Back to Journals » Clinical Ophthalmology » Volume 15
Phase 3 Efficacy (Worse-Eye Analysis) and Long-Term Safety Evaluation of OTX-101 in Patients with Keratoconjunctivitis Sicca
Authors Sheppard J, Bergmann M, Schechter BA, Luchs J ![]() , Ogundele A, Karpecki P
, Ogundele A, Karpecki P ![]()
Received 1 September 2020
Accepted for publication 28 October 2020
Published 12 January 2021 Volume 2021:15 Pages 129—140
DOI https://doi.org/10.2147/OPTH.S279364
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
John Sheppard,1 Mark Bergmann,2 Barry A Schechter,3 Jodi Luchs,4 Abayomi Ogundele,5 Paul Karpecki6
1Virginia Eye Consultants, Norfolk, VA, USA; 2Apex Eye, Cincinnati, OH, USA; 3Florida Eye Microsurgical Institute, Boynton Beach, FL, USA; 4Florida Vision Institute, West Palm Beach, FL, USA; 5Medical Affairs, Sun Pharmaceutical Industries, Inc, Princeton, NJ, USA; 6Kentucky Eye Institute, Lexington, KY, USA
Correspondence: John Sheppard
Virginia Eye Consultants, Suite #210, 241 Corporate Blvd, Norfolk, VA 23502, USA
Tel +1 757 226-8021
Email [email protected]
Background: OTX-101 is approved for treatment of keratoconjunctivitis sicca (KCS). We present results of a phase 3 worse-eye efficacy analysis and 1-year safety extension.
Methods: During the double-masked treatment phase, patients with bilateral KCS were randomized 1:1 to 12 weeks OTX-101 or vehicle 1 drop per eye twice daily. Efficacy assessments included Schirmer’s test and corneal and conjunctival staining. All patients who completed the treatment phase were eligible for enrollment in the open-label extension and received 1 drop OTX-101 twice daily for up to 52 weeks. Safety endpoints included adverse event (AE) monitoring, Snellen visual acuity (VA), intraocular pressure (IOP), slit-lamp examination (SLE), and dilated fundoscopy.
Results: Overall, 745 and 258 patients enrolled in the treatment and safety extension phases, respectively. At 12 weeks, number (%) of patients with Schirmer’s score increase of ≥ 10 mm from baseline was 76 (20.5%) vs. 42 (11.3%) for OTX-101 vs. vehicle (P=0.0005). OTX-101 significantly improved total conjunctival staining vs. vehicle at week 12 (least squares mean change from baseline − 1.65 [0.12] vs. − 1.12 [0.12], P=0.0013), and number (%) of patients with clear central corneas vs. vehicle at week 12 (222 [64.0%] vs. 199 [55.3%], P=0.0179). In the 1-year safety extension, AEs were mostly mild; instillation site pain was most common in 59 (22.9%) patients (17 [13.2%] vs. 42 [32.6%] patients receiving prior OTX-101 and vehicle). No safety concerns were raised by VA, IOP, SLE, and fundoscopy.
Conclusion: OTX-101 efficacy was confirmed in the eye with lower baseline Schirmer’s score. OTX-101 was well tolerated long term.
Clinical Trial: Registered at ClinicalTrials.gov on July 27, 2016. NCT02845674 https://clinicaltrials.gov/ct2/show/NCT02845674?term=OTX-101&draw=2&rank=1.
Keywords: cyclosporine A, dry-eye disease, keratoconjunctivitis sicca; KCS, OTX-101
Background
Dry eye disease, also known as keratoconjunctivitis sicca (KCS), is a multifactorial disease of the ocular surface.1 KCS is characterized by loss of homeostasis of the tear film and a vicious cycle where hyperosmolarity leads to inflammation resulting in ocular surface damage.1,2 Symptoms include stinging, grittiness, and visual disturbance, but presentation varies from patient to patient, and the relationship between disease signs and symptoms is not linear.1,3 Estimates of prevalence range from 5% to 33% depending on disease definition used and studied populations.4 KCS is more prevalent in women and Asians, and incidence increases with age.4 Disease management follows a stepwise approach, starting with patient education, environmental modifications, tear supplementation and conservation, and lid hygiene, and then progressing to nonpharmacologic devices and prescription medications if initial treatments are inadequate in providing relief.5
Ophthalmic cyclosporine A (CsA) formulations are one such prescription medication option. CsA targets the underlying inflammation responsible for ocular surface damage and the clinical signs and symptoms of KCS.6,7 It has potent immunomodulatory activity but is highly lipophilic, posing a challenge to ocular delivery.8 Ophthalmic CsA oil-in-water emulsions may cause vision blurring and irritation, and their relatively low ocular tissue penetration may delay the onset of efficacy.9 There is a need for novel CsA formulations with improved ocular bioavailability.
OTX-101 (CEQUA™; Sun Pharmaceutical Industries, Inc.; Cranbury, NJ) is a nanomicellar CsA 0.09% solution approved in the US to increase tear production in patients with KCS.10 The novel nanomicellar formulation is designed to improve CsA delivery to target ocular tissues.11 In a preclinical study in New Zealand white rabbits, OTX-101 0.05% administered topically to the eye achieved higher CsA concentration vs. CsA 0.05% emulsion by factor of 2.18 and 1.76 in the cornea and superior bulbar conjunctiva, respectively, with minimal systemic exposure.12
In both phase 2b/3 and phase 3 studies in patients with KCS, 12 weeks of treatment with OTX-101 0.09% led to significant improvement in the objective signs of tear production, conjunctival staining, and corneal staining vs. vehicle.13,14 Adverse events (AE) were mostly mild or moderate in severity. Pooled analyses confirmed the efficacy of OTX-101 for improving the objective signs of KCS and its favorable safety profile, with significant improvements in corneal staining vs. vehicle evident as early as 4 weeks and persisting through 12 weeks.15–17 We present a phase 3 worse-eye efficacy analysis and a 1-year open label extension evaluating long-term safety of OTX-101 0.09% in KCS.
Methods
Study Design
This phase 3, randomized, multicenter study included a 12-week, double-masked, vehicle-controlled treatment phase and a subsequent long-term safety extension phase of up to 52 weeks. Primary efficacy and safety results of the treatment phase have been previously described.14 The current analysis focuses on the worse-eye efficacy results of the treatment phase, and on the long-term safety results of the extension phase.
The study adhered to the International Council for Harmonisation guidelines and all applicable US federal regulations and local legal and regulatory requirements. The study protocol was approved by a central Institutional Review Board and all patients provided written informed consent prior to screening.
Men and women ≥18 years of age were eligible to enroll in the treatment phase if they had a self-reported history of KCS for ≥6 months prior to screening and clinical diagnosis of bilateral KCS. Additional key inclusion criteria were lissamine green conjunctival staining sum score excluding superior zones of 3 to 9 out of a possible score of 12 in the same eye, global symptom assessment in dry eye score ≥40 both at screening and baseline visits, corrected Snellen visual acuity (VA) score better than 20/200 in each eye, and willingness to discontinue any current dry eye treatment. Patients who successfully completed the treatment phase were eligible to enroll in the long-term safety extension if they had corrected Snellen VA better than 20/200 in each eye at week 12 of the treatment phase.
Key exclusion criteria for the treatment phase were previous treatment failure with or use within 3 months before screening of cyclosporine ophthalmic emulsion 0.05%, diagnosis of Sjögren’s disease >5 years before screening, and corneal refractive surgery within the 6 months before screening. Patients were excluded from the long-term safety extension phase if they experienced an AE that had not resolved by week 12 of the treatment phase or used any topical cyclosporine preparation (excluding OTX-101) after the week 12 visit of the treatment phase.
