Back to Journals » Drug Design, Development and Therapy » Volume 9
Pharmacological approaches to improving cognitive function in Down syndrome: current status and considerations
Authors Gardiner K
Received 9 September 2014
Accepted for publication 21 October 2014
Published 17 December 2014 Volume 2015:9 Pages 103—125
DOI https://doi.org/10.2147/DDDT.S51476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Katheleen J Gardiner
Linda Crnic Institute for Down Syndrome, Department of Pediatrics, Department of Biochemistry and Molecular Genetics, Human Medical Genetics and Genomics Program, Neuroscience Program, University of Colorado School of Medicine, Aurora, CO, USA
Abstract: Down syndrome (DS), also known as trisomy 21, is the most common genetic cause of intellectual disability (ID). Although ID can be mild, the average intelligence quotient is in the range of 40–50. All individuals with DS will also develop the neuropathology of Alzheimer’s disease (AD) by the age of 30–40 years, and approximately half will display an AD-like dementia by the age of 60 years. DS is caused by an extra copy of the long arm of human chromosome 21 (Hsa21) and the consequent elevated levels of expression, due to dosage, of trisomic genes. Despite a worldwide incidence of one in 700–1,000 live births, there are currently no pharmacological treatments available for ID or AD in DS. However, over the last several years, very promising results have been obtained with a mouse model of DS, the Ts65Dn. A diverse array of drugs has been shown to rescue, or partially rescue, DS-relevant deficits in learning and memory and abnormalities in cellular and electrophysiological features seen in the Ts65Dn. These results suggest that some level of amelioration or prevention of cognitive deficits in people with DS may be possible. Here, we review information from the preclinical evaluations in the Ts65Dn, how drugs were selected, how efficacy was judged, and how outcomes differ, or not, among studies. We also summarize the current state of human clinical trials for ID and AD in DS. Lastly, we describe the genetic limitations of the Ts65Dn as a model of DS, and in the preclinical testing of pharmacotherapeutics, and suggest additional targets to be considered for potential pharmacotherapies.
Keywords: Ts65Dn, pharmacotherapy, clinical trials, Hsa21
Introduction
Down syndrome (DS), also known as trisomy 21, is caused by an extra copy of the long arm of human chromosome 21 (Hsa21). While the phenotype of DS is complex and many aspects are variable in penetrance, common to all individuals are both some level of intellectual disability (ID) and the early development of the neuropathology of Alzheimer’s disease (AD).1 The incidence of DS remains at approximately one in 730 live births in the United States and approximately one in 1,000 within Europe.2,3 Given that the average level of cognitive ability is associated with intelligence quotient scores of 40–50, and that a significant percentage is more profoundly impaired,4,5 pharmacotherapies that could lessen or ameliorate the level of impairment would have substantial social consequences.
The major hypothesis that has been and is driving DS research is that the phenotype is a consequence of elevated levels of expression, due to dosage, of trisomic genes.6 Hsa21 genes that are conserved in mouse include ~160 encoding diverse protein functions, a large family (~45 members) encoding apparently redundant keratin associated proteins, and a number of provisionally annotated microRNAs. Hsa21 also encodes several hundred additional genes/gene models of unknown function that lack detectable nucleotide sequence conservation in the mouse genome.7 Many experiments have examined gene expression in DS-related tissues, including fetal brains, amniocytes, neurospheres, and cell lines derived from DS, as well as brain regions and other tissues and cells derived from some partial trisomy mouse models of DS (summarized by Vilardell et al8). Results, most often generated at the RNA level, have largely supported the gene dosage hypothesis; however, not all trisomic genes are elevated in any one experiment, and which genes are elevated and by precisely how much depends on the organism, genetic background, developmental time point and tissue or cell type.
Expression levels of non-Hsa21 genes are also altered in DS.8 This is expected given that among the known Hsa21 protein-coding genes are 20 transcription factors/modulators; ten proteins involved in the processing and/or modification of messenger (m)RNA, transfer (t)RNA, and ribosomal (r)RNA; nine proteins that function directly and indirectly in protein phosphorylation, methylation, and sumoylation; and 16 proteases, protease inhibitors, and proteins that regulate degradation by the ubiquitin pathway.9 Overexpression of genes in each of these classes would be expected to affect levels of expression and/or activity of many non-Hsa21 genes. Because the levels of Hsa21 proteins vary with time and place, the perturbations in the expression and activity of their non-Hsa21 targets also vary with the tissue, developmental time, and model system. Consideration of the number of Hsa21 genes, together with the complexities of their function, regulation, and expression, might suggest that ID in DS is too complex in its genetic basis to be ameliorated by pharmacotherapeutic intervention. However, over the last several years, there have been a number of reports of the successful rescue of learning and memory (L/M) deficits in a mouse model of DS, the Ts65Dn. Drugs and small molecules with diverse targets and mechanisms of action have been tested in a variety of L/M protocols, and for their effects on cellular and electrophysiological features, in mice that range in age from pre- and early postnatal to young and older adults (see Table 1). In this review, we discuss this substantial literature. We consider the challenges of integrating these results, the shortcomings of the Ts65Dn mouse model, and the vexing problem of planning effective human clinical trials based on data generated for the Ts65Dn model.
|
Table 1 Drugs tested for the rescue of abnormalities in learning/memory, adult neurogenesis, or long-term potentiation in the Ts65Dn |
Modeling DS in mouse and the Ts65Dn
The mouse is the model organism of choice for the preclinical evaluation of drug efficacy. A challenge for modeling of DS is that orthologs of Hsa21 genes are distributed among three mouse chromosomes. Of the ~160 Hsa21 protein-coding genes, ~100 map to the telomeric segment of mouse chromosome 16 (Mmu16), and ~20 and ~40 map to internal segments of Mmu17 and Mmu10, respectively (Figure 1).7 Many partial trisomy models have been created using several different methods, and each is trisomic for a unique subset of Hsa21 genes or their mouse orthologs. These have been reviewed10–12 and will not be discussed here. Instead, this review focuses on a single model, the Ts65Dn, which is the only DS model that has been used in the preclinical evaluation of drugs for L/M.
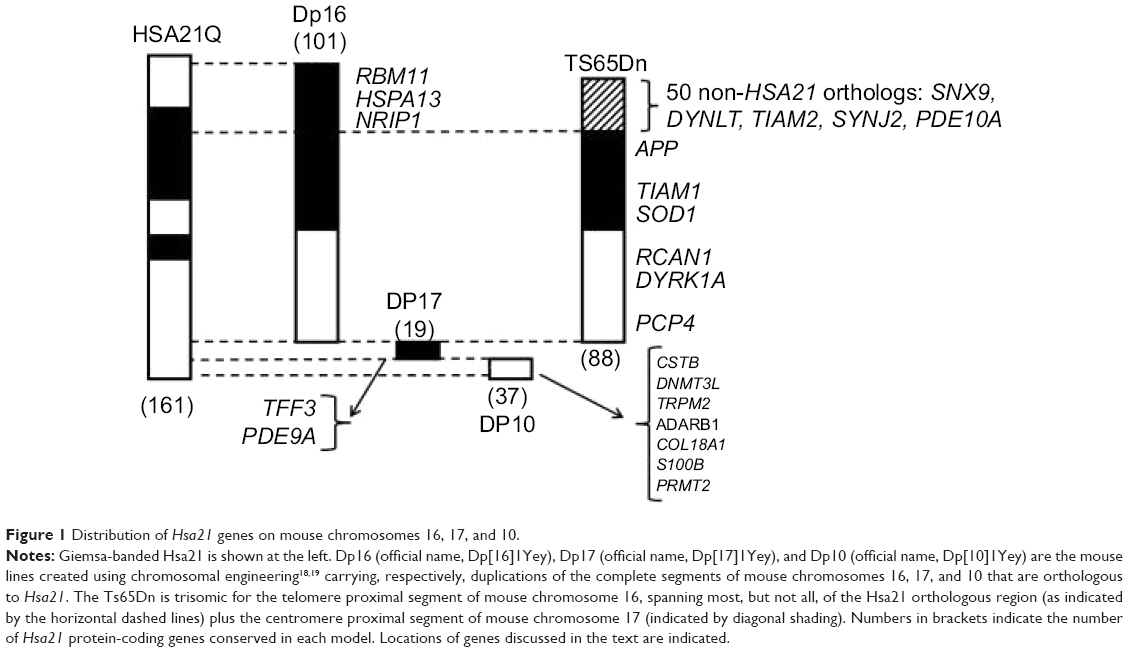 |
Figure 1 Distribution of Hsa21 genes on mouse chromosomes 16, 17, and 10. |
The Ts65Dn was the first viable mouse model of DS and has been available for 20 years.13,14 Because of this history and its long reign as the only viable segmental trisomy, the Ts65Dn remains the most popular model. The Ts65Dn carries a freely segregating marker chromosome composed of the telomere proximal region of Mmu16 translocated to the centromere and pericentromeric region of Mmu17. The Mmu16 segment contains a majority (88 of 102) of the Hsa21 orthologous protein-coding genes that map to Mmu16 – ie, ~55% of the (nonkeratin-associated proteins) Hsa21 protein-coding genes conserved in mouse.7 The Ts65Dn thus lacks trisomy for ~45% of Hsa21 orthologs – the remainder of those mapping to Mmu16 and all of those mapping to the Mmu17 and Mmu10 orthologous regions. The Mmu17 region that is trisomic in the Ts65Dn carries 50 protein-coding genes that are not orthologs of Hsa21 genes15,16 (the Hsa21 syntenic region is ~20 Mb distal on Mmu17). This segment is an artifact of the method used in construction, and the genes are not relevant to DS and possibly confound assessments of gene–phenotype correlations and drug responses in the Ts65Dn (see the “Discussion” section).
The Ts65Dn, although not an ideal model, has been extremely valuable for the study of a number of phenotypic features similar to those seen in people with DS, or speculated to contribute to features seen in people with DS (for reviews, see Rueda et al10 and Roubertoux and Carlier,17 as well as the references in Table 1). Particularly well documented are deficits in L/M tasks that require a functional hippocampus, including the Morris water maze (MWM) and several other maze tasks, object recognition tasks, and fear conditioning.10,17 Electrophysiological abnormalities include deficits in gamma-aminobutyric acid (GABA)ergic and glutamatergic transmission in the hippocampus and cerebellum; among them are age- and cell type-specific deficits in hippocampal long-term potentiation (LTP) and long-term depression (LTD). Also well documented are impairments in adult neurogenesis in the dentate gyrus, abnormalities of neuron number and dendritic spine morphologies, and decreased numbers of cerebellar granule cells. Although the Ts65Dn do not show accumulation of beta-amyloid, as in the neuropathology of AD, age-dependent loss of the functional markers choline acetyltransferase (ChAT), nerve growth factor (NGF), and neutotrophic tyrosine kinase receptor type 1 (TRKA) are observed in the basal forebrain cholinergic neurons (BFCN), and the loss of tyrosine hydroxylase (TH) in the locus coeruelus (LC). These supply cholinergic and noradrenergic input, respectively, to the hippocampus (and other brain regions), and their loss is relevant to premature cognitive decline and development of AD-like dementia that is commonly seen in individuals with DS over the age of 60 years (see Table 1).10,17 Each of these features has been explored by multiple research groups, showing these abnormalities to be robust and reproducible (see Table 1).10,17
Most recently, chromosome engineering has been used to create three lines of mice that are separately trisomic for the complete Mmu16 region (the Dp[16]1Yey, or Dp16), the Mmu17 region (the Dp[17]1Yey, or Dp17), and the Mmu10 region (the Dp[10]1Yey, or Dp10).18,19 A full trisomy mouse can be obtained by sequential crossing of the Dp16, Dp17, and Dp10; however, because of viability issues, time, and expense, this has not been practical to pursue.20 Thus, in spite of its limitations, and because of its long and productive history of use, the Ts65Dn remains the most popular choice among DS mouse models.
Over the last few years, a number of experiments have shown that L/M deficits in the Ts65Dn can be rescued pharmacologically (Table 1). To date, more than 20 drugs/small molecules have successfully rescued hippocampal deficits, most of them in adult mice. Based on these, and sometimes additional information, several clinical trials have been carried out and others are currently in progress. The enthusiasm for such trials is strong: drugs that could improve cognitive function – for example, by increasing the intelligence quotient by 10–20 points – would have enormous positive consequences for individuals with DS. It would increase their potential to participate in society more fully and more independently, with consequent positive impacts on their families and on society as a whole.
In this review, we discuss results from 29 drugs that have been investigated in the Ts65Dn, both successfully and unsuccessfully (Table 1). Subsets of these drugs have been included in recent reviews.10,11,21 Here, we discuss the complete set, why each was chosen, the details of how each was evaluated and, in particular, the similarities and differences in their outcomes. Also discussed is the current status of clinical trials for cognition in DS and the features of the Ts65Dn that may limit the predictive value of its use in preclinical evaluations.
