Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
Pharmacokinetic drug–drug interactions between 1,4-dihydropyridine calcium channel blockers and statins: factors determining interaction strength and relevant clinical risk management
Authors Zhou Y, Yu L, Zeng S, Huang Y, Xu H, Zhou Q ![]()
Received 7 October 2013
Accepted for publication 14 November 2013
Published 20 December 2013 Volume 2014:10 Pages 17—26
DOI https://doi.org/10.2147/TCRM.S55512
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Yi-Ting Zhou,1 Lu-Shan Yu,2 Su Zeng,2 Yu-Wen Huang,1 Hui-Min Xu,1 Quan Zhou1
1Department of Pharmacy, the Second Affiliated Hospital, School of Medicine, 2Department of Pharmaceutical Analysis and Drug Metabolism, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang Province, People's Republic of China
Background: Coadministration of 1,4-dihydropyridine calcium channel blockers (DHP-CCBs) with statins (or 3-hydroxy-3-methylglutaryl-coenzyme A [HMG-CoA] reductase inhibitors) is common for patients with hypercholesterolemia and hypertension. To reduce the risk of myopathy, in 2011, the US Food and Drug Administration (FDA) Drug Safety Communication set a new dose limitation for simvastatin, for patients taking simvastatin concomitantly with amlodipine. However, there is no such dose limitation for atorvastatin for patients receiving amlodipine. The combination pill formulation of amlodipine/atorvastatin is available on the market. There been no systematic review of the pharmacokinetic drug–drug interaction (DDI) profile of DHP-CCBs with statins, the underlying mechanisms for DDIs of different degree, or the corresponding management of clinical risk.
Methods: The relevant literature was identified by performing a PubMed search, covering the period from January 1987 to September 2013. Studies in the field of drug metabolism and pharmacokinetics that described DDIs between DHP-CCB and statin or that directly compared the degree of DDIs associated with cytochrome P450 (CYP)3A4-metabolized statins or DHP-CCBs were included. The full text of each article was critically reviewed, and data interpretation was performed.
Results: There were three circumstances related to pharmacokinetic DDIs in the combined use of DHP-CCB and statin: 1) statin is comedicated as the precipitant drug (pravastatin–nimodipine and lovastatin–nicardipine); 2) statin is comedicated as the object drug (isradipine–lovastatin, lacidipine–simvastatin, amlodipine–simvastatin, benidipine-simvastatin, azelnidipine–simvastatin, lercanidipine–simvastatin, and amlodipine–atorvastatin); and 3) mutual interactions (lercanidipine–fluvastatin). Simvastatin has an extensive first-pass effect in the intestinal wall, whereas atorvastatin has a smaller intestinal first-pass effect. The interaction with simvastatin seems mainly driven by CYP3A4 inhibition at the intestinal level, whereas the interaction with atorvastatin is more due to hepatic CYP3A4 inhibition. The interaction of CYP3A4 inhibitor with simvastatin has been more pronounced compared with atorvastatin. From the current data, atorvastatin seems to be a safer CYP3A4-statin for comedication with DHP-CCB. There is no convincing evidence that amlodipine is an unusual DHP-CCB, either as a precipitant drug or as an object drug, from the perspective of CYP3A4-mediated drug metabolism. Amlodipine may have interactions with CYP3A5 in addition to CYP3A4, which may explain its particular characteristics in comparison with other DHP-CCBs. The degree of DDIs between the DHP-CCB and statin and the clinical outcome depends on many factors, such as the kind of statin, physicochemical proprieties of the DHP-CCB, the dose of either the precipitant drug or the object drug, the sex of the patient (eg, isradipine–lovastatin), route of drug administration (eg, oral versus intravenous nicardipine–lovastatin), the administration schedule (eg, nonconcurrent dosing method versus concurrent dosing method), and the pharmacogenetic status (eg, CYP3A5-nonexpressers versus CYP3A5-expressers).
Conclusion: Clinical professionals should enhance risk management regarding the combination use of two classes of drugs by increasing their awareness of the potential changes in therapeutic efficacy and adverse drug reactions, by rationally prescribing alternatives, by paying attention to dose adjustment and the administration schedule, and by review of the appropriateness of physician orders. Further study is needed – the DDIs between DHP-CCBs and statins have not all been studied in humans, from either a pharmacokinetic or a clinical perspective; also, the strength of the different pharmacokinetic interactions of DHP-CCBs with statins should be addressed by systematic investigations.