At the start of the treatment phase, all enrolled patients received 1 drop of vehicle in each eye twice daily for 14 to 20 days to wash out previous ocular medications (Figure 1). The vehicle consisted of the OTX-101 nanomicellar formulation without CsA and contained the inactive ingredients polyoxyl hydrogenated castor oil, Octoxynol-40, polyvinylpyrrolidone, sodium phosphate monobasic dihydrate, sodium phosphate dibasic anhydrous, sodium hydroxide or hydrochloric acid for pH adjustment, and water for injection.10 Patients were then randomized 1:1 to either 1 drop of OTX-101 0.09% or vehicle in each eye twice daily for 12 weeks. Following completion of the 12-week treatment phase, all patients received 1 drop of OTX-101 0.09% in each eye twice daily for 40 weeks during the long-term safety extension. Patients who had received prior vehicle during the treatment phase could choose to participate in the study for an additional 12 weeks for a total of 52 weeks of OTX-101 treatment.
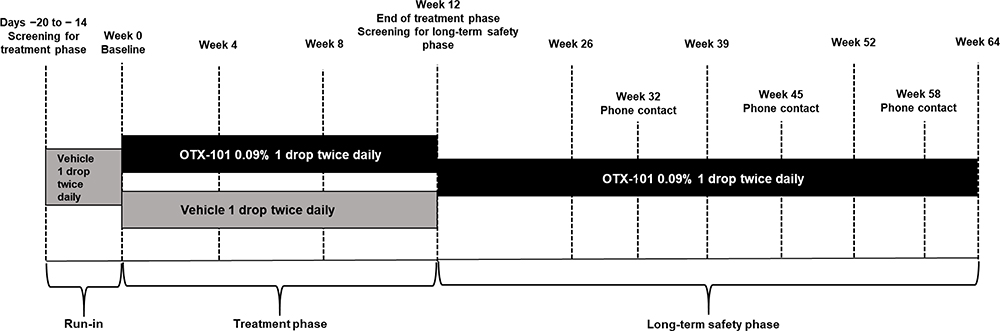 |
Figure 1 Study design. |
The treatment phase included 5 total visits (Figure 1). A screening visit was conducted 14 to 20 days before the baseline visit (day 0). Safety and efficacy assessments were performed at 3 additional visits at weeks 4 ± 3 days, 8 ± 3 days, and 12 ± 7 days. Both eyes were assessed at each visit, and the worse eye was defined as the eye with lower baseline Schirmer’s score for Schirmer’s test and conjunctival staining assessments and as the eye with lower baseline CFS score for CFS assessments. The long-term safety extension phase included 5 total visits and 3 phone contacts. The screening visit took place 14 to 20 days before the start of the long-term safety extension phase and could be conducted at the same day as the week 12 visit of the treatment phase. Safety assessments were performed during visits at weeks 26 ± 7 days, 39 ± 7 days, 52 ± 14 days, and 64 ± 14 days; and during phone contacts at weeks 32 ± 7 days, 45 ± 7 days, and 58 ± 7 days. Weeks were counted since baseline of the treatment phase and excluded the time period between screening and start of the long-term safety extension phase.
Efficacy Assessments
Secondary efficacy assessments during the treatment phase included worse-eye results for Schirmer’s scores, lissamine green conjunctival staining, and complete clearing of central corneal staining. Unanesthetized Schirmer’s test was performed at baseline and week 12/early discontinuation by placing strips in both eyes at the same time and recording the wet strip length after 5 minutes.
Conjunctival staining and corneal staining were performed at screening; baseline; and weeks 4, 8, and 12/early discontinuation. For conjunctival staining, 1 drop (10 µL) of 1% lissamine green solution was instilled in each eye, and after 1 to 4 minutes, staining was measured under low-to-moderate intensity white light using a slit-lamp. Scoring was performed separately for 6 zones of the conjunctiva (nasal, nasal superior, temporal superior, nasal inferior, temporal inferior, and temporal) using a 4-grade scale ranging from 0 (no staining) to 3 (densely concentrated macropunctate stain spots). The total conjunctival staining score was calculated as the sum of scores from all individual zones excluding the 2 superior zones. Worse-eye analysis of conjunctival staining in individual zones also excluded the superior zones.
Corneal fluorescein staining was performed by instilling 1 drop (10 µL) of 0.5% fluorescein solution into the conjunctival cul-de-sac followed by adequate blinking. Five zones of the cornea (central, nasal, temporal, superior, and inferior) were graded separately using the Expanded National Eye Institute/Industry Workshop Scale for Corneal Staining Score using 0.5 score increments from 0 (no staining) to 4 (severe diffuse macropunctate staining).
Safety Assessments
The primary endpoint was long-term safety of OTX-101 0.09% during the open-label extension phase, as assessed by corrected Snellen VA, slit-lamp examination (SLE), intraocular pressure (IOP), ophthalmoscopy and dilated fundus examination, and AE monitoring. AEs were defined as any untoward medical occurrence in a patient who received OTX-101.
Corrected Snellen VA, SLE, and IOP assessments were performed at every study visit other than screening. For corrected Snellen VA, patients were required to read ≥50% of the letters on a single line to accept the line, and the refraction used at the screening visit for the treatment phase was used for all VA assessments during the long-term safety phase. For IOP, measurements for the same patient were recorded using the same tonometer and the same observer each time, if possible. Ophthalmoscopy/dilated fundoscopy were performed at Screening and week 12 of the treatment phase, and at weeks 52 and 64/early discontinuation of the safety extension. AEs were monitored throughout the study and reported according to the Medical Dictionary for Regulatory Activities v19.0.
Statistical Analyses
The intent-to-treat (ITT) population included all patients enrolled in the treatment phase. The safety population included all patients enrolled in the long-term safety phase who received ≥1 dose of study drug.
Continuous variables were summarized with descriptive statistics (n, mean, median, standard deviation [SD], minimum, and maximum) while categorical variables were summarized with counts and percentages.
Comparisons of mean change from baseline used a restricted maximum likelihood repeated measures mixed model, with baseline as a covariate, and treatment group as a fixed factor. Missing Schirmer’s test results at week 12 were imputed from baseline measurements. For comparisons of binary response measures, P-values were calculated with the SAS PROC GENMOD procedure, using visits as repeated measures, with treatment as a fixed effect using an unstructured correlation matrix. Estimated rate and 95% confidence interval were provided for both treatment groups and for the difference between groups.
Results
Patient Disposition and Baseline Demographics
A total of 745 patients enrolled in the phase 3 study—372 randomized to OTX-101 0.09% and 373 randomized to vehicle—as previously described (Supplementary Figure 1).14 One patient randomized to OTX-101 withdrew consent before receiving any study medication and was not included in the efficacy analyses. The treatment phase was successfully completed by 347/372 (93.3%) patients randomized to OTX-101 and 361/373 (96.8%) patients randomized to vehicle. Of these, 258 patients—129 from each prior treatment group—continued to the long-term safety phase; 111/129 (86.0%) from the prior OTX-101 group and 79/129 (61.2%) from the prior vehicle group completed the study (Figure 2). Major reasons for discontinuation (n [%] of patients in prior OTX-101 vs. prior vehicle group) included AEs (8 [6.2%] vs. 14 [10.9%]), patient’s decision (4 [3.1%] vs. 12 [9.3%]), and administrative error and sponsor’s decision to end study treatment (0 vs. 11 [8.5%] each).