Ts65Dn and preclinical evaluation of drug efficacy
As with other mouse models of human diseases, there are no standards for the assessment of drug efficacy in the Ts65Dn, so the numbers, types, and stringencies of evaluations vary among studies. For the drugs in Table 1, the rescue of L/M in at least one task has been assessed, and in several cases, effects on adult neurogenesis, LTP/LTD, and age-related neurodegeneration in the BFCN and/or LC have been evaluated. To achieve some consistency in the discussion, the results of only these four categories are included here, although many reports included additional types of assessments (see references in Table 1).
The results in Table 1, even from the same types of experiments, were obtained using protocols that varied in their details. The age of the mice evaluated ranged from embryonic and early postnatal to 16 months. While the majority of experiments used male mice, a few used female mice, some used a mix of male and female mice, but the authors did not separate the sexes in their analyses, and some did not report the sex (see Table 1). Some drugs were administered as acute single doses and others were chronic, with treatments extending from a few days/weeks to several months. Drug delivery methods include stressful subcutaneous and intraperitoneal injections, and benign ingestion through food, water, or milk. When different laboratories tested the same drug, concentrations often differed. Some L/M evaluations used more stressful tests, such as water mazes and electric shocks, which may result in exaggerated poor performance caused, at least in part, by differential reaction to stressors, rather than by solely impaired L/M,22 while others used nonaversive, nonwater mazes, and one used olfaction. Lastly, not all drug treatments provided complete rescue of impairment, and some studies did not provide all statistical analyses needed for such determinations. For example, comparing the performance of drug-treated Ts65Dn mice to that of vehicle-treated Ts65Dn mice merely asks if the drug improved L/M; a comparison to untreated control mice is necessary to determine how well L/M was rescued. Together, such variations in experimental protocols and analyses make comparisons among drug responses, and their extension to predictions for DS, less than straightforward.
The drugs in Table 1 are clearly diverse in their targets and mechanisms. Only three (memantine, epigallocatechin-3-gallate [EGCG], and ethosuximide) have a known/predicted direct link to the overexpression of one or more genes encoded by Hsa21. Most drugs, however, have a rational basis for their choice, frequently from studies that show rescue of a Ts65Dn-relevant phenotypic abnormality in a non-DS mouse model (eg, rescue of neurodegeneration in an AD mouse model) (see references in Table 1). The entries in Table 1 are in chronological order to capture the development of thought and information in the field of treatment of ID in DS.
Successful drug treatments in the Ts65Dn
Estrogen and minocycline
The first demonstration of L/M rescue in the Ts65Dn used estrogen and was reported more than 10 years ago.23 The choice of estrogen was based on the observation of premature menopause and early onset cognitive decline in women with DS. Older (aged 11–14 months) female Ts65Dn mice were treated with estrogen (via implanted time-release capsules) for 2 months, and then tested in the discriminating water T-maze, where they were required to learn which arm of the maze contained the escape ladder. Untreated Ts65Dn mice were impaired in this task, learning more slowly and not reaching the same level of efficiency as control mice. Estrogen significantly improved both aspects of initial learning, although not to the levels of the control mice. Estrogen also improved the speed of learning a new location of the escape ladder and reversed the age-related loss of BFCN functional markers, ChAT and NGF, increasing them to levels that were not significantly different from controls. Subsequent studies using male Ts65Dn mice at 6 months of age and with a different L/M test (the water radial arm maze [WRAM]), showed that 3 weeks of estrogen treatment failed to rescue the learning deficit.24 Although the two experimental protocols differed in most parameters, the results suggest sex differences in estrogen responses.
The same group looked at the efficacy of minocycline, a derivative of tetracycline.25 In addition to antibiotic properties, minocycline is of interest because it is considered to have neuroprotective effects, modulating the activity of interleukin 1 beta (inflammation) and inhibiting caspases (apoptosis).25 In the 10-month-old male Ts65Dn, 3 months of treatment with minocycline improved performance in the WRAM, although again not to the levels of controls; it also rescued age-related loss of ChAT in the BFCN.26
GABAA receptor antagonists
A number of studies have shown electrophysiological abnormalities of repressed LTP and enhanced LTD in the Ts65Dn.27–29 Coordinated regulation of LTP and LTD is considered a cellular model of L/M, and this imbalance between excitatory and inhibitory neurotransmission in the Ts65Dn is therefore proposed to contribute to impaired L/M. Based on these observations, the GABAA receptor (GABAAR) antagonists picrotoxin, bilobalide, and pentylenetetrazole (PTZ) were tested for their efficacy in rescuing the performance of 3–4-month-old Ts65Dn in novel object recognition (NOR).30 While acute injection of picrotoxin was ineffective, chronic treatment composed of 2 weeks of daily injection was sufficient for each drug to rescue learning in NOR. Performance in the spontaneous alternation T-maze was also tested and found to be normal after PTZ treatment. Most importantly, the beneficial effects continued up to 2 months after PTZ treatment had stopped; mice continued to learn successfully and LTP remained normal.
In subsequent experiments, 4-month-old male Ts65Dn mice were treated with PTZ for 4 weeks and then exposed to a battery of behavioral tests that spanned a further 3 weeks with continued PTZ treatment.31 Two results are particularly noteworthy: PTZ rescued performance in the MWM, thus extending the results from Fernandez et al30 but in one specific test of equilibrium (the aluminum rod), PTZ caused significantly worse performance. This latter raises the possibility that there are some negative side effects of PTZ even at lower doses. In more recent experiments, 2 weeks of daily injections of PTZ were shown to be effective in rescuing deficits in NOR in both younger (2–3-month-old) and older (12–15-month-old) male Ts65Dn mice.32 It was further shown that rescue persisted 2 weeks after injections were terminated and, interestingly, that PTZ was effective when given during the light (inactive) phase, but not when given during the dark phase.
The proposal of PTZ for clinical trials in DS has been controversial because it is a known convulsant.34 Although seizures in rodents have been seen only at doses significantly higher than those used to rescue learning in the Ts65Dn, children with DS are at increased risk of seizures and therefore could be more sensitive to PTZ.1,33 PTZ is not currently US Food and Drug Administration (FDA) approved. While it was used for many years, without recorded adverse events, in elderly patients with uncharacterized cognitive impairments, approval was withdrawn when evidence of efficacy in that population could not be demonstrated (discussed in Wetmore and Garner34).
To lessen the probability of negative side effects in people with DS, other more specific GABAAR antagonists are now being studied. GABAAR exhibit considerable heterogeneity in terms of their composition and functional properties because 19 different subunits are assembled as heteropentamers (reviewed in Deidda et al35). Receptors containing the α5 subunit are receiving attention both because a mouse model lacking this subunit shows enhanced performance in the MWM and because α5 antagonists enhance L/M in control mice.36 Furthermore, the α5 subunit is highly expressed in hippocampus but less commonly present in other brain regions,37 allowing better regional specificity in targeting drug effects and perhaps lessening the probability of adverse responses in other brain regions. Within the hippocampus, the highest concentration of α5-containing GABAAR is found at the molecular layer of the dentate gyrus, a distribution that is not altered in the AD brain,38 and therefore possibly similar in the DS brain. These observations suggest that antagonists specific to the α5 subunit would be less likely to elicit negative consequences in DS. Based on these data, 3-month-old male Ts65Dn were treated with a single injection of α5 inverse agonist (α5IA) (5 mg/kg) and tested in the NOR and the MWM.39 Performance in the NOR and acquisition in the MWM were rescued; however, treated Ts65Dn mice failed to show improvement in the probe trial (retention) portion of the MWM, where the platform is removed from the pool and the amount of time the mouse spends near the former platform location, versus elsewhere in the pool, is measured. Unlike the control mice, the Ts65Dn mice did not spend more time in the training quadrant. Additional experiments demonstrated that acute injection of a tenfold higher dose of α5IA did not induce convulsions and did not exacerbate the convulsion rate in either control mice or the Ts65Dn mice, which were pretreated with PTZ at doses that typically induce convulsions in 50% of animals.
A comprehensive evaluation of the effects of chronic treatment with another α5 antagonist, RO4938581, was recently reported.40 Male Ts65Dn mice, 3–4 months old, were treated daily for 6 weeks and throughout behavioral testing. Normalization of LTP and neurogenesis were shown. Learning in the MWM was clearly improved, but it is not clear that performance in the probe trial for RO4938581-treated Ts65Dn mice reached levels seen in control mice, and the statistical analysis of this comparison was not provided. A failure of rescue in this evaluation would be consistent with results shown by Braudeau et al.39
It is of interest that the α5 subunit is most commonly found in GABAAR that also contain a γ subunit, GABRG2.37 GABRG2 is dephosphorylated, and inactivated, by the Ca++–calmodulin-dependent protein phosphatase, calcineurin (CaN). In turn, CaN activity is directly modulated by the Hsa21 protein, RCAN1, and indirectly modulated by the Hsa21 protein, PCP4. The consequent predicted perturbations in the dynamics of CaN activity in the Ts65Dn mice would specifically impact α5-containing GABAAR activity, exacerbating LTD.41
Memantine, an NMDA receptor antagonist
Memantine is an uncompetitive antagonist of the N-methyl-d-aspartate (NMDA) receptor and has been shown to rescue L/M in mouse models of AD and stroke.42 It is also currently approved by the US FDA for use in moderate to severe AD. Unlike the majority of drugs listed in Table 1, the mechanism of action of memantine has known relationships with proteins encoded by Hsa21. The activity of the N-methyl-d-aspartate receptor (NMDAR), and therefore of excitatory neurotransmission, is regulated by the phosphorylation/dephosphorylation of receptor subunits, with the protein phosphatase, CaN playing a major role.43 As discussed in Siddiqui et al44 there are nine Hsa21-encoded proteins, all trisomic in the Ts65Dn, that directly or indirectly interact with the NMDAR. Among these is the regulator of CaN, RCAN1. While it was first shown that the elevated expression of RCAN1 inhibited CaN activity, it is now clear that the precise regulation of CaN activity is complex and governed by both the relative proportions of RCAN1 splice variants and the phosphorylation state of RCAN1 which is affected by the Hsa21 kinase DYRK1A.45,46 RCAN1 activity is further modulated by levels of reactive oxygen species that are affected by the Hsa21 proteins, superoxide dismutase (SOD1), the amyloid precursor protein (APP), and the transcription factor, BACH1. Activity of CaN, as a Ca++–calmodulin regulated phosphatase, is also influenced by the expression level of the Hsa21 calmodulin-binding protein, PCP4. Additional Hsa21 proteins participate in the regulation of NMDAR activity and membrane localization. The guanine nucleotide exchange factor (TIAM1), the multidomain endocytic protein (ITSN1), and APP interact with NMDAR and the phosphoinositol phosphatase (SYNJ1) tethers TIAM1 at the membrane to facilitate its interaction with NMDAR. Given these many contributions, it is not obvious how NMDARs are affected either during baseline activity or in dynamic responses to stimulation. Nevertheless, LTP is inhibited in the Ts65Dn, where all these Hsa21 genes are present in triplicate. Ts65Dn mice display exaggerated responses to the locomotor stimulatory effects of another NMDAR antagonist, MK-801.47 Overall, these data are consistent with altered dynamics of NMDAR responses.
Costa et al47 were the first to propose and test memantine in the Ts65Dn. The researchers showed that acute injection of memantine rescued L/M deficits in context fear conditioning (CFC) in male Ts65Dn at both 4–6 months and 10–14 months of age. Subsequently, it was shown that 9 weeks of chronic memantine treatment of 9-month-old male Ts65Dn mice rescued impaired performance on the MWM, at least in terms of acquisition (no probe trial was reported).48 Slightly different results were obtained when male Ts65Dn mice were treated with memantine from 4–9.5 months of age.49 This indeed improved performance on the WRAM, but not to the levels seen in untreated control mice. Continued treatment with memantine through several additional weeks of training also improved Ts65Dn performance on the NOR. However, while Ts65Dn performance on the NOR was not different from that of memantine-treated controls, memantine appeared to have impaired the performance of the control mice on the NOR relative to that of the untreated controls (no statistical evaluation of that comparison was provided). In a final behavioral evaluation, 1 week after memantine treatment was terminated, Ts65Dn performance on the NOR had returned to the initial untreated impaired levels. Thus, unlike GABAAR antagonists, memantine does not produce lasting positive effects.
As described above, hippocampal LTD in the Ts65Dn is increased relative to controls.27 Given the clear indications of impaired NMDAR responses, Scott-McKean and Costa50 hypothesized that elevated LTD might be mediated by NMDAR-dependent mechanisms. Consistent with this, the authors demonstrated that LTD induced by NMDA in Ts65Dn hippocampal slices was significantly greater than that in controls, while LTD induced by dihydroxyphenylglycine (a metabolic glutamate receptor agonist) in the Ts65Dn was equivalent to that in controls. Importantly, preincubation of the slices with memantine resulted in the stimulation of normal levels of LTD with exposure to NMDA.