Keywords: CYP3A4, 1,4-dihydropyridine, drug–drug interactions, HMG-CoA reductase inhibitors, myopathy, polypharmacy, physicochemical phenomena, prescription auditing
Introduction
Multimorbidity, defined as the coexistence of two or more chronic diseases, is a common phenomenon, especially in older people. Multimorbid patients usually take multiple concomitant drugs (polypharmacy). Adverse drug reactions and medication errors are all potential consequences of polypharmacy.1–3 Polypharmacy is not a problem in itself but carries a risk of drug–drug interactions (DDIs) in the event of a lack of coordination among care providers.4 Real or potential DDIs are one of the key elements included in the appropriateness review process before dispensing, as required by the Joint Commission International.5 For each DDI, the object drug is defined as the medication whose pharmacokinetics and/or pharmacodynamics may be modified by the drug interaction process. The precipitant drug is defined as the medication responsible for affecting the pharmacologic action or the pharmacokinetic properties of the object drug.4
The coadministration of 1,4-dihydropyridine calcium channel blockers (DHP-CCBs) with statins (or 3-hydroxy-3-methylglutaryl-coenzyme A [HMG-CoA] reductase inhibitors) is common for patients with hypercholesterolemia and hypertension. Lovastatin, simvastatin, and atorvastatin are widely used for the treatment of hypercholesterolemia and prevention of cardiovascular diseases. These three statins, along with DHP-CCBs, are extensively metabolized by cytochrome P450 (CYP)3A4.6,7 The commonly used DHP-CCBs include amlodipine, benidipine, felodipine, isradipine, lacidipine, lercanidipine, nifedipine, nimodipine, and nicardipine. In 2011, the US Food and Drug Administration (FDA) Drug Safety Communication set a new dose limitation for Zocor® (simvastatin) (Merck and Co, Inc, Whitehouse Station, NJ, USA) in patients taking simvastatin concomitantly with amlodipine. The daily dose of simvastatin should not exceed 20 mg because amlodipine can raise the levels of simvastatin in the body and increase the risk of myopathy.8 However, there is no dose limitation for lovastatin or atorvastatin in patients receiving amlodipine. Meanwhile, a combination pill formulation of amlodipine/atorvastatin (Caduet®) (Pfizer, Inc, New York, NY, USA) has been available in the market. Up to now, there has been no systematic review of the pharmacokinetic DDI profile of DHP-CCBs administered concomitantly with statins, the underlying mechanisms of DDIs of different degree, or of corresponding clinical risk management.
In June 2013, a cardiac physician in our hospital wrote a new discharge prescription containing simvastatin 40 mg with amlodipine 5 mg and he was notified by the auditing pharmacist about the drug-related problem and potential risk of myopathy. The physician seemed ignorant of the amlodipine–simvastatin interaction information. Since a dose reduction of simvastatin was impractical for the patient who required intensive lipid-lowering therapy, atorvastatin 20 mg was recommended by the pharmacist as an alternative, equivalent dose for simvastatin 40 mg. The case gave us a profound lesson, ie, even specialist physicians have areas in their field about which they are ignorant. A study in a primary care unit of a tertiary hospital showed that the incidence of inappropriate concomitant use of amlodipine and simvastatin (40 mg/day) was 5%. This implied that general practitioners might not know that the concomitant use of amlodipine and high dosage of simvastatin could be a risk factor for myopathy.9
Therefore, we here present an updated review on this issue, to enhance the awareness of DDI potential with the combination use of DHP-CCBs and statins and to endeavor to answer the scientific questions arising in clinical practice.
Methods
Relevant literature was identified by performing a PubMed search covering the period from January 1987 (the year lovastatin was launched) to September 2013, using the medical search headings (MeSH) terms “calcium channel blockers and HMG-CoA reductase inhibitors and pharmacokinetics” or “simvastatin and amlodipine and drug interaction” and additional filters (languages: English). Eighty-nine articles were detected. The inclusion criteria included studies in the field of drug metabolism and pharmacokinetics that described DDI between DHP-CCBs and statins currently available in the market. Eleven articles were finally included under this search strategy and inclusion/exclusion criteria.
We conducted a further review of the pharmacokinetic interaction studies associated with CYP3A4-metabolized statins, using the MeSH terms “lovastatin and simvastatin and atorvastatin and drug interaction and pharmacokinetics.” A further 56 articles were detected, and five of these articles, meeting the inclusion criteria “directly comparing degree of DDIs associated with lovastatin, simvastatin, or atorvastatin,” were finally included. In addition, three studies directly comparing DDI potential with regard to DHP-CCBs were selected from 56 articles that were retrieved in PubMed, using the search terms “dihydropyridine calcium channel blockers and CYP3A4 inhibition.” The full text of each included article was critically reviewed, and valuable information was summarized by data interpretation.