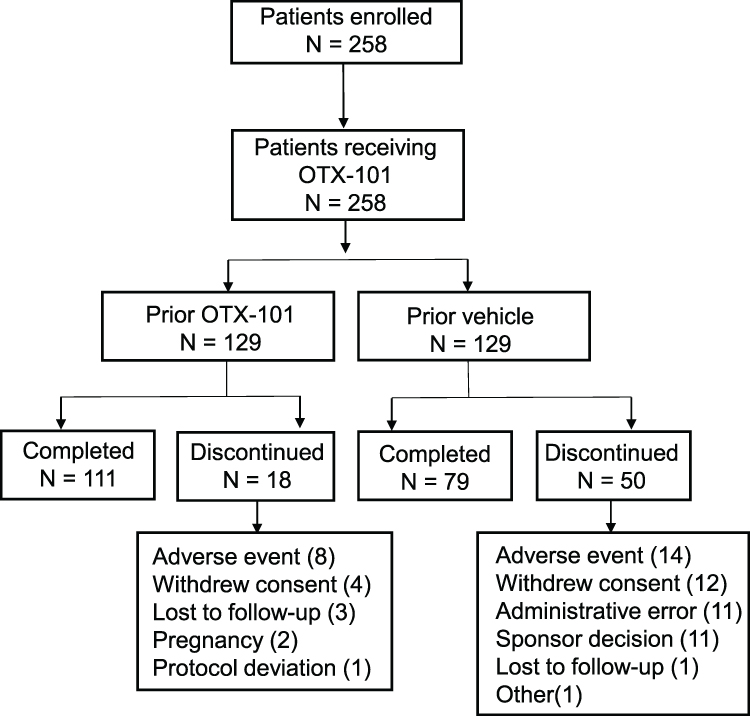 |
Figure 2 Patient disposition of the long-term safety phase. |
The mean (SD) age was 59.0 (14.4) years for patients enrolled in the treatment phase (Supplementary Table 1) and 60.0 (14.9) years for patients continuing to the long-term safety phase (Table 1). Throughout the study, the patient population was predominately female (84.1% and 83.7% for the treatment and long-term safety phase, respectively) and white (82.7% and 85.7% for the treatment and long-term safety phase, respectively).
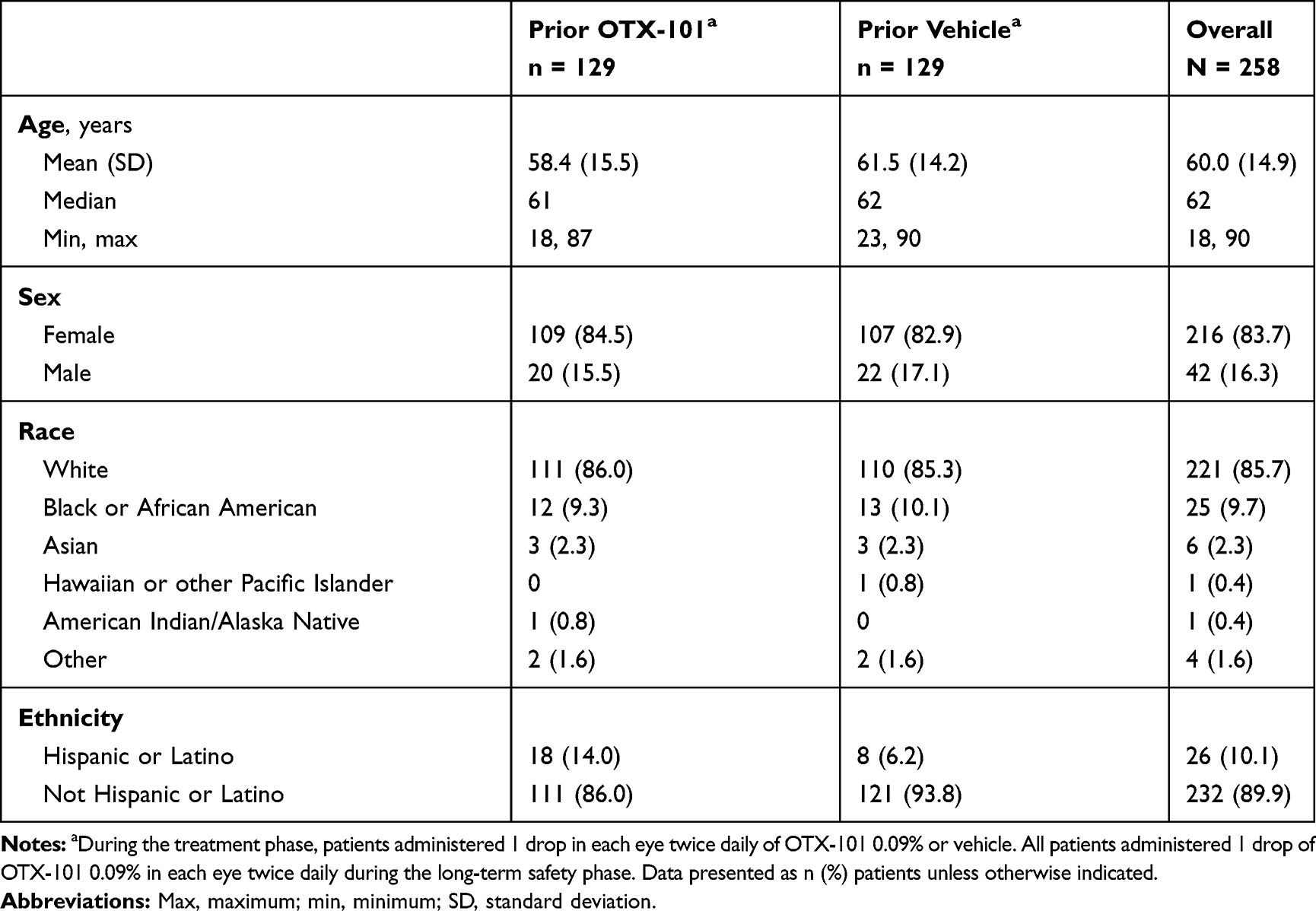 |
Table 1 Baseline Demographics of Patients Enrolled in the Long-Term Safety Phase |
Efficacy
Schirmer’s Test
Baseline mean (SD) Schirmer’s scores in the worse eye were 9.7 (7.2) and 10.2 (7.4) mm for the OTX-101 and vehicle groups, respectively (Table 2). Mean (SD) change from baseline in Schirmer’s scores at week 12 was significantly higher in the OTX-101 group (4.0 [7.8] mm) vs. the vehicle group (2.2 [6.8] mm, P = 0.0017). The number (%) of patients with an increase in Schirmer’s scores of ≥10 mm at week 12 relative to baseline was also higher in patients receiving OTX-101 vs. vehicle (76 [20.5%] vs. 42 [11.3%], respectively; P = 0.0005; Figure 3).