Fluoxetine
The potential for efficacy of the serotonin reuptake inhibitor, fluoxetine, in DS was supported by multiple lines of evidence. Hippocampal volume and cell numbers are decreased in both the Ts65Dn and people with DS.10 Because the hippocampus is one of the two brain regions in which neurogenesis continues in adulthood, impaired adult neurogenesis could contribute to observed hippocampal structural and functional abnormalities. It had been shown previously in rodents that chronic treatment with fluoxetine, and other antidepressants, stimulates the production of new neurons and their incorporation into functional networks.51 The first Ts65Dn study with fluoxetine examined adult male mice (2–5 months of age) and demonstrated that adult neurogenesis is indeed impaired in untreated mice, and that 2 weeks of fluoxetine injections rescued this deficit.52 The next study used neonatal mice, treating a mix of male and female Ts65Dn from postnatal day (P)3 to P15. Untreated mice had decreased proliferation in the dentate gyrus, and repressed levels of the serotonin 5HT2A receptor, BDNF, and NGF. Fluoxetine rescued all of these abnormalities, except for NGF levels.53 Furthermore, 1 month after fluoxetine treatments had been terminated (at P45), Ts65Dn showed normal learning in CFC.
In addition to actions on the serotonin system, fluoxetine has been shown to decrease GABAergic inhibition.54–56 Based on these data, and on the observation of overinhibition in the Ts65Dn and the efficacy of PTZ, Heinen et al57 treated Ts65Dn at 5–7 months of age with fluoxetine for a total of 6 weeks. Both male and female mice were used, but the analysis did not include separation by sex. Fluoxetine-treated mice showed no improvement on the MWM and no change in the BFCN levels of ChAT. If sex differences were significant, they could have obscured previous observations of male-specific efficacies. More importantly, however, the authors reported a very high rate of seizures and death (three of nine mice and four of nine mice, respectively) in the Ts65Dn animals treated with fluoxetine. These results are in stark contrast with those of a prior report where mice of the same age were used.52 Possible reasons for the negative results are a much higher concentration of fluoxetine (equivalent to 80 mg/kg consumed in drinking water versus 5 mg/kg intraperitoneal injection, in Clark et al52) and treatment for a much longer time (6 weeks versus 15 days).
In another study of adult Ts65Dn mice plus fluoxetine, the effects of drug concentration were explicitly explored.58 The authors first showed that higher levels of fluoxetine (40 mg/kg and 20 mg/kg, based on the daily consumption of fluoxetine in drinking water and the weight of the mice) were indeed associated with seizures and death. Thus, for behavioral studies, a lower dose was chosen (10 mg/kg), which was equivalent to approximately one-eighth of that used in Heinen et al57 and approximately twofold higher than that used in Clark et al.52 Begenisic et al58 treated 2-month-old male and female mice for 8 weeks and found that impaired performances in OL and spontaneous alternation in a four-arm maze were both rescued, and that levels of LTP and GABA release from synaptosomes were both normalized. There were no instances of adverse effects.
Most recently, fluoxetine has been used in prenatal treatment. Pregnant Ts65Dn mice were injected daily with 10 mg/kg fluoxetine from embryonic day 10 to birth.59 When tested at P45, after no postnatal fluoxetine treatment, learning in CFC in Ts65Dn offspring from fluoxetine-treated dams was normal. No information was provided on the sex of the mice analyzed. Contrary to prior reports, no deficit was seen on the NOR, even in untreated Ts65Dn mice, possibly a result of the younger age of the mice used here. Prenatal fluoxetine treatment also rescued the abnormalities in neurogenesis seen in untreated Ts65Dn, both at birth and at 45 days. This report is a powerful demonstration of the positive modulation of trisomic brain development.
Acetylcholinesterase inhibitors
The use of acetylcholinesterase inhibitors to treat the Ts65Dn mice has been motivated by the potential to prevent or reverse the loss of functional markers in the BFCN. In the first study, physostigmine was used to treat male Ts65Dn mice at three ages: 4 months; 10 months; and 16 months.60 Mice were tested for the rescue of deficits in the four-arm spontaneous alternation task. This task was chosen because, unlike water mazes and CFC, it is low stress and nonaversive, and it does not use a reward system that could bias outcomes if differences in motivation exist. Performance in this task is known to deteriorate with age in rodents, and this deterioration can be alleviated in rats with cholinesterase inhibitors.60 Untreated Ts65Dn were impaired in the task at all three ages. Acute injection of physostigmine (10 minutes prior to testing) rescued the impairment, but only in 4-month-old mice. Measurement of acetylcholine release showed no abnormality at baseline in Ts65Dn; however, while release increased in the control mice during task performance, it increased only marginally in Ts65Dn at 4 months, and not at all in older Ts65Dn.
In a second study, galantamine was used with an olfactory test of L/M.61 This test was also chosen because it is nonaversive, and because it exploits a normal sensory drive that is particularly strong in rodents. Furthermore, there are data showing that people with DS have impaired olfaction, even at young ages,62 and that the impairment increases with age. When young adult Ts65Dn mice (3–6 months of age) were treated for a total of 10 days with galantamine, including during training, the deficits were completely rescued. Both male and female (although predominately male) mice were used, but sex differences were not discussed.
B-adrenergic receptor agonists
Salehi et al63 demonstrated that levels of TH in the LC of Ts65Dn mice were normal at 3 months, but were decreased at 6 months relative to controls. Reasoning that this abnormality would compromise norepinephrine (NE) input to the hippocampus, and possibly contribute to the observed impairment in hippocampal tasks such as CFC,47 the authors tested the effects on CFC performance of 6-month-old male Ts65Dn mice after two different treatments – injection of the NE prodrug L-DOPS that is metabolized to NE, or the β1 adrenergic receptor partial agonist, xamoterol.64 Both drugs increased freezing in CFC of the Ts65Dn mice to levels seen in normal controls. Further investigation of the molecular basis of the rescue is warrented, however, because the Ts65Dn are impaired in CFC at 3 months of age, where TH levels in the LC were still normal. Thererfore, impairment in CFC is not directly caused by decreases in TH.
Other work also showed that the Ts65Dn mice progressively lost NE markers in the LC with age, and that these mice are more sensitive than normal control littermates to a noradrenergic neurotoxin.65 Based on this and the prior demonstration that the Ts65Dn are impaired in CFC, male Ts65Dn (5–6 months old) were treated with the β2 adrenergic agonist, formoterol, 4 hours prior to training in CFC (and presumably also 4 hours prior to testing).66 Effects of formoterol in the periphery, and thus restriction of its effects to the brain, were prevented by prior injection of the same mice with the β adrenergic antagonist, nadolol, that does not cross the blood–brain barrier. With this treatment, the CFC deficit in the Ts65Dn mice was partially rescued, but the interpretation is ambiguous because the increase in freezing was only evident at later time points in the testing interval (minutes 4 and 5 out of a total of 5 minutes). In addition, although the authors did not remark on its possible significance, the formoterol data indicate a significant decrease in Ts65Dn activity levels – ie, in speed and the total distance traveled in the open field assay, reducing them to levels of the control mice. Therefore, it remains possible that in the absence of other information, the marginal increase in freezing in CFC is related to a decrease in overall activity, and not to contextual learning. For one group of mice, after CFC testing, formoterol injection was continued for 10 days and followed by injection of bromodeoxyuridine (BrdU) for the assay of neurogenesis. Formoterol did increase the number of BrdU cells in the Ts65Dn mice, but not the number of new neurons.
Lithium
Lithium is a common treatment for mood disorders that has been in use since the 1970s. While the molecular mechanisms underlying responses are not known in detail, it has been shown that lithium acts in part to inhibit the activity of GS3KB and to modulate signaling in the Wnt/β-catenin pathway.67,68 It also inhibits activities of inositol phosphatases. However, the ability of lithium to compete with Mg++ in protein binding implies its effects could be very broad. Lithium has neuroprotective properties and has also been shown to increase adult neurogenesis in control rodents.67,68
In the Ts65Dn, 12-month-old female mice were treated with lithium in food for 1 month.69 An increase in neurogenesis was documented in both controls and Ts65Dn mice, with levels in Ts65Dn reaching those of untreated control mice. In other work, when younger (~5-month-old) male Ts65Dn mice were treated for 1 month with lithium, a complete rescue of neurogenesis was seen in the dentate gyrus.70 LTP was normalized, levels of phosphorylated β-catenin were increased, and performance in CFC, NOR, and OL were completely rescued. Importantly, however, performance in the spontaneous alternation T-maze was not corrected. These observations emphasize the need for the comprehensive analysis of L/M when comparisons among drug treatments are wanted, and when considering potential efficacy in clinical trials.
To further investigate the molecular basis of lithium effects, the in vitro properties of neural precursors derived from the subventricular zone were examined in P0 Ts65Dn (sex unknown).71 Proliferation was impaired and levels of pGSK3B-ser9 were decreased – ie, activity of GSK3B would be increased. Treatment of the cultures with lithium increased the levels of Ser9 phosphorylation and normalized proliferation.
Melatonin
Melatonin (N-acetyl-5-methoxytryptamine) is well studied for its role in the regulation of circadian rhythms – in particular, sleep patterns.72,73 However, recent experiments treating mouse models of AD have indicated that melatonin has many beneficial effects as an antioxidant, an antiapoptotic, and an anti-inflammatory agent, and it offers protection from neurodegeneration and cognitive deficits.72 It has also been used in clinical trials for AD.73 Based on these data, Corrales et al74 treated ~5 month-old male Ts65Dn mice with melatonin (in drinking water) for 4 months prior to and 1 month during behavioral testing. In both control and Ts65Dn mice, melatonin improved acquisition in the hidden platform MWM. No probe test data were provided; the absence of this important analysis leaves the effects of melatonin on memory unknown. Melatonin also decreased the amount of time for both controls and Ts65Dn to find the visible platform in the MWM, but only the controls were indicated as showing a significant decrease. Levels of ChAT in the BFCN were measured. While initial levels were significantly lower in untreated Ts65Dn compared with controls (consistent with prior studies),26 and levels increased with melatonin, these increases apparently did not reach significance compared with untreated Ts65Dn; moreover, a statistical comparison with untreated controls was not provided. In a further work,75 the same authors treated 6-month-old male Ts65Dn mice with melatonin for 5–6 months. This treatment rescued impairment in LTP and neurogenesis.
Sonic hedgehog
Brains from individuals with DS and the Ts65Dn mice have small cerebella and reduced granule cell density. Roper et al76 first showed that an agonist of sonic hedgehog, SAG 1.1, rescued decreased cell numbers in the granule cell layer of the cerebellum. This work was particularly important because the correction, measured at P6, was obtained with a single injection of SAG 1.1 at P0. In recent work, the authors analyzed 4 month old Ts65Dn (males and females) that had again been given a single SAG 1.1 injection at P0.77 Similar to the data obtained at P6, cerebellar cellular deficits were corrected, although not deficits in cerebellar LTD. It also rescued impairment in the MWM, but it failed to rescue performance in the Y-maze. Consistent with success in the hippocampal-based MWM, SAG 1.1 also rescued hippocampal LTP in the Ts65Dn mice.
Epigallocatechin-3-gallate (EGCG)
EGCG is a catechin found in green tea. In a review of the activity of commercially available protein kinase inhibitors against a panel of 28 kinases, EGCG was shown to be highly specific for two of these kinases – the Hsa21-encoded kinase, DYRK1A, and the MAPKAPK5 kinase (aka PRAK), inhibiting each by ~90%.78 To a lesser extent (~50%), EGCG also inhibited ERK2, ROCK2, and PDK1. DYRK1A was already a gene of considerable interest for contributions to the DS neurological phenotype79 and EGCG provided the means to rescue phenotypic features in DYRK1A single-gene transgenic mice.80–82 The effects of EGCG on the Ts65Dn mice were first studied by demonstrating the normalization of LTP in hippocampal slices preincubated with EGCG.83 Most recently, it was demonstrated that when 3-month-old male Ts65Dn mice were treated with EGCG for 30 days, performance in the acquisition phase of the MWM improved to the level of the control mice.84 EGCG also reduced the thigmotactic behavior of the Ts65Dn mice in the probe trial, although the amount of time spent in the training quadrant was not provided. EGCG also rescued Ts65Dn behavior in NOR, but notably it caused severe impairment in the control mice in this task.
Reduction of activity of DYRK1A by EGCG in the Ts65Dn occurs in the context of elevated expression of other multiple Hsa21 genes. In particular, among DYRK1A substrates are the Hsa21-encoded proteins APP, SYNJ1 (a phosphoinositol–phosphatase), and RCAN1.85,86 DYRK1A also has indirect functional interactions with RCAN1 because DYRK1A phosphorylates substrates that are dephosphorylated by the protein phosphatase CaN, which is regulated by RCAN1 activity. In addition, DYRK1A phosphorylation activates the transcription factor GLI1, which functions in sonic hedgehog signaling;87 GLI1 activity is also increased by editing of its pre-mRNA, carried out by the Hsa21-encoded protein ADAR2.88 The ADAR2 gene maps to Mmu10 and is therefore not trisomic in the Ts65Dn (but is in full trisomy Hsa21). Decreasing DYRK1A expression in the presence of elevated expression of these Hsa21-encoded substrates and functional interactors could cause additional imbalances relevant to DS phenotypic features.
