101410. doi:10.1016/j.inat.2021.101410
10. Chaudhari HD, Thakkar G, Darji P, Khokhani P. Prenatal ultrasound diagnosis of holoprosencephaly and associated anomalies. Case Rep. 2012;2012:bcr0320126129.
11. Wong H, Lam Y, Tang M, Cheung L, Ng L, Yan K. First‐trimester ultrasound diagnosis of holoprosencephaly: three case reports. Ultrasound Obstet Gynecol. 1999;13(5):356–359. doi:10.1046/j.1469-0705.1999.13050356.x
12. Ionescu CA, Calin D, Navolan D, et al. Alobar holoprosencephaly associated with a rare chromosomal abnormality: case report and literature review. Medicine. 2018;97(29):e11521. doi:10.1097/MD.0000000000011521
13. Paladini D, Volpe P. Ultrasound of congenital Fetal Anomalies: Differential Diagnosis and Prognostic Indicators. CRC press; 2024.
14. Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007;2(1):8. doi:10.1186/1750-1172-2-8
15. Kauvar EF, Muenke M. Holoprosencephaly: recommendations for diagnosis and management. Curr Opin Pediatr. 2010;22(6):687–695. doi:10.1097/MOP.0b013e32833f56d5
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.