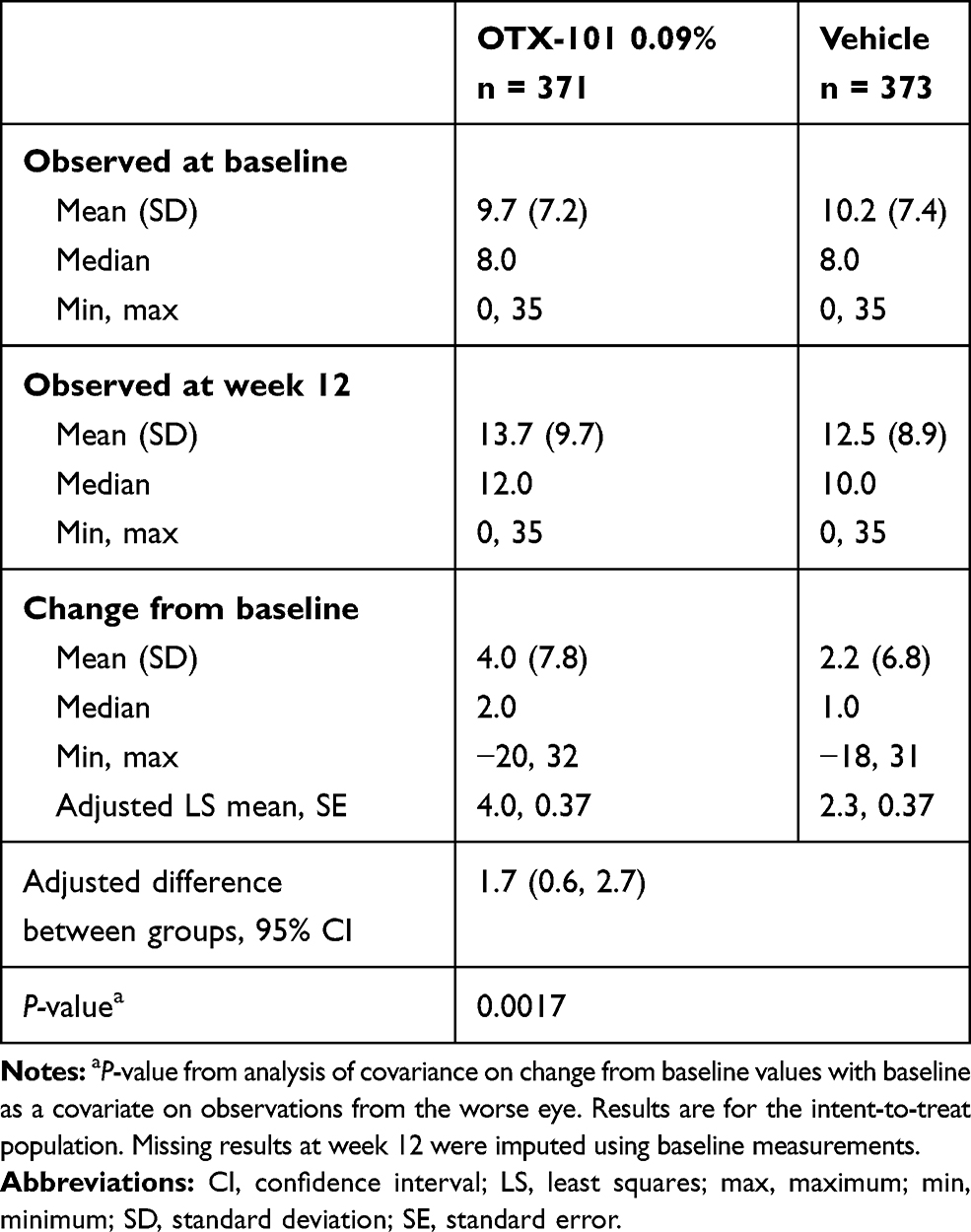 |
Table 2 Observed and Change from Baseline in Worse-Eye Schirmer’s Scores |
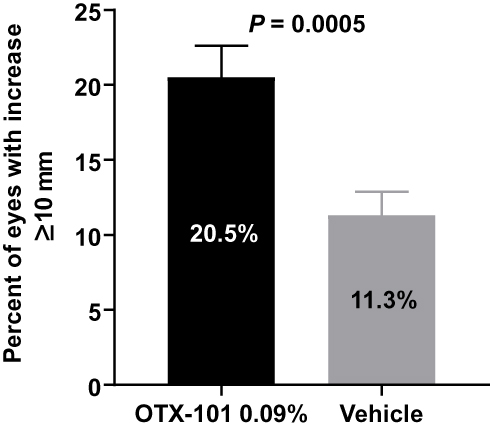 |
Figure 3 Percentage of worse eyes with Schirmer’s score increase from baseline ≥10 mm at week 12. Analysis included 371 patients receiving OTX-101 and 373 patients receiving vehicle. Abbreviation: CI, confidence interval. |
Conjunctival Staining
Mean (SD) total conjunctival staining scores in the worse eye at baseline were 5.50 (1.92) and 5.53 (1.96) for the OTX-101 and vehicle groups, respectively. OTX-101 significantly improved total conjunctival staining vs. vehicle in the worse eye at weeks 8 (least squares [LS] mean change from baseline [SE] of −1.48 [0.11] vs. −0.87 [0.11], respectively; P <0.0001) and week 12 (LS mean change from baseline [SE] of −1.65 [0.12] vs. −1.12 [0.12], respectively; P = 0.0013) (Figure 4). In individual zones in the worse eye, conjunctival staining scores were significantly improved in the OTX-101 vs. vehicle group at weeks 8 and 12 for the temporal zone (P = 0.0072 and 0.0022, respectively), inferior temporal zone (P = 0.0002 and 0.0059, respectively), and inferior nasal zone (P = 0.0006 and 0.0047, respectively). There were no significant differences between OTX-101 and vehicle in LS mean change from baseline in the nasal zone.
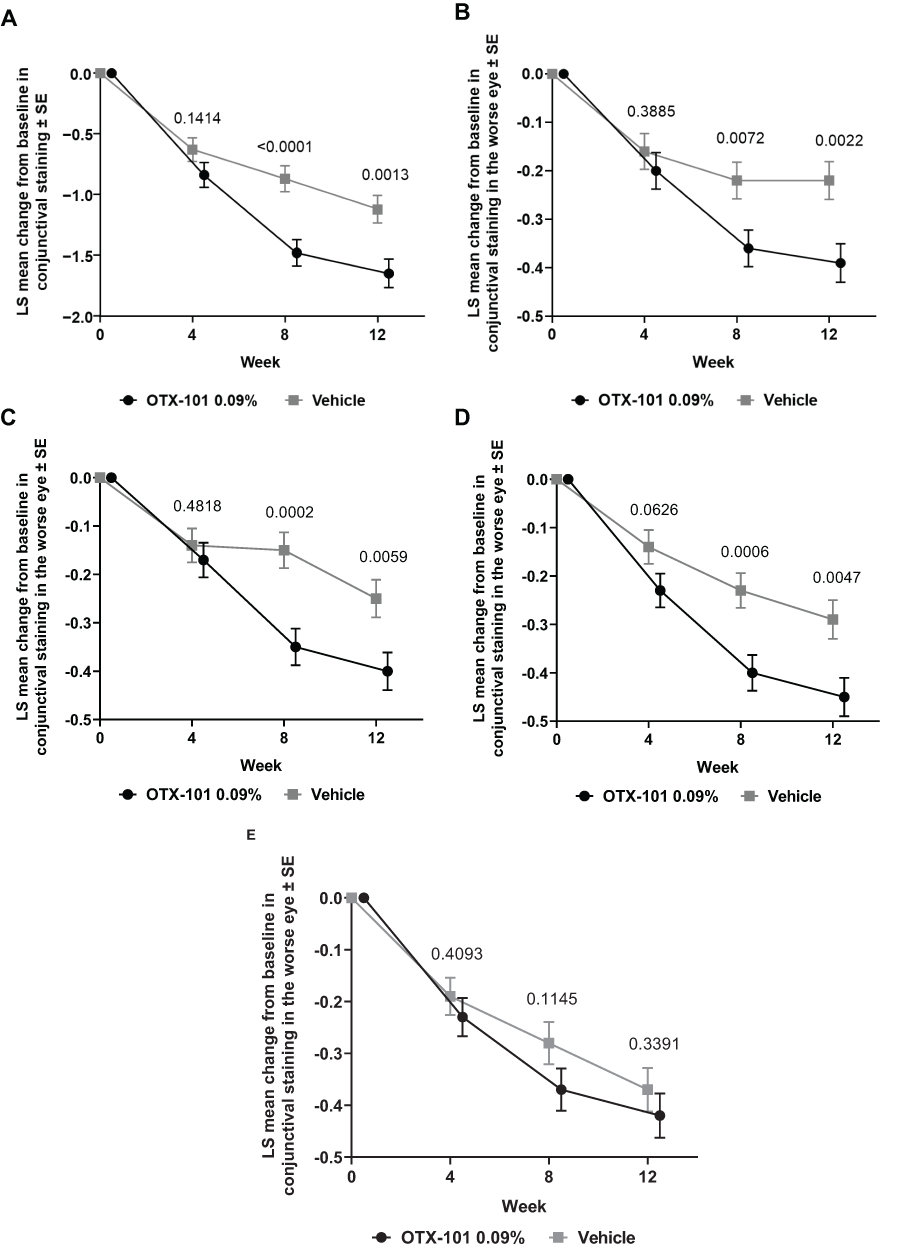 |
Figure 4 Least squares mean change from baseline in conjunctival staining in the worse eye for (A) total (temporal, nasal, and inferior zones), (B) temporal, (C) inferior temporal, (D) inferior nasal, and (E) nasal zones. Abbreviations: LS, least squares; SE, standard error. |
Percent of Patients with Complete Central Corneal Clearing
At baseline, the number (%) of patients with clear central corneas (central corneal fluorescein staining [CFS] score of 0) in the worse eye was 107 (28.8%) and 105 (28.2%) for the OTX-101 and vehicle groups, respectively (Figure 5). The number (%) of patients with clear central corneas in the worse eye was significantly higher in patients receiving OTX-101 vs. vehicle at week 4 (196 [55.1%] vs. 169 [45.6%], P = 0.0101) and week 12 (222 [64.0%] vs. 199 [55.3%], P = 0.0179), and numerically higher at week 8 (209 [59.5%] vs. 194 [53.2%], P = 0.0839).