It is unlikely that the benefits of EGCG treatment in the Ts65Dn are limited to inhibition of the activity of the DYRK1A kinase. EGCG has been shown to be a powerful antioxidant and antitumor agent, and it is possible that most of its targets and effectors are unknown.89,90 EGCG is currently in clinical trials for obesity, several types of malignancy, Huntington’s disease, Fragile X, and AD, as well as for DS (http://clinicaltrials.gov).
Antioxidants
Oxidative stress and mitochondrial dysfunction are both considered hallmarks of DS tissues and contributors to neurological phenotypes across the DS lifespan.1,91 Cortical neurons derived from DS fetal brains show elevated levels of reactive oxygen species, and survival is enhanced by treatment with antioxidants.92 Several Hsa21 genes, among them SOD1, BACH1, ETS2, and S100B, are known to contribute to the regulation of oxidative stress when overexpressed.9 Consistent with these observations, it was shown that levels of oxidative stress are elevated in the brains of adult Ts65Dn, and when male Ts65Dn mice were fed a diet high in vitamin E (0.4 g per kg) from the age of 4 months to ~8 months and 10 months, the levels of oxidative stress were decreased.93 Performance in the WRAM was improved at 8 months, and at 10 months; loss of the TRKA receptor in the BFCN had been prevented.
In an attempt to prevent the developmental consequences of elevated oxidative stress, pregnant Ts65Dn female mice and their offspring were treated with a vitamin E-enriched diet (0.002%, or tenfold lower than the amounts used in Lockrow et al94).94 Offspring were continued on the enriched diet and tested on the MWM at 10 weeks of age. Learning was improved, as the escape latency was significantly decreased in the treated Ts65Dn mice, although apparently not to the level of untreated control littermates.
Inhibition of Aβ production
Elevated levels of the Aβ peptide are present in the DS brain from young ages and are postulated to cause, or at least contribute to, the common occurrence of AD in older individuals with DS. Levels of the parent protein APP and the α- and β-secretase APP products were shown to be elevated in brains of 4-month-old female Ts65Dn mice compared with controls, although neither Aβ accumulation nor plaques are seen in the Ts65Dn mice.95 After 4 days of injection with a β-secretase inhibitor, N-[2S-(3,5-difluorophenyl)acetyl]-L-alanyl-2-phenyl-1,1-dimethylethyl ester-glycine, it was noted that while the total levels of APP were not altered, the levels of Aβ fragments were significantly decreased to the levels seen in control brains. This treatment also rescued performance in the MWM in both acquisition and retention.95
Neuropeptides
Two peptide sequences (NAPVSIPQ and SALLRSIPA, abbreviated NAP and SAL, respectively) derived from the neurotrophic factors activity-dependent neuroprotective protein (ADNP) and activity-dependent neurotrophic factor (ADNF) have been shown in in vitro and in vivo systems to protect from Aβ toxicity, Tau hyperphosphorylation, and fetal alcohol exposure.96 Initial experiments with Ts65Dn mice involved treating 10-month-old mice for 9 days with NAP + SAL.97 During days 4–9, mice were tested on the MWM. Treated Ts65Dn showed improved performance, to the levels of untreated controls, in both acquisition and retention. After a further 10 days with no additional treatment, however, treated Ts65Dn mice no longer remembered the platform location, while control mice performed as previously. In a second set of experiments, pregnant Ts65Dn and control females were injected with NAP/SAL daily from embryonic day (E) 8 through E12.98 Offspring were evaluated in the MWM between 8 months and 10 months of age. Ts65Dn offspring of the treated dams showed improved speed in acquisition, although apparently not to the levels of the control mice. No probe trial data were provided. The limited analysis makes it difficult to evaluate the efficacy of prenatal versus aged treatment.
Peptide 6 is an 11-amino acid sequence derived from ciliary neurotrophic factor, which has been shown to enhance neurogenesis.99 Blanchard et al100 treated 11–15-month-old female Ts65Dn mice for 1 month with peptide 6. While improvements in the MWM was stated, the data are slim. Improvements in terms of time to reach the hidden platform were seen in the Ts65Dn mice only on day 3 of training, and overall there was no improvement. During the probe trial, treated Ts65Dn indeed spent more time in the training quadrant than did the untreated mice, but since this time was still less than 25%, it is considered random. Untreated control mice also spent <25% time in the training quadrant, which leaves open the question of the robustness of training in this experiment.
Choline
Considerable evidence has accumulated to suggest that prenatal and perinatal supplementation with choline can have long-term benefits for L/M,101 including the enhancement of adult neurogenesis.102 Given the observed loss of functional markers in the BFCN of Ts65Dn mice, and in the brains of DS, Strupp et al103–105 examined the effects of choline supplementation in both prenatal and perinatal Ts65Dn. Dams were maintained on an enriched choline diet (4.5-fold higher than standard chow) throughout pregnancy and until litters were weaned. Offspring were maintained on standard choline diets until testing at 13–17 months of age. Notably, however, offspring were also maintained in an enriched environment and on a reverse light cycle, so that they were tested during the equivalent of their active period. These two features are in contrast to typical experiments where mice are maintained in a generally deprived environment and tested during their normal sleep cycle. These differences must be considered when weighing the positive effects of choline versus other drug treatments. Performance on the WRAM of Ts65Dn offspring of choline-treated dams was completely rescued. Hippocampal neurogenesis was assessed by counting doublecortin (DCX)-positive cells in the dentate gyrus. Choline-treated Ts65Dn mice had a larger number of DCX cells when compared with untreated Ts65Dn mice, but still significantly fewer than controls. They also showed increased levels of ChAT and other markers in subsets of the BFCN.
GABAB receptor antagonist
The Hsa21 gene, KCNJ6, encodes an inwardly-rectifying K+ channel, G protein-coupled inwardly-rectifying potassium channel (GIRK)2, that couples to GABAB receptors. Levels of GIRK2 are elevated in Ts65Dn hippocampus and have been proposed to contribute to abnormal synaptic plasticity.106 Best et al107,108 investigated GABAB-induced potassium currents in the Ts65Dn and demonstrated that they are elevated in the hippocampus, thus suggesting that they likely contribute to the observed imbalance between excitatory and inhibitory neurotransmission. Building on this work, inhibition of the GABAB receptor was examined using CGP55845, a GABAB receptor antagonist.109 Treating 2–3-month-old male Ts65Dn mice, both acutely and chronically, rescued performances in NOR and OL to levels not distinguishable from untreated control mice. Effects on performance in CFC were less clear; while the level of freezing in the Ts65Dn treated with the drug was greater than without, the difference did not reach significance. Similar to several other drug treatments, performance in the spontaneous alternation T-maze was also not rescued.
Failed drug treatments in Ts65Dn
Six additional drugs listed in Table 2 failed to improve Ts65Dn performance in one or more tests of L/M. The acetylcholinesterase inhibitor, donepezil, had shown minimal positive effects in small clinical trials with adults and with children with DS.110 Based on the hypothesis that donepezil might prevent the loss of ChAT seen in the BFCN of Ts65Dn mice after ~6 months of age, and therefore it would prevent or rescue some cognitive impairment, 4-month-old male Ts65Dn mice were treated for 7 weeks with donepezil; no improvement was seen in acquisition in the MWM.31 This contrasts with the positive effects of the other anticholinesterases, physostigmine and galantamine,60,61 discussed earlier, that rescued learning deficits in Ts65Dn of similar age (in the case of physostigmine with a single injection60). However, the structure of donepezil is not related to those of other acetylcholinesterases and, in addition, the effects of donepezil in other mouse models have been sensitive to dose and have not been completely consistent.111 In particular, in AD mouse models where donepezil was effective, doses of 0.3 mg/kg (tenfold lower than in Rueda et al31) were more effective than doses of 0.6 mg/kg and 1.0 mg/kg.
|
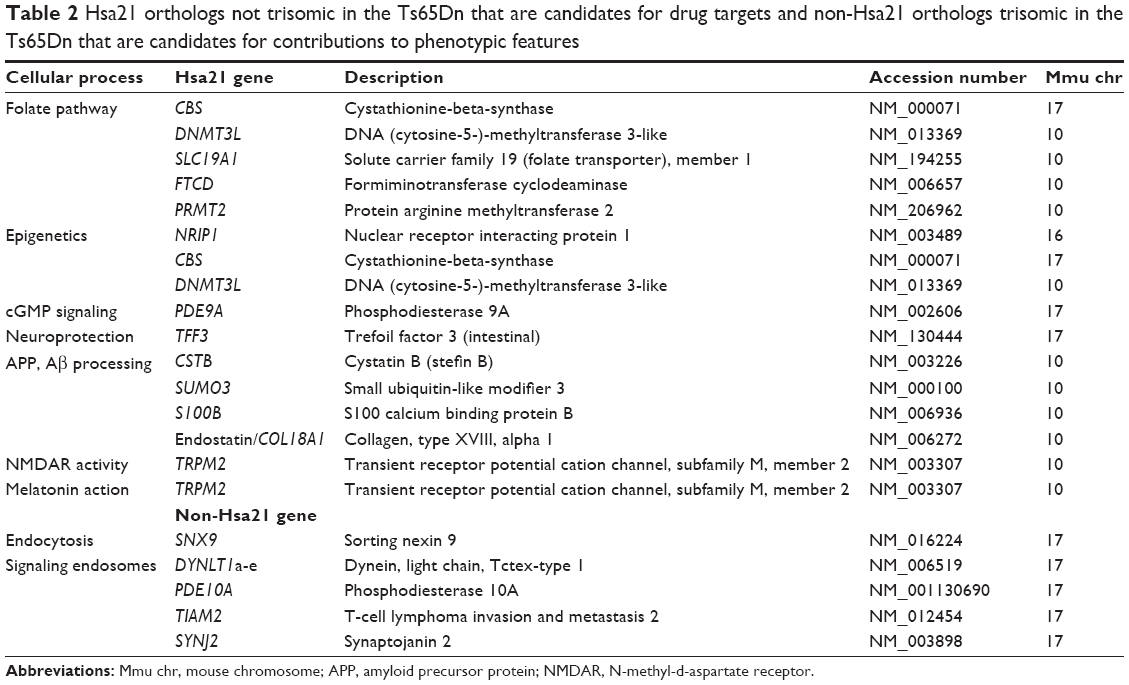
Table 2 Hsa21 orthologs not trisomic in the Ts65Dn that are candidates for drug targets and non-Hsa21 orthologs trisomic in the Ts65Dn that are candidates for contributions to phenotypic features |
In the late 1990s and early 2000s, piracetam, a cyclic analog of GABA, received considerable attention as a nootropic (reviewed in Malykh et al112) based on both animal and human studies. The targets and mechanism of action of piracetam remain unknown, and its benefits failed to be substantiated in larger, placebo-controlled, human clinical trials.112 Piracetam also failed to improve Ts65Dn performance on the MWM.113 The piracetam analog, SGS-111, was shown to have antioxidant properties and prevent apoptosis in cultured cortical neurons from DS fetal brains.114 Based on this evidence, SGS-111 was tested in the Ts65Dn, but it was shown to be ineffective in rescuing acquisition in the MWM.115
The evidence cited above for contributions of the Hsa21 gene, KCNJ6, to abnormal synaptic plasticity is compelling.106 It is further supported by the observations that a transgenic mouse overexpressing the KCNJ6 gene from a genomic construct is impaired in CFC and displays elevated LTD.116 To explore this, 4.5–5-month-old male Ts65Dn mice were treated with ethosuximide, an antiepileptic known to inhibit the activity of several GIRK channels, including GIRK2 encoded by the Hsa21 KCNJ6 gene.117 After a total of 10 weeks of treatment, impaired performance in neither the MWM nor CFC was rescued. Given the diversity of the known functions of Hsa21 genes, it should not be surprising that normalization of the activity of a single Hsa21 gene, in the context of overexpression of many remaining trisomic genes, fails to rescue learning deficits.
Based on the evidence of elevated activity of the NMDA receptor in the Ts65Dn mice, and the relatively high levels of the NMDA receptor subunit NR2B in the hippocampus, 3–6-month-old male Ts65Dn mice were injected for 2 weeks with the NMDAR 2B subunit-specific antagonist, Ro256981. Not only was performance in the Y-maze not improved, but performance in the Barnes maze actually deteriorated.118
Comparison of drug effects in the Ts65Dn
Of the 43 reports listed in Table 1, four did not include L/M in the evaluation, and eight of the 39 that did, failed to rescue L/M in the assay(s) used. The 31 reports of the successful rescue of L/M involved 23 different drugs targeting a diverse set of 19 proteins, complexes, or cellular processes. In 14 of the 31 successful reports, rescue was less than complete in that 1) performance improved, but did not reach the level of untreated control mice; or 2) performance was rescued in one test of L/M, but not in another in the same study; or 3) results were inconsistent among tests of the same drug carried out by different laboratories. Incomplete rescue does not diminish the significance of the results; rather, it potentially provides additional opportunities for insight.
L/M responses to drug treatments, of course, are first influenced by the targets and mechanisms of action of the drugs employed. However, responses may be modulated – leading to partial rescue – by details of the experimental design, which may include the use of older mice, suboptimal drug doses, and increased difficulty of the L/M task. Stress could also be a factor, depending on the specific L/M task and the method used for drug delivery. Among the drugs showing only partial rescue in specific L/M tasks, minocycline, memantine, and vitamin E were tested in the WRAM, while melatonin, NAP + SAL, and peptide 6 only partially rescued acquisition in the MWM. Water mazes are generally aversive tasks for mice, and the Ts65Dn mice are more sensitive than are controls to the stresses associated with the MWM.22,119 The stresses of drug delivery by daily injections are not implicated in these cases, because each drug was provided in food, water, or by means of implanted timed release subcutaneous capsules. Partial rescue was observed in one instance of another stressful task, CFC, where the drug, formoterol, was delivered by daily injection.66 On the other hand, there are drugs – among them, PTZ, memantine, and EGCG – that completely rescued performance in the MWM, and performance in CFC (with memantine).47 Thus, effects of stressors are not yet clear.
In several cases, a drug rescued L/M in one task but not in another. The effect of task complexity may be evident in the memantine experiments. When memantine was delivered in drinking water (a benign delivery method), learning was completely rescued in NOR, but only partially rescued in the WRAM, a task that places greater demand on working memory than does NOR and other water tasks such as the MWM. These mice were treated with memantine for 6 months, as compared to an acute injection that successfully rescued CFC and NOR.47,49 Therefore, the difficulty of WRAM versus CFC may be a contributing factor in this case. In a different scenario, SAG 1.1 rescued both acquisition and retention on the MWM, but it failed to rescue performance in the Y-maze. Drugs influencing (or not) performance in the spontaneous alternation Y-maze, and the related T-maze, are worth further consideration. Typically, neither is as aversive as the water tasks (if the mice are handled carefully); and in the spontaneous versions most often used with the Ts65Dn, they test only the preference for novelty and the ability to remember prior choices. In addition to SAG 1.1, both lithium and the GABAB receptor antagonist CGP55845 failed to rescue performance in the T-maze, although each rescued performance in other tasks, including NOR, OL and the aversive CFC. Conversely, spontaneous alternation T-maze or Y-maze performances were rescued with fluoxetine, xamoterol, and PTZ, and performance on a related four-arm alternation maze was rescued with physostigmine.58,60,30,64 Taken together, these data are interesting in terms of what they may indicate about drug targets and their distribution within the brain, and regarding what brain regions are involved in each task, as related to cognitive deficits in DS.
Additional limits of the current data are that some other drugs were tested only in a single task, and that the various studies differed in terms of the age of mice used, the dose used, and the details of L/M protocols. While additional, more comprehensive experiments could clarify the contributions of these issues, the data overall suggest that single drugs are unlikely to produce the desired efficacy, and that combinations of drugs should be explored.
Impaired adult neurogenesis has been linked to deficits in L/M in specific tasks. Of interest, ablation of adult neurogenesis in rats by hippocampal irradiation resulted in impaired performance in CFC (but not in a discrimination version of the T-maze).120 Rescue of impaired adult neurogenesis plus L/M in the Ts65Dn mice was investigated with five different drugs with inconsistent results. When neurogenesis was rescued by treating adult mice with lithium or by treating prenatal and perinatal mice with fluoxetine, performance in CFC was also rescued. However, performance on the T-maze was not rescued by lithium, while performance on the Y-maze was rescued by fluoxetine.53,70 (It should be noted that both of these experiments tested for spontaneous alternation; not discrimination as used in Hernández-Rabaza et al120.) Similarly, RO4938581 rescued both neurogenesis and performance on the MWM, but conversely, prenatal choline treatment only partially rescued adult neurogenesis, but it completely rescued performance on the WRAM. Moreover, formoterol failed to rescue neurogenesis, but it partially rescued CFC. While impaired adult neurogenesis is well established in the Ts65Dn mice, the requirement for normal adult neurogenesis for successful learning in CFC and various mazes needs further study.
Two drugs/drug targets were tested by multiple groups with inconsistent results. Of four reports using fluoxetine, one reported severe negative outcomes of seizures and death.57 This study, however, used a much higher dose of fluoxetine than the other three, and indeed, such a high dose has since been shown by a second group to have adverse effects.58 Three drugs targeting cholinesterase activity were tested. Both physostigmine and galantamine successfully rescued performance in the four-arm alternation and an olfactory test, respectively, while donepezil failed to rescue performance on the MWM.60,61
It is also notable that physostigmine was successful in mice at 4 months of age, but not in mice at 10 months or 16 months of age.60 This could reflect an age-dependent level of neurodegeneration and loss of acetylcholine, which is too advanced at 10 months for rescue by a single injection of an anticholinesterase. Conversely, when treatments with minocycline and vitamin E were started at earlier ages – 7 months and 4 months, respectively – and maintained for several months, both prevented neurodegeneration at 10 months. However, when started at 5 months, melatonin only partially rescued neurodegeneration at 10 months, while prenatal choline only partially prevented it at 13–17 months.74,104,105 For L/M amelioration, older age at the start of treatment is not an absolute exclusion – PTZ, memantine, and xamoterol each completely rescued L/M and estrogen, partially, when mice were treated at >9–12 months of age.23,32,47,64
The age for pharmacological treatment is an important consideration not only for AD in DS, but also for ID in DS. For two drugs, fluoxetine and SAG 1.1, treatment solely in the early postnatal time periods resulted in successful L/M at young adult ages of 6 weeks and 16 weeks, respectively.53,77 Indeed, for rescue on the MWM (but not on the spontaneous alternation Y-maze), SAG 1.1 treatment involved only a single P0 injection.77 Three drugs were tested by the treatment of only pregnant dams: fluoxetine and NAP/SAL were injected during gestation;59,98 and choline was provided in food to dams during gestation and through weaning.105 In another treatment, vitamin E was provided in food both to dams during gestation and to the offspring through to the age of L/M testing.94 In each case, the prenatal drug exposure resulted in at least the partial rescue of L/M in adult offspring. As promising as such results are, they should be interpreted with caution. Adverse effects of fluoxetine on the developing fetus are a concern,121,122 and as Reeves et al have discussed,76,77 activation of the sonic hedgehog pathway is seen in many malignancies and thus cannot be assumed to be completely benign either. While choline is an essential nutrient, its biochemistry is linked to folate and one-carbon metabolism; as will be discussed, several genes in this pathway map to the Mmu10 syntenic region and therefore their effects in trisomy cannot be assayed in the Ts65Dn mice. Lastly, clinical trials with vitamin E failed to prevent cognitive decline (see the following sections).
In spite of the partial rescues, it is of interest that so many drugs of such diverse targets can all be successful in rescuing complex tasks of L/M. Just how diverse the targets and mechanisms of action are, however, remains to be fully elucidated. For example, memantine, in addition to antagonizing NMDA receptor activation, also acts as a dopamine receptor agonist123 and indirectly increases dopamine levels.124 Memantine and fluoxetine act synergistically in models of depression to rescue behavior in the forced swim test.125 NMDA receptor activation modulates GABA receptor activity through phosphorylation of the γ subunit and fluoxetine alters GABA transmission in the hippocampus.126,127
Current status of clinical trials for ID in DS
Several clinical trials for cognitive deficits in DS have been completed recently and/or are in progress. Such trials are challenging. Outcome measures are complex and not standardized, and improvements in a specific task(s) are not straightforward enough to translate to improvements in important quality-of-life factors such as confidence, independence, or job satisfaction. These challenges are not unique to ID in DS. They are being addressed in other causes of ID (eg, Fragile X), and proposed solutions may be applicable to DS.128
Some very small trials (fewer than ten participants) have tested the cholinesterase inhibitors, donepezil, and rivastigmine, based on the observations discussed earlier regarding abnormal levels of acetylcholine in the DS brain.129–131 While some positive outcomes were reported for individual participants, overall, these treatments were considered ineffective, and the trials were hampered by low power.
The efficacy of folate supplementation was tested in a larger trial where infants with DS, aged 3–30 months, received leucovorin (folinic acid) or placebo daily for 12 months.132 The choice of folate was based on the fact that seven Hsa21 genes are involved in folate metabolism, and evidence for their elevated expression and resulting perturbed concentrations of folate pathway components have been observed in DS. The primary outcome measure was a significant increase in global development age. While only a small effect was seen with folate, this effect was significantly increased in children who were also receiving thyroxine for hypothyroidism. Only two of the seven genes predicted to affect folate metabolism are trisomic in the Ts65Dn mice, making it an inadequate model for preclinical evaluations of folate-related supplements. The Hsa21 connection to the thyroid hormone may involve NRIP1, which is also not trisomic in the Ts65Dn mice.133 A larger trial with folate plus thyroxine is in progress (see http://clinicaltrials.gov).
Other trials are based, at least in part, on positive preclinical evaluations in Ts65Dn mice. A 2-year trial evaluated the effects of daily antioxidant intake for 53 individuals with DS and dementia.134 No difference in cognitive function and decline was seen between the treated and placebo groups.134 Also to evaluate treatment in older individuals with DS (>40 years old), the first large randomized, double-blind trial135 evaluated memantine, which is already in use for moderate to severe AD in the typical population, for the prevention of the development or worsening of dementia. Eighty-eight participants received memantine and 85 received placebo for 52 weeks. There were no differences in outcomes between the groups; both groups declined in terms of cognitive level and function.135 It cannot be excluded that the neurodegeneration was too advanced in this age group for antioxidants and memantine to provide a benefit.
A smaller randomized, placebo-controlled trial tested memantine in 20 younger adults with DS (18–32 years old).136 After 16 weeks of treatment, there were no differences in the primary outcome measures assessing episodic and spatial memory between the memantine group and the placebo controls; however, there was a marginally significant outcome in one secondary memory measurement.136 Results of a small trial with the green tea component, EGCG, were recently reported.84 Fifteen participants aged 14–29 years were treated with EGCG for 3 months. Compared with the placebo group, those treated with EGCG showed a significant improvement in a test of visual memory.
Clinical trials currently recruiting participants include a Phase II trial with another GABAA α5 antagonist, RG1662 (Hoffman-La Roche Ltd., Basel, Switzerland), that started in May 2014 (http://clinicaltrials.gov). This is an international trial with centers in the US, France, Italy, and Spain already recruiting, and additional centers are planned in several other countries. The expected end date is August 2015. Treatment will run for 26 weeks. Also in the recruiting stage (in Australia) is a trial using PTZ (BTD-001; Balance Therapeutics, Inc., San Bruno, CA, USA). Participants in both of these trials will be in the age range of 13–30 years. Moreover, Elan Corporation Ltd (Dublin, Ireland) has started recruiting for a trial using scyllo-inositol as a method for inhibiting the aggregation of Aβ and, therefore, for preventing cognitive decline.
Validity of the Ts65Dn in preclinical drug evaluations
Clinical trials are expensive in terms of time and resources, as well as the goodwill of the individuals they are designed to help, and their families. So far, clinical trials for ID and AD in DS have not shown the positive outcomes predicted from evaluations in Ts65Dn mice. The validity of a mouse model for DS (and, indeed, any disease system) needs careful consideration if the rather poor results obtained to date in the clinical trials overall are to be improved.137 For almost 20 years, the Ts65Dn has been the primary mouse model for DS, and its study has vastly increased our understanding and predictions of the cellular, molecular, physiological, and behavioral deficits in DS. However, as a model for testing drug efficacy, the Ts65Dn needs to be judged critically and dispassionately. There are two features of the Ts65Dn that need to be considered: 1) the Ts65Dn is trisomic for orthologs of only 88 of 161 Hsa21 protein-coding genes (ie, it is not trisomic for 73 protein-coding genes [45%] that are trisomic in the large majority of people with DS); and 2) it is trisomic for 50 protein-coding genes that are not orthologs of Hsa21 genes and are not trisomic in individuals with DS. This gene content raises several questions: 1) would the phenotypic features be modified if the 73 missing orthologs were trisomic?; 2) do the 50 non-Hsa21 orthologs make any contribution to the phenotypic features of the Ts65Dn? and 3) would drug responses be altered in either case? The answers to these questions are not known at this time, but a brief consideration of some gene functions is useful. Even with our limited knowledge regarding the functions and interactions of Hsa21 genes (a complete list of Hsa21 protein-coding genes and their distribution among mouse chromosomes and the Ts65Dn can be found in Ahmed et al16), many that are not trisomic in the Ts65Dn can be predicted to contribute to some aspect of ID or AD in DS; Table 2 lists 13 examples.