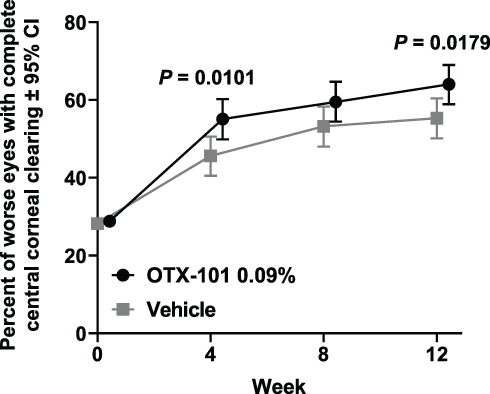 |
Figure 5 Percentage of worse eyes with complete clearing of the central cornea. Abbreviation: CI, confidence interval. |
Safety
Duration of Exposure
Median (SD) duration of exposure to OTX-101 during the long-term safety phase was 9.47 (2.40) months for the prior OTX-101 group, 9.53 (3.24) months for the prior vehicle group, and 9.50 (2.86) months overall.
Clinical Safety Evaluations
Clinically significant abnormal findings from the SLE are summarized in Table 3. Clinically significant findings for SLE were reported as AEs in 4 patients from the prior OTX-101 group and 6 patients from the prior vehicle group. Of these, 3 patients in the prior vehicle group experienced mild conjunctival hyperemia occurring bilaterally that was considered related to study treatment but did not result in interruption or permanent withdrawal of study treatment. All other clinically significant abnormal SLE findings were not considered treatment-related.
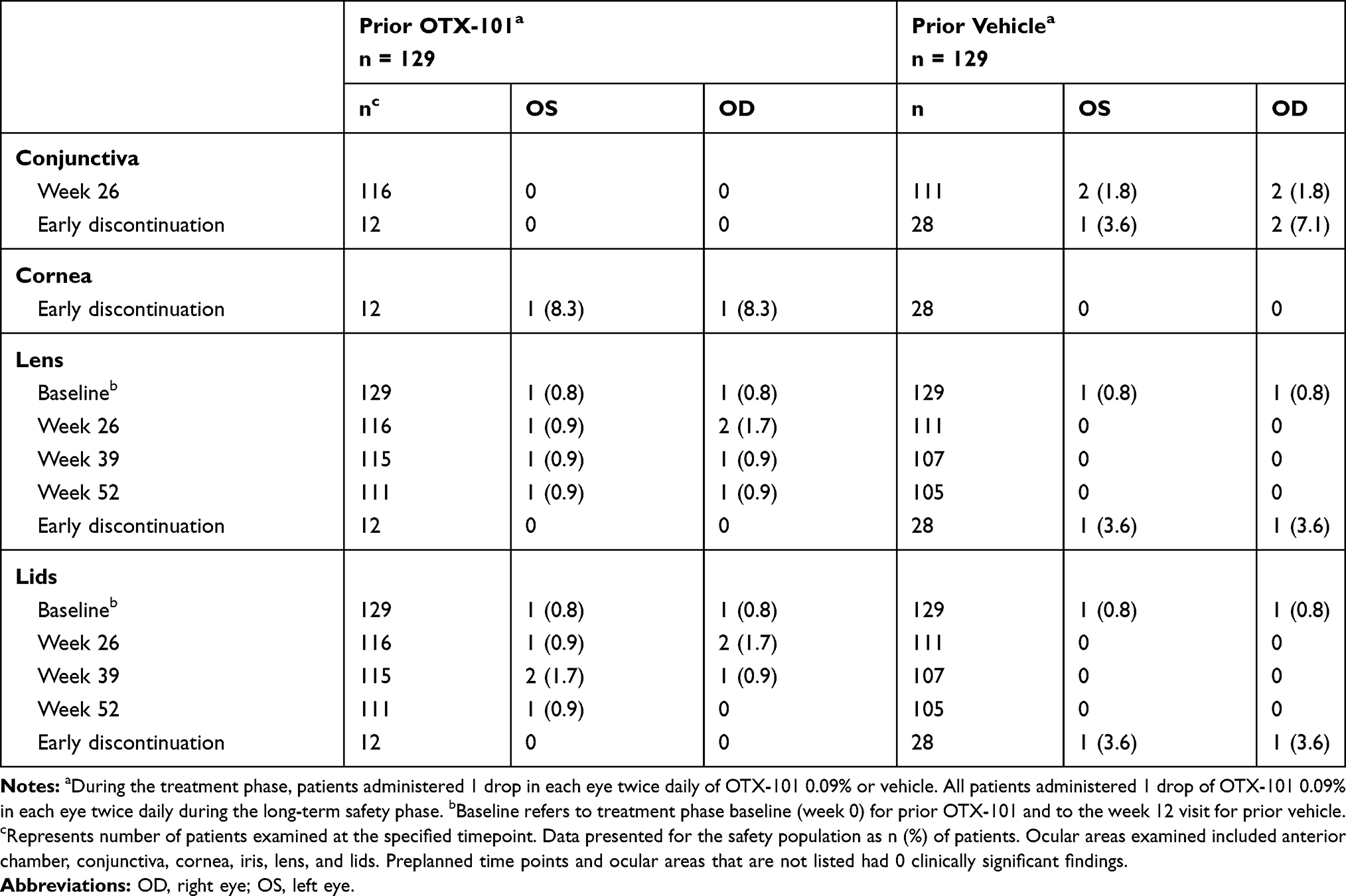 |
Table 3 Clinically Significant Abnormal Findings from Slit Lamp Examinations During the Long-Term Safety Phase |
There were no patterns or relationships apparent from dilated fundoscopy observations. All abnormal clinically significant findings (Table 4) were mild, not considered treatment related, and did not require any action regarding changes in study medication.
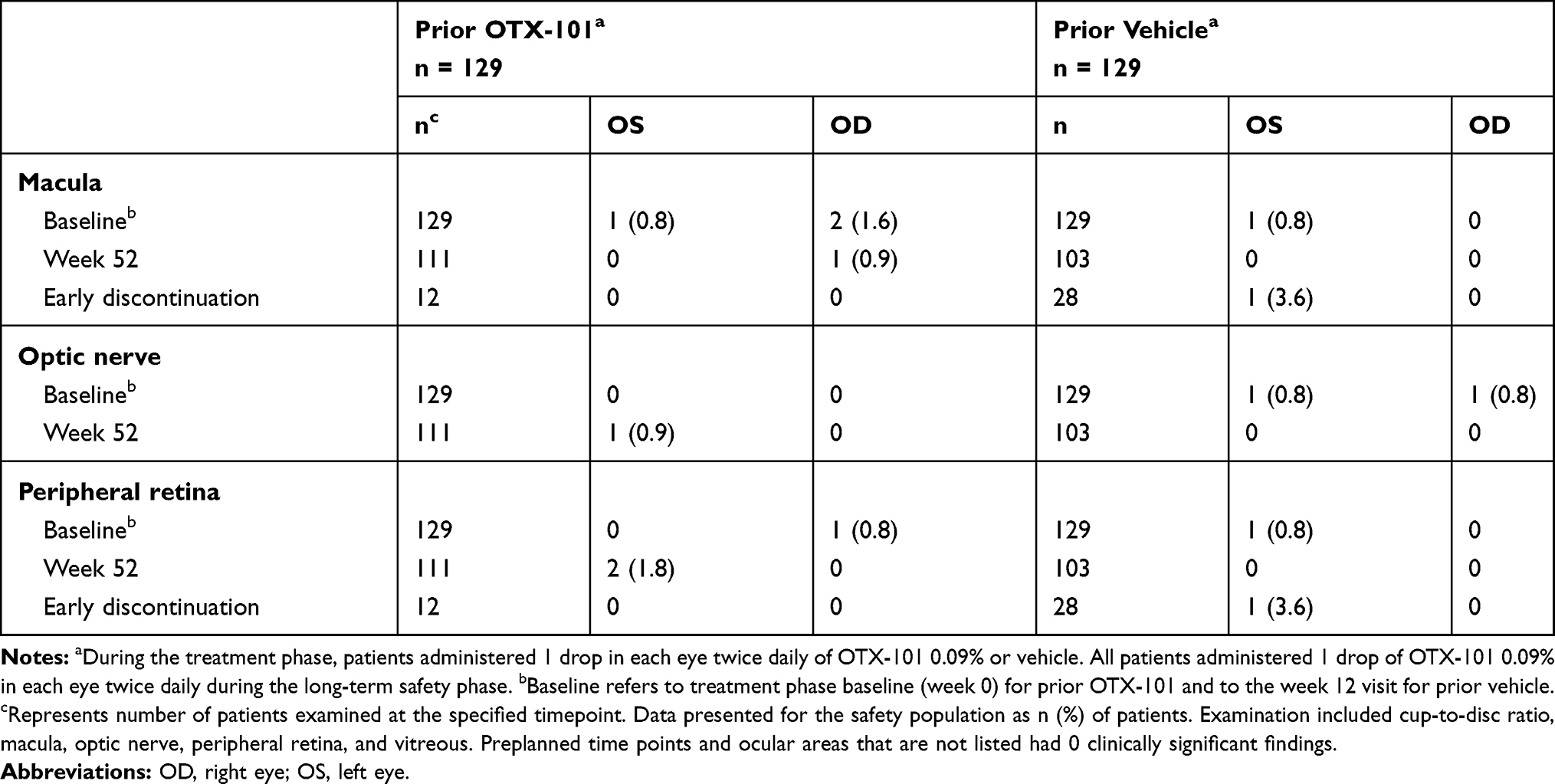 |
Table 4 Clinically Significant Abnormal Findings from Dilated Fundoscopy During the Long-Term Safety Phase |
Overall, Snellen VA assessments during the long-term safety phase did not exhibit any consistent clinically significant changes or trends (Supplemental Table 2). Two events of decreased VA were reported as AEs in 2 patients in the prior OTX-101 group. Both events were mild, transient, and not considered clinically significant.
All observed IOP values were within normal limits for both the right and left eye (Supplemental Table 2). Increased IOP was reported as an AE for 3 patients, 2 in the prior OTX-101 group and 1 in the prior vehicle group. These AEs were graded as mild, not considered related to study treatment, and resolved or were resolving at the time of last follow-up.
Adverse Events
A total of 149 (57.8%) patients (68 [52.7%] and 81 [62.8%] in the prior OTX-101 and vehicle groups, respectively) reported AEs during the long-term safety phase. The most common AE was instillation site pain in 59 (22.9%) patients (17 [13.2%] and 42 [32.6%] in the prior OTX-101 and vehicle groups, respectively) (Table 5). AEs, including instillation site pain, were mostly mild or moderate (Table 6). Severe instillation site pain in 1 (0.8%) patient in the prior vehicle group was the only severe AE suspected to be related to study drug. No serious AEs were ocular or considered to be related to study drug.
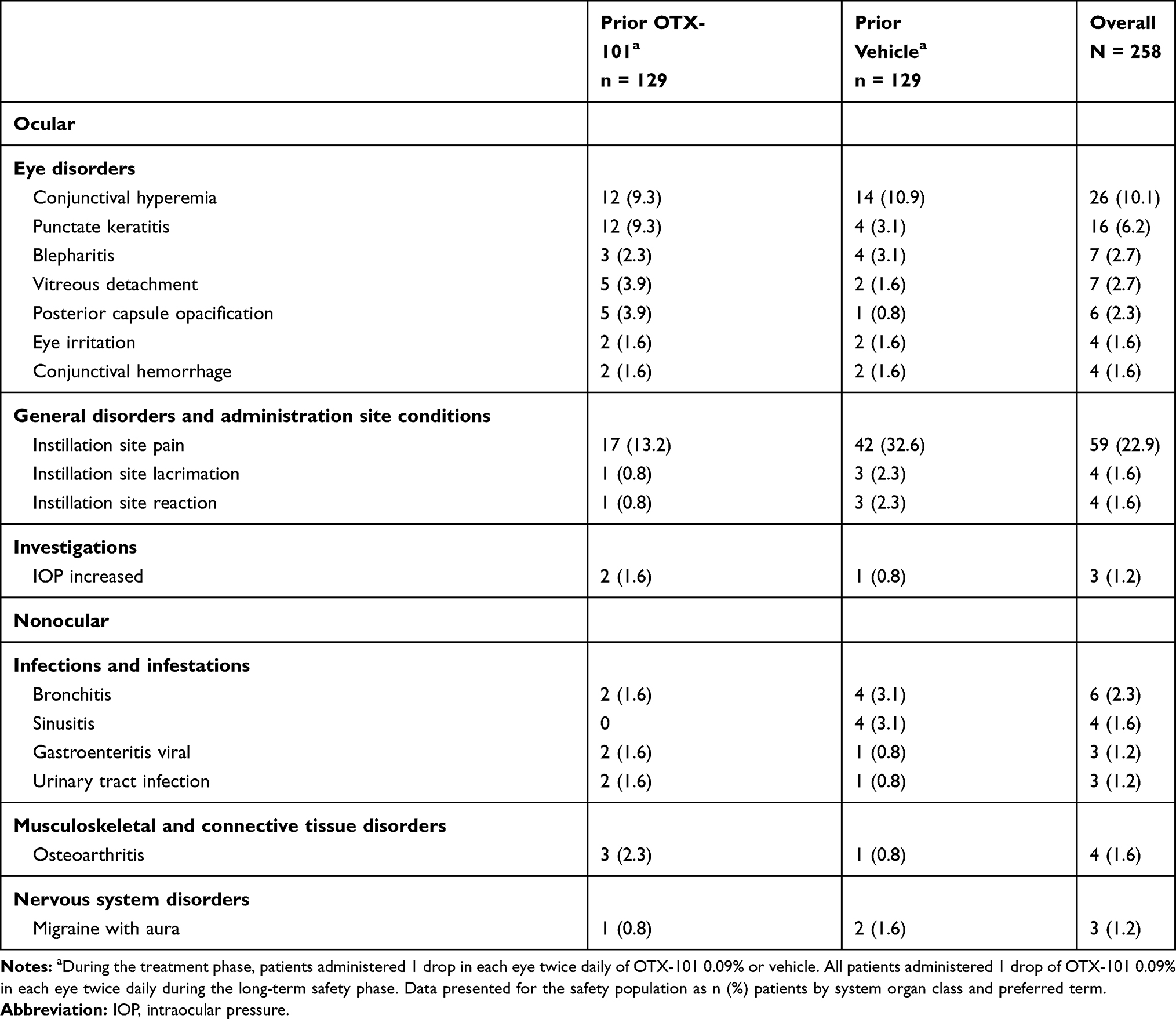 |
Table 5 Treatment-Emergent Adverse Events in ≥1% of Patients During the Long-Term Safety Phase |
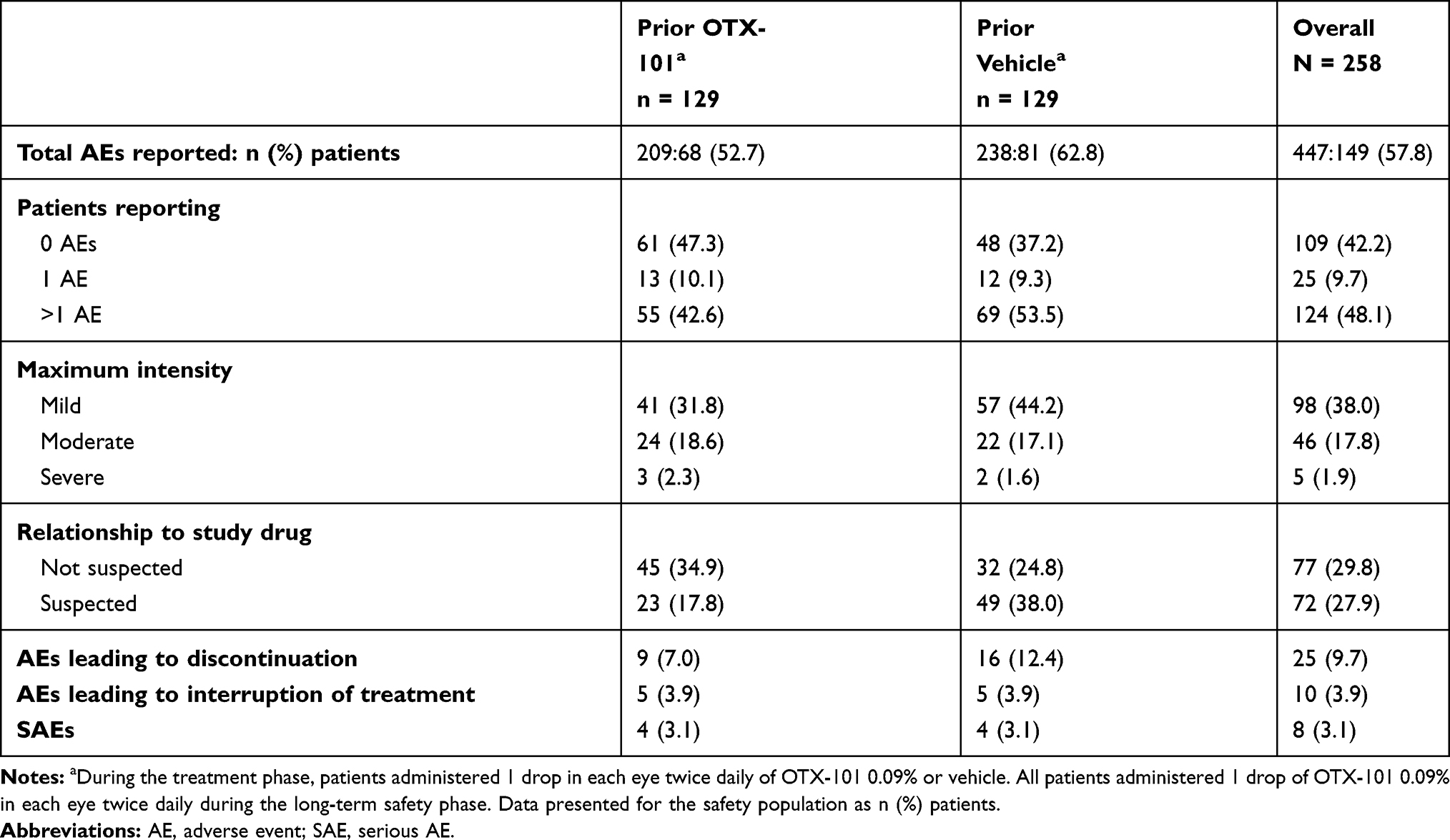 |
Table 6 Summary of Treatment-Emergent Adverse Events During the Long-Term Safety Phase |
Discussion
In this follow up, worse-eye analysis confirmed the rapid efficacy of OTX-101 for improving the objective signs of KCS seen in the phase 3 study and pooled analyses. Twelve weeks of treatment with OTX-101 0.09% significantly improved tear production, as demonstrated by significantly higher percent of eyes with increase from baseline in Schirmer’s score ≥10 mm compared with vehicle. This was the primary endpoint met in the phase 3 study, and the result favoring OTX-101 efficacy was verified when examining the eyes with lower Schirmer’s score in the ITT population. Significant improvement in total conjunctival staining vs. vehicle was evident as early as week 8 and persisted through the study. The percent of eyes with complete corneal clearing was significantly improved vs. vehicle at weeks 4 and 12.
The 1-year open-label extension phase is the longest follow up of OTX-101 in patients with KCS. Results from this long-term safety phase suggest a favorable tolerability profile of OTX-101 consistent with the one previously evaluated during the treatment phase. AEs were predominately mild and clinical safety evaluations did not raise any long-term safety concerns.