It has been repeatedly observed in cell lines and in tissues from both DS and DS mouse models that gene dosage does not result in exactly 50% increases in trisomic gene expression.8 Rather, there is considerable interindividual variation in the level of expression perturbation, with some genes showing <50% and others showing >>50% elevation. There are also gene, tissue, and development timing specificities in the observed levels of aberrations. Interindividual variation in the levels of gene expression is a natural phenomenon and is expected, at least in part, from allelic variation. Such variation is likely to contribute to the observed variation in phenotypic severity seen in DS, including in the level of cognitive impairment. It has also been proposed that the expression and phenotypic variation in DS could be due to epigenetic perturbation – ie, trisomy-induced aberrations in patterns of DNA methylation and histone modification.138 Indeed, altered patterns in DNA methylation in DS tissues have been reported.139 A number of Hsa21 genes could be relevant to epigenetics – among them, DNMT3L, which interacts with DNA methyl transferases, as well as the transcriptional regulators, HMGN1 and NRIP1, which bind histones and regulate chromatin configuration. Ten Hsa21 proteins, based on known functions, and Hsa21-encoded microRNAs, were proposed to potentially influence epigenetic phenomena.138 The same authors further proposed that these genes be used as a first group of targets for drugs. They did not discuss the challenges inherent in the design and simultaneous use of multiple drugs each directed at a different target, nor the potential advantages in considering targets that are downstream of the Hsa21 individual perturbations – eg, DNA methylase activities or histone modifications. It is noteworthy that several of the genes they propose as targets (eg, DNMT3L and NRIP1) are not trisomic in the Ts65Dn.
The cyclic nucleotide, cyclic guanosine monophosphate (cGMP), functions as an intracellular second messenger that has established roles in synaptic plasticity.140 Several phosphodiesterases regulate cGMP levels through degradation. Among them, the Hsa21-encoded protein, PDE9A, has the highest affinity for cGMP and is highly expressed in the hippocampus.140 Relevance to DS stems from the recent demonstrations that inhibition of PDE9A successfully rescued learning impairments in AD mouse models and prevented Aβ-induced neurotoxicity.141 The overexpression of PDE9A in DS could be an exacerbating influence both for cognitive deficits in DS, as well as for AD in DS. The PDE9A gene maps to the Mmu17 orthologous region of Hsa21, and therefore its effects cannot be assayed in the Ts65Dn.
Endostatin is an antiangiogenic fragment derived from the C-terminal of the Hsa21-encoded protein collagen 18A1. Endostatin is found with Aβ in plaques, and with cerebral vasculature in the AD brain and apoptotic neurons.142 It is induced following stroke and traumatic brain injury.143 It is not clear from these observations if endostatin is exacerbating neurodegeneration and damage, or if it is possibly serving a compensatory role. More recently, Al Ahmad et al144 studied the functions of endostatin in vitro in PC12 cells and demonstrated that endostatin inhibits NGF-induced differentiation, migration, and neurite outgrowth. Notably, these effects were dependent upon endostatin concentration, although the concentrations used could not be directly extended to predictions in the DS brain. In drosophila, endostatin functions in the regulation of neurotransmitter release, and it is required for maintenance of presynaptic homeostasis.145 The possible contribution of elevated COL18A1 and endostatin to phenotypic features in DS has not been studied. Of note, COL18A1 maps to the Mmu10 orthologous region.
Three trefoil factor genes, TFF1–TFF3, are clustered within distal Hsa21q and map to the Mmu17 orthologous region. TFF3 has been studied as a neuropeptide. In control mice, acute injection of TFF3 prior to training in NOR was shown to enhance acquisition and retention.146 While it has not been examined with respect to DS, it is notable that in the three DS mouse models trisomic for TFF3, the Tc1 (which carries a partially deleted Hsa21 encoding ~120 Hsa21 protein-coding genes) shows a more mild phenotype than Ts65Dn, while the Dp17 shows enhanced LTP, and the Ts1Yah (which is trisomic for a subset of nine genes from the Mmu17 region) shows enhanced learning on the MWM.16,147,148 Therefore, unlike predictions for most other Hsa21 genes, overexpression of TFF3 in trisomy may have positive or compensatory contributions to the DS phenotype.
Hsa21 components of the folate pathway have already been discussed in relation to a successful preliminary trial; one (cystathionine beta-synthetase) maps to the Mmu17 region and four to the Mmu10 region. The NRIP1 gene may play a role in the same clinical trial through the regulation of thyroid hormone expression.
While triplication of APP is promoted as the cause of AD in DS, it is important to note that there is considerable evidence to suggest that at least four Mmu10 genes – CSTB, SUMO3, S100B, and COL18A1/endostatin – may modulate this process. Each has been shown to affect the processing of APP and/or neuroinflammation, and/or to accumulate in AD plaques,149–152 which is discussed further.153 Furthermore, S100B is sensitive to the redox environment, showing either neuroprotective or neurotoxic properties, and it causes L/M deficits and neurodegeneration when overexpressed. Elevated levels of the Mmu10 gene, TRPM2, in DS might directly affect the efficacy of melatonin signaling and indirectly affect memantine efficacy through the impact on NMDA receptor activity.154,155
The majority of these non-Ts65Dn Hsa21 proteins are encoded by the Mmu10 region, and therefore it is notable that Dp10 mice have shown no impairment in CFC, NOR, MWM, or LTP at 2–4 months of age.19 These mice do, however, have very abnormal protein profiles at 8 months of age.153 These facts suggest that the abnormal protein profiles may include compensatory responses and protective effects, and/or that these mice develop age-related L/M impairments coincident with protein abnormalities. These possibilities need to be investigated. That genes mapping to the Mmu17 region also need to be considered is emphasized by a recent report showing that L/M deficits and LTP abnormalities present in Dp16:Dp17 mice that are trisomic for both the Mmu17 and the Mmu16 regions are not rescued by the same genetic manipulations that rescue them in the Dp16.156 This is an especially salient observation; a similar phenotype can have differing underlying molecular genetic causes and, therefore, may also have differing responses to pharmacotherapies.
The 50 non-Hsa21 orthologs trisomic in the Ts65Dn have not been considered by DS researchers for their potential influences on phenotypic features. These genes are, however, overexpressed in the Ts65Dn,15 and thus there is no logical motivation for assuming that they make no contribution. Among them are sorting nexin 9, which complexes with the Hsa21 DS cell adhesion molecule (DSCAM) involved in axon guidance, and that causes abnormal synaptic vesicle recycling when overexpressed.157,158 Several dynein light chain genes are also present; their overexpression perturbs early endosome signaling,159 similar to that seen in the Ts65Dn, which is notably only partially rescued with APP gene copy number reduction.160 Other Mmu17 genes trisomic in the Ts65Dn are paralogs of the Hsa21 genes and include the guanine nucleotide exchange factor TIAM2, the phosphoinositol phosphatase SYNJ2, and the cGMP degrading PDE10A. If their functions and expression patterns even partially overlap with the Hsa21 genes, they may be increasing perturbations seen in the Ts65Dn over what occurs in trisomy Hsa21 alone.
Lastly, Troca-Martin et al161 have proposed the mechanistic target of rapamycin (MTOR) pathway inhibitor, rapamycin, as a potential treatment for cognitive deficits in DS. This is based on the well-established role of the MTOR pathway in L/M, and on the observation of elevated phosphorylation of MTOR and AKT in the Ts1Cje mouse model of DS. The Ts1Cje is trisomic for a subset of ~70 protein-coding genes trisomic in the Ts65Dn. Rapamycin is US FDA approved and is being investigated in clinical trials for other IDs, including Fragile X, tuberous sclerosis, and Rett syndrome (http://clinicaltrials.gov). Similar aberrations in AKT and MTOR were not seen in the Ts65Dn, or in the Tc1 DS mouse model;16,162 however, several abnormalities in levels of MTOR pathway components are present in the Dp10 and Dp17 models153 (source: Gardiner). In addition, when the Ts65Dn are exposed to CFC, the dynamic responses in the MTOR pathway seen in control littermates are either absent or abnormal in the Ts65Dn (submitted to PLoSOne; currently under review). However, significant perturbations in the mitogen-activated protein kinase (MAPK) pathway, that is also robustly associated with L/M, have also been reported in the Ts65Dn and Tc1,16,162 and are present in the Dp10 and Dp17 (source: Gardiner et al). Thus, as with other proposed drug treatments, multiple options must be considered.
Conclusion
The reproducible rescue of L/M deficits in the Ts65Dn, with a large and diverse panel of drugs, is a valid reason for optimism that effective human clinical trials for cognitive deficits in DS will be possible. That these preclinical evaluations were successful in a broad age range of adult (as well as in pre and perinatal) Ts65Dn mice is of major significance. There are an estimated 300,000 people with DS in the US, thus forming a very significant target treatment population. While prevention of cognitive deficits is an important goal, prenatal treatments come with their own more stringent and challenging requirements for safety demonstrations. The details of the Ts65Dn experiments used in preclinical evaluations are important; different drugs rescued L/M in different tasks, or they produced only partial rescue, with and without attendant rescue of neurogenesis. This suggests that combinations of drugs may be both necessary and advantageous. This approach is common in cancer therapy and viral infections, and recent methods for the rapid identification of optimally effective combinations of drugs could be easily applied to in vitro DS systems.163,164 In spite of reasonable cause for optimism, however, the genetic limitations of the Ts65Dn are clear and may have consequences for outcomes of clinical trials. If phenotypic features and drug responses are affected in DS by Hsa21 genes that are not trisomic in the Ts65Dn, as well as by the non-Hsa21 genes that are trisomic, then preclinical evaluations may be less reliable and their effective translation to human clinical trials less successful than outcomes in the Ts65Dn suggest. Solutions to this challenge are not clear. The use of the full trisomy mouse that can be created by crossing the three duplication lines20 is very expensive, especially when testing of multiple drugs and multiple L/M tasks is the objective. Exploration of drug responses in human, neurally relevant, DS-derived cell lines could complement studies with additional mouse models of DS. Elucidation of the molecular basis of drug responses has received little attention to date, but it could be used to identify features common to successful drugs, leading to potentially more efficacious drug targets.
Acknowledgments
This work was supported by the Linda Crnic Institute for Down Syndrome and the National Institutes of Health (HD056235).
Disclosure
The author reports no conflicts of interest in this work.
References
Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–121. | ||
Irving C, Basu A, Richmond S, Burn J, Wren C. Twenty-year trends in prevalence and survival of Down syndrome. Eur J Hum Genet. 2008;16(11):1336–1340. | ||
CDC, Centers for Disease Control and Prevention. Improved national prevalence for 18 major birth defects. MMWR. 2006;54:6–12. | ||
Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with Down syndrome. Ment Retard Dev Disabil Res Rev. 2000;6(2):84–95. | ||
Silverman W. Down syndrome: cognitive phenotype. Ment Retard Dev Disabil Res Rev. 2007;13(3):228–236. | ||
Wiseman FK, Alford KA, Tybulewicz VL, Fisher EM. Down syndrome – recent progress and future prospects. Hum Mol Genet. 2009;18(R1):R75–R83. | ||
Sturgeon X, Gardiner KJ. Transcript catalogs of human chromosome 21 and orthologous chimpanzee and mouse regions. Mamm Genome. 2011;22(5–6):261–271. | ||
Vilardell M, Rasche A, Thormann A, et al. Meta-analysis of heterogeneous Down Syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genomics. 2011;12:229. | ||
Sturgeon X, Le T, Ahmed MM, Gardiner KJ. Pathways to cognitive deficits in Down syndrome. Prog Brain Res. 2012;197:73–100. | ||
Rueda N, Flórez J, Martínez-Cué C. Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012;2012:584071. | ||
Das I, Reeves RH. The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis Model Mech. 2011;4(5):596–606. | ||
Liu C, Belichenko PV, Zhang L, et al. Mouse models for Down syndrome-associated developmental cognitive disabilities. Dev Neurosci. 2011;33(5):404–413. | ||
Davisson MT, Schmidt C, Reeves RH, et al. Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res. 1993;384:117–133. | ||
Reeves RH, Irving NG, Moran TH, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11(2):177–184. | ||
Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm Genome. 2011;22(11–12):674–684. | ||
Ahmed MM, Dhanasekaran AR, Tong S, et al. Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain. Hum Mol Genet. 2013;22(9):1709–1724. | ||
Roubertoux PL, Carlier M. Mouse models of cognitive disabilities in trisomy 21 (Down syndrome). Am J Med Genet C Semin Med Genet. 2010;154C(4):400–416. | ||
Li Z, Yu T, Morishima M, et al. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet. 2007;16(11):1359–1366. | ||
Yu T, Liu C, Belichenko P, et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 2010;1366:162–171. | ||
Yu T, Li Z, Jia Z, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010;19(14):2780–2791. | ||
Das D, Phillips C, Hsieh W, Sumanth K, Dang V, Salehi A. Neurotransmitter-based strategies for the treatment of cognitive dysfunction in Down syndrome. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:140–148. | ||
Stasko MR, Costa AC. Experimental parameters affecting the Morris water maze performance of a mouse model of Down syndrome. Behav Brain Res. 2004;154(1):1–17. | ||
Granholm AC, Ford KA, Hyde LA, et al. Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiol Behav. 2002;77(2–3):371–385. | ||
Hunter CL, Bimonte-Nelson HA, Nelson M, Eckman CB, Granholm AC. Behavioral and neurobiological markers of Alzheimer’s disease in Ts65Dn mice: effects of estrogen. Neurobiol Aging. 2004;25(7):873–884. | ||
Plane JM, Shen Y, Pleasure DE, Deng W. Prospects for minocycline neuroprotection. Arch Neurol. 2010;67(12):1442–1448. | ||
Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down’s syndrome. Ann Neurol. 2004;56(5):675–688. | ||
Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology. 1999;38(12):1917–1920. | ||
Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci. 2004;24(37):8153–8160. | ||
Costa AC, Grybko MJ. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci Lett. 2005;382(3):317–322. | ||
Fernandez F, Morishita W, Zuniga E, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10(4):411–413. | ||
Rueda N, Flórez J, Martínez-Cué C. Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neurosci Lett. 2008;433(1):22–27. | ||
Colas D, Chuluun B, Warrier D, et al. Short-term treatment with the GABAA receptor antagonist pentylenetetrazole produces a sustained pro-cognitive benefit in a mouse model of Down’s syndrome. Br J Pharmacol. 2013;169(5):963–973. | ||
Arya R, Kabra M, Gulati S. Epilepsy in children with Down syndrome. Epileptic Disord. 2011;13(1):1–7. | ||
Wetmore DZ, Garner CC. Emerging pharmacotherapies for neurodevelopmental disorders. J Dev Behav Pediatr. 2010;31(7):564–581. | ||
Deidda G, Bozarth IF, Cancedda L. Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front Cell Neurosci. 2014;8:119. | ||
Collinson N, Kuenzi FM, Jarolimek W, et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 2002;22(13):5572–5580. | ||
Sur C, Quirk K, Dewar D, Atack J, McKernan R. Rat and human hippocampal alpha5 subunit-containing gamma-aminobutyric AcidA receptors have alpha5 beta3 gamma2 pharmacological characteristics. Mol Pharmacol. 1998;54(5):928–933. | ||
Howell O, Atack JR, Dewar D, McKernan RM, Sur C. Density and pharmacology of alpha5 subunit-containing GABA(A) receptors are preserved in hippocampus of Alzheimer’s disease patients. Neuroscience. 2000;98(4):669–675. | ||
Braudeau J, Delatour B, Duchon A, et al. Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J Psychopharmacol. 2011;25(8):1030–1042. | ||
Martínez-Cué C, Martínez P, Rueda N, et al. Reducing GABAA α5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome. J Neurosci. 2013;33(9):3953–3966. | ||
Wang J, Liu S, Haditsch U, et al. Interaction of calcineurin and type-A GABA receptor gamma 2 subunits produces long-term depression at CA1 inhibitory synapses. J Neurosci. 2003;23(3):826–836. | ||
Lipton SA. Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr Drug Targets. 2007;8(5):621–632. | ||
Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature. 1994;369(6477):235–239. | ||
Siddiqui A, Lacroix T, Stasko MR, Scott-McKean JJ, Costa AC, Gardiner KJ. Molecular responses of the Ts65Dn and Ts1Cje mouse models of Down syndrome to MK-801. Genes Brain Behav. 2008;7(7):810–820. | ||
Wu Y, Song W. Regulation of RCAN1 translation and its role in oxidative stress-induced apoptosis. FASEB J. 2013;27(1):208–221. | ||
Jung MS, Park JH, Ryu YS, et al. Regulation of RCAN1 protein activity by Dyrk1A protein-mediated phosphorylation. J Biol Chem. 2011;286(46):40401–40412. | ||
Costa AC, Scott-McKean JJ, Stasko MR. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology. 2008;33(7):1624–1632. | ||
Rueda N, Llorens-Martín M, Flórez J, et al. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of Down syndrome. J Alzheimers Dis. 2010;21(1):277–290. | ||
Lockrow J, Boger H, Bimonte-Nelson H, Granholm AC. Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav Brain Res. 2011;221(2):610–622. | ||
Scott-McKean JJ, Costa AC. Exaggerated NMDA mediated LTD in a mouse model of Down syndrome and pharmacological rescuing by memantine. Learn Mem. 2011;18(12):774–778. | ||
Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20(24):9104–9110. | ||
Clark S, Schwalbe J, Stasko MR, Yarowsky PJ, Costa AC. Fluoxetine rescues deficient neurogenesis in hippocampus of the Ts65Dn mouse model for Down syndrome. Exp Neurol. 2006;200(1):256–261. | ||
Bianchi P, Ciani E, Guidi S, et al. Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for Down syndrome. J Neurosci. 2010;30(26):8769–8779. | ||
Maya Vetencourt JF, Sale A, Viegi A, et al. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. 2008;320(5874):385–388. | ||
Choi HC, Kim YI, Song HK, Kim JE, Kim DS, Kang TC. Effects of selective serotonin reuptake inhibitors on GABAergic inhibition in the hippocampus of normal and pilocarpine induced epileptic rats. Brain Res. 2010;1357:131–141. | ||
Caiati MD, Cherubini E. Fluoxetine impairs GABAergic signaling in hippocampal slices from neonatal rats. Front Cell Neurosci. 2013;7:63. | ||
Heinen M, Hettich MM, Ryan DP, Schnell S, Paesler K, Ehninger D. Adult-onset fluoxetine treatment does not improve behavioral impairments and may have adverse effects on the Ts65Dn mouse model of Down syndrome. Neural Plast. 2012;2012:467251. | ||
Begenisic T, Baroncelli L, Sansevero G, et al. Fluoxetine in adulthood normalizes GABA release and rescues hippocampal synaptic plasticity and spatial memory in a mouse model of Down syndrome. Neurobiol Dis. 2014;63:12–19. | ||
Guidi S, Stagni F, Bianchi P, et al. Prenatal pharmacotherapy rescues brain development in a Down’s syndrome mouse model. Brain. 2014;137(Pt 2):380–401. | ||
Chang Q, Gold PE. Age-related changes in memory and in acetylcholine functions in the hippocampus in the Ts65Dn mouse, a model of Down syndrome. Neurobiol Learn Mem. 2008;89(2):167–177. | ||
de Souza FM, Busquet N, Blatner M, Maclean KN, Restrepo D. Galantamine improves olfactory learning in the Ts65Dn mouse model of Down syndrome. Sci Rep. 2011;1:137. | ||
Nijjar RK, Murphy C. Olfactory impairment increases as a function of age in persons with Down syndrome. Neurobiol Aging. 2002;23(1):65–73. | ||
Salehi A, Faizi M, Colas D, et al. Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Sci Transl Med. 2009;1(7):7ra17. | ||
Faizi M, Bader PL, Tun C, et al. Comprehensive behavioral phenotyping of Ts65Dn mouse model of Down syndrome: activation of β1-adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiol Dis. 2011;43(2):397–413. | ||
Lockrow J, Boger H, Gerhardt G, Aston-Jones G, Bachman D, Granholm AC. A noradrenergic lesion exacerbates neurodegeneration in a Down syndrome mouse model. J Alzheimers Dis. 2011;23(3):471–489. | ||
Dang V, Medina B, Das D, et al. Formoterol, a long-acting β2 adrenergic agonist, improves cognitive function and promotes dendritic complexity in a mouse model of Down syndrome. Biol Psychiatry. 2014;75(3):179–188. | ||
Pasquali L, Busceti CL, Fulceri F, Paparelli A, Fornai F. Intracellular pathways underlying the effects of lithium. Behav Pharmacol. 2010;21(5–6):473–492. | ||
Wada A. Lithium and neuropsychiatric therapeutics: neuroplasticity via glycogen synthase kinase-3beta, beta-catenin, and neurotrophin cascades. J Pharmacol Sci. 2009;110(1):14–28. | ||
Bianchi P, Ciani E, Contestabile A, Guidi S, Bartesaghi R. Lithium restores neurogenesis in the subventricular zone of the Ts65Dn mouse, a model for Down syndrome. Brain Pathol. 2010;20(1):106–118. | ||
Contestabile A, Greco B, Ghezzi D, Tucci V, Benfenati F, Gasparini L. Lithium rescues synaptic plasticity and memory in Down syndrome mice. J Clin Invest. 2013;123(1):348–361. | ||
Trazzi S, Fuchs C, De Franceschi M, Mitrugno VM, Bartesaghi R, Ciani E. APP-dependent alteration of GSK3β activity impairs neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2014;67:24–36. | ||
Singh M, Jadhav HR. Melatonin: functions and ligands. Drug Discov Today. 2014;19(9):1410–1418. | ||
Wade AG, Farmer M, Harari G, et al. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: a 6-month, randomized, placebo-controlled, multicenter trial. Clin Interv Aging. 2014;9:947–961. | ||
Corrales A, Martínez P, García S, et al. Long-term oral administration of melatonin improves spatial learning and memory and protects against cholinergic degeneration in middle-aged Ts65Dn mice, a model of Down syndrome. J Pineal Res. 2013;54(3):346–358. | ||
Corrales A, Vidal R, García S, et al. Chronic melatonin treatment rescues electrophysiological and neuromorphological deficits in a mouse model of Down syndrome. J Pineal Res. 2014;56(1):51–61. | ||
Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc Natl Acad Sci U S A. 2006;103(5):1452–1456. | ||
Das I, Park JM, Shin JH, et al. Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci Transl Med. 2013;5(201):201ra120. | ||
Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371(Pt 1):199–204. | ||
Hämmerle B, Elizalde C, Galceran J, Becker W, Tejedor FJ. The MNB/DYRK1A protein kinase: neurobiological functions and Down syndrome implications. J Neural Transm Suppl. 2003;(67):129–137. | ||
Park J, Song WJ, Chung KC. Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell Mol Life Sci. 2009;66(20):3235–3240. | ||
Becker W, Sippl W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011;278(2):246–256. | ||
Tejedor FJ, Hämmerle B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011;278(2):223–235. | ||
Xie W, Ramakrishna N, Wieraszko A, Hwang YW. Promotion of neuronal plasticity by (-)-epigallocatechin-3-gallate. Neurochem Res. 2008;33(5):776–783. | ||
De la Torre R, De Sola S, Pons M, et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol Nutr Food Res. 2014;58(2):278–288. | ||
Ryoo SR, Cho HJ, Lee HW, et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer’s disease. J Neurochem. 2008;104(5)1333–1344. | ||
Adayev T, Chen-Hwang MC, Murakami N, Wang R, Hwang YW. MNB/DYRK1A phosphorylation regulates the interactions of synaptojanin 1 with endocytic accessory proteins. Biochem Biophys Res Commun. 2006;351(4):1060–1065. | ||
Shimokawa T, Rahman MF, Tostar U, et al. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013;10(2):321–333. | ||
Morita K, Lo Celso C, Spencer-Dene B, Zouboulis CC, Watt FM. HAN11 binds mDia1 and controls GLI1 transcriptional activity. J Dermatol Sci. 2006;44(1):11–20. | ||
Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011;82(12):1807–1821. | ||
Patra SK, Rizzi F, Silva A, Rugina DO, Bettuzzi S. Molecular targets of (-)-epigallocatechin-3-gallate (EGCG): specificity and interaction with membrane lipid rafts. J Physiol Pharmacol. 2008;59 Suppl 9:217–235. | ||