The Tear Film & Ocular Surface Society Dry Eye Workshop II recommends the use of topical ophthalmic cyclosporine in the treatment of KCS as supported by Level 1 evidence, but cautions that currently there is no cure for KCS and treatment with ophthalmic cyclosporine needs to be continued for an extended period of time.5 According to a comprehensive review of studies published in March 2018 evaluating CsA ophthalmic emulsion 0.05% in KCS, very few studies include long-term follow-up of patients treated with CsA.18 The safety results presented here are relevant to a real-world setting and stand out among the scarcity of long-term CsA data.
Limitations of the current study include the lack of vehicle comparator and lack of efficacy assessments during the safety extension phase. Another limitation is the lack of Schirmer’s test assessments between the baseline and week 12 visits. Significant improvement in tear production vs. vehicle was evident at week 12, but the time at which this improvement first emerges during treatment is not known. An additional limitation is the exclusion of patients with previous treatment failure or recent use of cyclosporine ophthalmic emulsion 0.05%. We did not include these patients as we did not feel it was ethically appropriate to include patients who had a history of failed treatment with the active ingredient in OTX-101.
CsA oil-in-water emulsion 0.05% (Restasis®, Allergan, Irvine, CA) was the first ophthalmic CsA formulation approved in the US for increasing tear production in patients with KCS.19 The most common AE associated with Restasis use is ocular burning in 17% of patients. Other common AEs reported in 1% to 5% of patients include conjunctival hyperemia, eye pain, foreign body sensation, and visual blurring.19
Conclusion
Overall, OTX-101 demonstrated efficacy in improving the objective KCS signs of tear production and conjunctival staining or corneal staining in the worse eye (ie, with lower or equal baseline Schirmer’s score or CFS score respectively) during the 12-week treatment phase. OTX-101 was well tolerated in patients with KCS during a 1-year safety extension phase. Results presented here support the long-term use of OTX-101 for the treatment of KCS.
Abbreviations
AE, adverse event; CFS, corneal fluorescein staining; CI, confidence interval; CsA, cyclosporine A; IOP, intraocular pressure; KCS, keratoconjunctivitis sicca; logMAR, logarithm of the minimum angle of resolution; LS, least squares; max, maximum; min, minimum; OD, right eye; OS, left eye; SAE, serious adverse event; SD, standard deviation; SE, standard error; SLE, slit-lamp examination; VA, visual acuity.
Data Sharing Statement
Data and other documents will be made available after publication, with no end date, to anyone who submits a reasonable request to the study sponsor (Sun Pharmaceutical Industries, Inc., Princeton, NJ).
Ethics Approval and Consent to Participate
The study adhered to the International Council for Harmonisation guidelines and all applicable US federal regulations and local legal and regulatory requirements. The study protocol was approved by Sterling Institutional Review Board (Atlanta, GA, US) and all patients provided informed consent prior to screening.
Acknowledgments
The authors would like to thank Charles Darby for his assistance with statistical analysis. Writing and editorial support for manuscript preparation were provided by Jennifer Meyering, RN, MS, CMPP, of AlphaBioCom, LLC, and funded by Sun Pharmaceutical Industries, Inc. All authors met the International Council of Medical Journal Editors criteria and received neither honoraria nor payment for authorship. Portions of this manuscript were presented at the 2019 Academy of Managed Care annual meeting and SECO 2020 as poster presentations with interim findings.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was sponsored and funded by Ocular Technologies, SARL (now a wholly owned subsidiary of Sun Pharmaceutical Industries, Inc.). Ocular Technologies, SARL, participated in the design, conduct, monitoring, data collection, data management, and data analysis of the study.
Disclosure
JS reports personal fees from Allergan; Alcon/Novartis; Avedro; Bausch + Lomb; Clementia Pharma; EyeGate, Ocular Therapeutix; Science Based Health; Senju; Shire; Sun Pharmaceutical Industries, Inc.; TearLab; TearScience; Topcon; and TopiVert; and other fees from Alcon/Novartis, Kala Pharmaceuticals, Novaliq, Noveome, Ocular Therapeutix, Shire, and TearLab. MB receives fees from ICON Clinical Research, InSite Vision, Kala Pharmaceuticals, Oculos Clinical Research, RevitaLid, and Shire. BS receives fees from Johnson & Johnson; Shire; Valeant; and Sun Pharmaceutical Industries, Inc. JL receives personal fees from Alcon; Allergan; Bausch + Lomb; Eyevance, Icare, Shire; TearLab; and Sun Pharmaceutical Industries, Inc; and equity interest from Calhoun Vision, CLXO, Insightful Solutions, Ocular Sciences, Omega Ophthalmics, Trefoil Therapeutics, and RPS. AO is an employee of Sun Pharmaceutical Industries, Inc. PK reports consultant fees from Aerie; Akorn; Alcon; Allergan; Avellino Labs; Bausch & Lomb; Beaver Visitech; Bio-Tissue; Blephex; Bruder; Cambium; DGH Technology; EyeBrain/Neurolens; EyeGate Pharma; Eyevance; Focus Labs; Imprimis; Ivantis; Jobson Medical Information/Web MD; Johnson & Johnson Vision; Konan Medical; LensTech; Novartis; Oasis Medical; Ocular Sciences; Oculus; OcuMedic; OcuSoft; Oyster Point Medical; Reichert/Ametek; Science Based Health; Sentiss; Shire; Sight Sciences; Silk Technologies; Sun Pharmaceutical Industries, Inc.; Tarsus Medical; TearFilm Innovations; Tear Lab; Total Eyecare Partners; Topcon; Visant Medical; Visionix; and Vital Tears. The authors report no other conflicts of interest in this work.
References
1. Craig JP, Nichols KK, Akpek EK, et al. TFOS DEWS II definition and classification report. Ocul Surf. 2017;15(3):276–283. doi:10.1016/j.jtos.2017.05.008
2. Craig JP, Nelson JD, Azar DT, et al. TFOS DEWS II report executive summary. Ocul Surf. 2017;15(4):802–812. doi:10.1016/j.jtos.2017.08.003
3. Wolffsohn JS, Arita R, Chalmers R, et al. TFOS DEWS II diagnostic methodology report. Ocul Surf. 2017;15(3):539–574. doi:10.1016/j.jtos.2017.05.001
4. Stapleton F, Alves M, Bunya VY, et al. TFOS DEWS II epidemiology report. Ocul Surf. 2017;15(3):334–365. doi:10.1016/j.jtos.2017.05.003
5. Jones L, Downie LE, Korb D, et al. TFOS DEWS II management and therapy report. Ocul Surf. 2017;15(3):575–628. doi:10.1016/j.jtos.2017.05.006
6. Messmer EM. The pathophysiology, diagnosis, and treatment of dry eye disease. Dtsch Arztebl Int. 2015;112(5):
7. Rhee MK, Mah FS. Clinical utility of cyclosporine (CsA) ophthalmic emulsion 0.05% for symptomatic relief in people with chronic dry eye: a review of the literature. Clin Ophthalmol. 2017;11
8. Lallemand F, Schmitt M, Bourges JL, Gurny R, Benita S, Garrigue JS. Cyclosporine A delivery to the eye: a comprehensive review of academic and industrial efforts. Eur J Pharm Biopharm. 2017;117:
9. Kuwano M, Ibuki H, Morikawa N, Ota A, Kawashima Y. Cyclosporine A formulation affects its ocular distribution in rabbits. Pharm Res. 2002;19(1):108–111. doi:10.1023/A:1013671819604
10. CEQUATM (cyclosporine opthalmic solution 0.09%). Full prescribing information. Cranbury, NJ: Sun Pharmaceutical Industries, Inc.; 2018.
11. Cholkar K, Gilger BC, Mitra AK. Topical, aqueous, clear cyclosporine formulation design for anterior and posterior ocular delivery. Transl Vis Sci Technol. 2015;4(3):1. doi:10.1167/tvst.4.3.1
12. Weiss SL, Kramer WG. Ocular distribution of cyclosporine following topical administration of OTX-101 in New Zealand white rabbits. J Ocul Pharmacol Ther. 2019;35(7):395–402. doi:10.1089/jop.2018.0106
13. Tauber JSB, Bacharach J, Toyos MM, Smyth-Medina R, Weiss SL, Luchs JI. A Phase II/III, randomized, double-masked, vehicle-controlled, dose-ranging study of the safety and efficacy of OTX-101 in the treatment of dry eye disease. Clin Ophthalmol. 2018;12:1921–1929. doi:10.2147/OPTH.S175065
14. Goldberg DF, Malhotra RP, Schechter BA, Justice A, Weiss SL, Sheppard JD. A Phase 3, randomized, double-masked study of OTX-101 ophthalmic solution 0.09% in the treatment of dry eye disease. Ophthalmology. 2019;126(9):1230–1237. doi:10.1016/j.ophtha.2019.03.050
15. Malhotra R, Devries DK, Luchs J, et al. Effect of OTX-101, a novel nanomicellar formulation of Cyclosporine A, on Corneal staining in patients with keratoconjunctivitis sicca: a pooled analysis of Phase 2b/3 and Phase 3 studies. Cornea. 2019;38(10):1259–1265. doi:10.1097/ICO.0000000000001989
16. Smyth-Medina R, Johnston J, Devries DK, et al. Effect of OTX-101, a novel nanomicellar formulation of Cyclosporine A, on conjunctival staining in patients with keratoconjunctivitis sicca: a pooled analysis of Phase 2b/3 and 3 clinical trials. J Ocul Pharmacol Ther. 2019;35(7):388–394. doi:10.1089/jop.2018.0154
17. Sheppard J, Kannarr S, Luchs J, et al. Efficacy and safety of OTX-101, a novel nanomicellar formulation of Cyclosporine A, for the treatment of keratoconjunctivitis sicca: pooled analysis of a Phase 2b/3 and Phase 3 study. Eye Contact Lens. 2020;46(Suppl 1):S14–S19. doi:10.1097/ICL.0000000000000636
18. Deveney T, Asbell PA. Patient and physician perspectives on the use of cyclosporine ophthalmic emulsion 0.05% for the management of chronic dry eye. Clin Ophthalmol. 2018;12:569–576. doi:10.2147/OPTH.S115098
19. RESTASIS® (cyclosporine ophthalmic emulsion) 0.05% for topical ophthalmic use. Full prescribing information. Irvine, CA: Allergan; 2017.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.