Coskun PE, Busciglio J. Oxidative Stress and Mitochondrial Dysfunction in Down’s Syndrome: Relevance to Aging and Dementia. Curr Gerontol Geriatr Res. 2012;2012:383170. | ||
Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature. 1995;378(6559):776–779. | ||
Lockrow J, Prakasam A, Huang P, Bimonte-Nelson H, Sambamurti K, Granholm AC. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp Neurol. 2009;216(2):278–289. | ||
Shichiri M, Yoshida Y, Ishida N, et al. α-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic Biol Med. 2011;50(12):1801–1811. | ||
Netzer WJ, Powell C, Nong Y, et al. Lowering beta-amyloid levels rescues learning and memory in a Down syndrome mouse model. PLoS One. 2010;5(6):e10943. | ||
Shiryaev N, Pikman R, Giladi E, Gozes I. Protection against tauopathy by the drug candidates NAP (davunetide) and D-SAL: biochemical, cellular and behavioral aspects. Curr Pharm Des. 2011;17(25):2603–2612. | ||
Incerti M, Toso L, Vink J, et al. Prevention of learning deficit in a Down syndrome model. Obstet Gynecol. 2011;117(2 Pt 1):354–361. | ||
Incerti M, Horowitz K, Roberson R, et al. Prenatal treatment prevents learning deficit in Down syndrome model. PLoS One. 2012;7(11):e50724. | ||
Chohan MO, Li B, Blanchard J, et al. Enhancement of dentate gyrus neurogenesis, dendritic and synaptic plasticity and memory by a neurotrophic peptide. Neurobiol Aging. 2011;32(8):1420–1434. | ||
Blanchard J, Bolognin S, Chohan MO, Rabe A, Iqbal K, Grundke-Iqbal I. Rescue of synaptic failure and alleviation of learning and memory impairments in a trisomic mouse model of down syndrome. J Neuropathol Exp Neurol. 2011;70(12):1070–1079. | ||
McCann JC, Hudes M, Ames BN. An overview of evidence for a causal relationship between dietary availability of choline during development and cognitive function in offspring. Neurosci Biobehav Rev. 2006;30(5):696–712. | ||
Glenn MJ, Gibson EM, Kirby ED, Mellott TJ, Blusztajn JK, Williams CL. Prenatal choline availability modulates hippocampal neurogenesis and neurogenic responses to enriching experiences in adult female rats. Eur J Neurosci. 2007;25(8):2473–2482. | ||
Moon J, Chen M, Gandhy SU, et al. Perinatal choline supplementation improves cognitive functioning and emotion regulation in the Ts65Dn mouse model of Down syndrome. Behav Neurosci. 2010;124(3):346–361. | ||
Velazquez R, Ash JA, Powers BE, et al. Maternal choline supplementation improves spatial learning and adult hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2013;58:92–101. | ||
Ash JA, Velazquez R, Kelley CM, et al. Maternal choline supplementation improves spatial mapping and increases basal forebrain cholinergic neuron number and size in aged Ts65Dn mice. Neurobiol Dis. 2014;70:32–42. | ||
Cramer NP, Best TK, Stoffel M, Siarey RJ, Galdzicki Z. GABAB-GIRK2-mediated signaling in Down syndrome. Adv Pharmacol. 2010;58:397–426. | ||
Best TK, Cramer NP, Chakrabarti L, Haydar TF, Galdzicki Z. Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp Neurol. 2012;233(2):749–757. | ||
Best TK, Siarey RJ, Galdzicki Z. Ts65Dn, a mouse model of Down syndrome, exhibits increased GABAB-induced potassium current. J Neurophysiol. 2007;97(1):892–900. | ||
Kleschevnikov AM, Belichenko PV, Faizi M, et al. Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J Neurosci. 2012;32(27):9217–9227. | ||
Heller JH, Spiridigliozzi GA, Doraiswamy PM, Sullivan JA, Crissman BG, Kishnani PS. Donepezil effects on language in children with Down syndrome: results of the first 22-week pilot clinical trial. Am J Med Genet A. 2004;130A(3):325–326. | ||
Yuede CM, Dong H, Csernansky JG. Anti-dementia drugs and hippocampal-dependent memory in rodents. Behav Pharmacol. 2007;18(5–6):347–363. | ||
Malykh AG, Sadaie MR. Piracetam and piracetam-like drugs: from basic science to novel clinical applications to CNS disorders. Drugs. 2010;70(3):287–312. | ||
Moran TH, Capone GT, Knipp S, Davisson MT, Reeves RH, Gearhart JD. The effects of piracetam on cognitive performance in a mouse model of Down’s syndrome. Physiol Behav. 2002;77(2–3):403–409. | ||
Rueda N, Flórez J, Martínez-Cué C. Effects of chronic administration of SGS-111 during adulthood and during the pre- and post-natal periods on the cognitive deficits of Ts65Dn mice, a model of Down syndrome. Behav Brain Res. 2008;188(2):355–367. | ||
Pelsman A, Hoyo-Vadillo C, Gudasheva TA, Seredenin SB, Ostrovskaya RU, Busciglio J. GVS-111 prevents oxidative damage and apoptosis in normal and Down’s syndrome human cortical neurons. Int J Dev Neurosci. 2003;21(3):117–124. | ||
Cooper A, Grigoryan G, Guy-David L, Tsoory MM, Chen A, Reuveny E. Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc Natl Acad Sci U S A. 2012;109(7):2642–2647. | ||
Vidal V, García S, Martínez P, et al. Lack of behavioral and cognitive effects of chronic ethosuximide and gabapentin treatment in the Ts65Dn mouse model of Down syndrome. Neuroscience. 2012;220:158–168. | ||
Hanson JE, Weber M, Meilandt WJ, et al. GluN2B antagonism affects interneurons and leads to immediate and persistent changes in synaptic plasticity, oscillations, and behavior. Neuropsychopharmacology. 2013;38(7):1221–1233. | ||
Paul CM, Magda G, Abel S. Spatial memory: Theoretical basis and comparative review on experimental methods in rodents. Behav Brain Res. 2009;203(2):151–164. | ||
Hernández-Rabaza V, Llorens-Martín M, Velázquez-Sánchez C, et al. Inhibition of adult hippocampal neurogenesis disrupts contextual learning but spares spatial working memory, long-term conditional rule retention and spatial reversal. Neuroscience. 2009;159(1):59–68. | ||
Alwan S, Friedman JM. Safety of selective serotonin reuptake inhibitors in pregnancy. CNS Drugs. 2009;23(6):493–509. | ||
Cantor Sackett J, Weller RA, Weller EB. Selective serotonin reuptake inhibitor use during pregnancy and possible neonatal complications. Curr Psychiatry Rep. 2009;11(3):253–257. | ||
Seeman P, Caruso C, Lasaga M. Memantine agonist action at dopamine D2High receptors. Synapse. 2008;62(2):149–153. | ||
Nakaya K, Nakagawasai O, Arai Y, et al. Pharmacological characterizations of memantine-induced disruption of prepulse inhibition of the acoustic startle response in mice: involvement of dopamine D2 and 5-HT2A receptors. Behav Brain Res. 2011;218(1):165–173. | ||
Rogóz Z, Skuza G, Maj J, Danysz W. Synergistic effect of uncompetitive NMDA receptor antagonists and antidepressant drugs in the forced swimming test in rats. Neuropharmacology. 2002;42(8):1024–1030. | ||
Méndez P, Pazienti A, Szabó G, Bacci A. Direct alteration of a specific inhibitory circuit of the hippocampus by antidepressants. J Neurosci. 2012;32(47):16616–16628. | ||
Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011;70(3):385–409. | ||
Berry-Kravis E, Hessl D, Abbeduto L, Reiss AL, Beckel-Mitchener A, Urv TK; Outcome Measures Working Groups. Outcome measures for clinical trials in fragile X syndrome. J Dev Behav Pediatr. 2013;34(7):508–522. | ||
Kishnani PS, Heller JH, Spiridigliozzi GA, et al. Donepezil for treatment of cognitive dysfunction in children with Down syndrome aged 10–17. Am J Med Genet A. 2010;152A(12):3028–3035. | ||
Johnson N, Fahey C, Chicoine B, Chong G, Gitelman D. Effects of donepezil on cognitive functioning in Down syndrome. Am J Ment Retard. 2003;108(6):367–372. | ||
Heller JH, Spiridigliozzi GA, Crissman BG, McKillop JA, Yamamoto H, Kishnani PS. Safety and efficacy of rivastigmine in adolescents with Down syndrome: long-term follow-up. J Child Adolesc Psychopharmacol. 2010;20(6):517–520. | ||
Blehaut H, Mircher C, Ravel A, et al. Effect of leucovorin (folinic acid) on the developmental quotient of children with Down’s syndrome (trisomy 21) and influence of thyroid status. PLoS One. 2010;5(1):e8394. | ||
Park SW, Huang WH, Persaud SD, Wei LN. RIP140 in thyroid hormone-repression and chromatin remodeling of Crabp1 gene during adipocyte differentiation. Nucleic Acids Res. 2009;37(21):7085–7094. | ||
Lott IT, Doran E, Nguyen VQ, Tournay A, Head E, Gillen DL. Down syndrome and dementia: a randomized, controlled trial of antioxidant supplementation. Am J Med Genet A. 2011;155A(8):1939–1948. | ||
Hanney M, Prasher V, Williams N, et al; MEADOWS trial researchers. Memantine for dementia in adults older than 40 years with Down’s syndrome (MEADOWS): a randomised, double-blind, placebo-controlled trial. Lancet. 2012;379(9815):528–536. | ||
Boada R, Hutaff-Lee C, Schrader A, et al. Antagonism of NMDA receptors as a potential treatment for Down syndrome: a pilot randomized controlled trial. Transl Psychiatry. 2012;2:e141. | ||
Perrin S. Preclinical research: Make mouse studies work. Nature. 2014;507(7493):423–425. | ||
Dekker AD, De Deyn PP, Rots MG. Epigenetics: the neglected key to minimize learning and memory deficits in Down syndrome. Neurosci Biobehav Rev. 2014;45:72–84. | ||
Jones MJ, Farré P, McEwen LM, et al. Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in Down syndrome. BMC Med Genomics. 2013;6:58. | ||
Sanderson TM, Sher E. The role of phosphodiesterases in hippocampal synaptic plasticity. Neuropharmacology. 2013;74:86–95. | ||
Kroker KS, Mathis C, Marti A, Cassel JC, Rosenbrock H, Dorner-Ciossek C. PDE9A inhibition rescues amyloid beta-induced deficits in synaptic plasticity and cognition. Neurobiol Aging. 2014;35(9):2072–2078. | ||
Deininger MH, Fimmen BA, Thal DR, Schluesener HJ, Meyermann R. Aberrant neuronal and paracellular deposition of endostatin in brains of patients with Alzheimer’s disease. J Neurosci. 2002;22(24):10621–10626. | ||
Hou Q, Ling L, Wang F, Xing S, Pei Z, Zeng J. Endostatin expression in neurons during the early stage of cerebral ischemia is associated with neuronal apoptotic cell death in adult hypertensive rat model of stroke. Brain Res. 2010;1311:182–188. | ||
Al Ahmad A, Lee B, Stack J, et al. Endostatin binds nerve growth factor and thereby inhibits neurite outgrowth and neuronal migration in vitro. Brain Res. 2010;1360:28–39. | ||
Wang T, Hauswirth AG, Tong A, Dickman DK, Davis GW. Endostatin is a trans-synaptic signal for homeostatic synaptic plasticity. Neuron. 2014;83(3):616–629. | ||
Shi HS, Yin X, Song L, Guo QJ, Luo XH. Neuropeptide Trefoil factor 3 improves learning and retention of novel object recognition memory in mice. Behav Brain Res. 2012;227(1):265–269. | ||
Morice E, Andreae LC, Cooke SF, et al. Preservation of long-term memory and synaptic plasticity despite short-term impairments in the Tc1 mouse model of Down syndrome. Learn Mem. 2008;15(7):492–500. | ||
Pereira PL, Magnol L, Sahún I, et al. A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum Mol Genet. 2009;18(24):4756–4769. | ||
Yang DS, Stavrides P, Mohan PS, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. 2011;7(7):788–789. | ||
Li Y, Wang H, Wang S, Quon D, Liu YW, Cordell B. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc Natl Acad Sci U S A. 2003;100(1):259–264. | ||
Lu J, Esposito G, Scuderi C, et al. S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors. PLoS One. 2011;6(7):e22126. | ||
Wilcock DM, Griffin WS. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation. 2013;10:84. | ||
Block A, Dhanasekaran AR, Ahmed MD, Gardiner KJ. Abnormal protein profiles in hippocampus of mouse models of Down syndrome: similarities with Alzheimers disease. J Alzheimers Dis Parkinsonism. 2014;4:138–149. | ||
Naziroğlu M, Çgelik Ö, Özgül C, et al. Melatonin modulates wireless (2.45 GHz)-induced oxidative injury through TRPM2 and voltage gated Ca(2+) channels in brain and dorsal root ganglion in rat. Physiol Behav. 2012;105(3):683–692. | ||
Xie YF, Belrose JC, Lei G, et al. Dependence of NMDA/GSK-3β mediated metaplasticity on TRPM2 channels at hippocampal CA3-CA1 synapses. Mol Brain. 2011;4:44. | ||
Zhang L, Meng K, Jiang X, et al. Human chromosome 21 orthologous region on mouse chromosome 17 is a major determinant of Down syndrome-related developmental cognitive deficits. Hum Mol Genet. 2014;23(3):578–589. | ||
Worby CA, Simonson-Leff N, Clemens JC, Kruger RP, Muda M, Dixon JE. The sorting nexin, DSH3PX1, connects the axonal guidance receptor, Dscam, to the actin cytoskeleton. J Biol Chem. 2001;276(45):41782–41789. | ||
Shin N, Lee S, Ahn N, et al. Sorting nexin 9 interacts with dynamin 1 and N-WASP and coordinates synaptic vesicle endocytosis. J Biol Chem. 2007;282(39):28939–28950. | ||
Duguay D, Bélanger-Nelson E, Mongrain V, Beben A, Khatchadourian A, Cermakian N. Dynein light chain Tctex-type 1 modulates orexin signaling through its interaction with orexin 1 receptor. PLoS One. 2011;6(10):e26430. | ||
Salehi A, Delcroix JD, Belichenko PV, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42. | ||
Troca-Marín JA, Alves-Sampaio A, Montesinos ML. An increase in basal BDNF provokes hyperactivation of the Akt-mammalian target of rapamycin pathway and deregulation of local dendritic translation in a mouse model of Down’s syndrome. J Neurosci. 2011;31(26):9445–9455. | ||
Ahmed MM, Sturgeon X, Ellison M, Davisson MT, Gardiner KJ. Loss of correlations among proteins in brains of the Ts65Dn mouse model of down syndrome. J Proteome Res. 2012;11(2):1251–1263. | ||
Jaynes J, Ding X, Xu H, Wong WK, Ho CM. Application of fractional factorial designs to study drug combinations. Stat Med. 2013;32(2):307–318. | ||
Honda Y, Ding X, Mussano F, Wiberg A, Ho CM, Nishimura I. Guiding the osteogenic fate of mouse and human mesenchymal stem cells through feedback system control. Sci Rep. 2013;3:3420. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.