Results and discussion
There were three circumstances described related to pharmacokinetic DDIs in the combination use of DHP-CCBs and statins: 1) statin is comedicated as a precipitant drug (pravastatin–nimodipine and lovastatin–nicardipine); 2) statin is comedicated as an object drug (isradipine–lovastatin, lacidipine–simvastatin, amlodipine–simvastatin, benidipine–simvastatin, azelnidipine–simvastatin, lercanidipine–simvastatin, and amlodipine–atorvastatin); and 3) mutual interactions (lercanidipine–fluvastatin). Table 1 lists the DDI potential in combination use of DHP-CCBs and statins, along with a “not recommended” category and a “recommended, monitor therapy” category.10–18
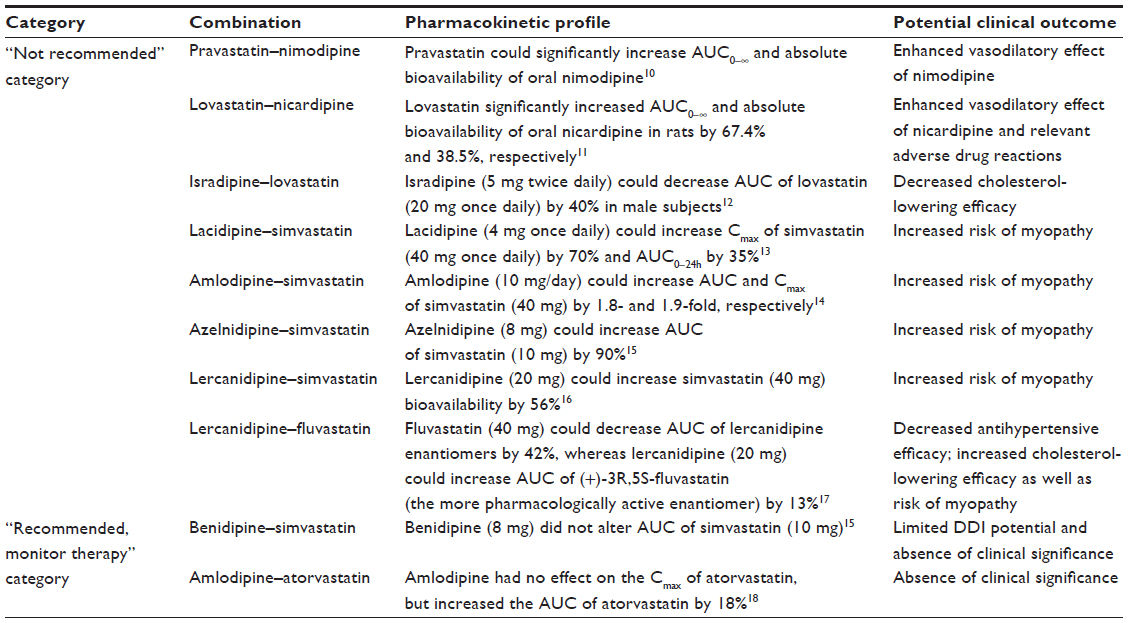 | Table 1 DDI potential in combination use of DHP-CCBs and statins |
We found that the degree of DDIs between DHP-CCB and statin and clinical outcome depend on many factors, such as the kind of statin, physicochemical proprieties of the DHP-CCB, dose of either the precipitant drug or object drug, sex of the patient (eg, isradipine–lovastatin combination), the route of drug administration (eg, oral nicardipine–lovastatin versus intravenous [IV] nicardipine–lovastatin), administration schedule (eg, nonconcurrent dosing method versus concurrent dosing method), and pharmacogenetic status (eg, CYP3A5 nonexpressers versus CYP3A5 expressers).
Circumstance 1: statin is comedicated as a precipitant drug
Pravastatin-nimodipine
Unlike other the DHP-CCBs primarily used for the treatment of hypertension, nimodipine is indicated for the improvement of neurological outcome as it reduces the incidence and severity of ischemic deficits in patients with subarachnoid hemorrhage.19 The metabolism of nimodipine is almost completely mediated by CYP3A4.20 Because of a high first-pass metabolism, the bioavailability of nimodipine averages 13% after oral administration.19 Pravastatin is known to be a statin that is not primarily metabolized by the CYP system, and it is not an object drug susceptible to CYP inhibitors or substrates. However, a CYP3A4 inhibition assay indicated that pravastatin could impair CYP3A4 enzyme activity in a concentration-dependent manner, with a 50% maximal inhibition concentration (IC50) of 14 μM.10 The combination use of nimodipine and pravastatin is common in clinical practice.
Lee et al investigated the effects of pravastatin on the pharmacokinetics of nimodipine in rats. Nimodipine was administered to rats intravenously (3 mg/kg) and orally (12 mg/kg) with pravastatin (0.3 and 1 mg/kg). The addition of pravastatin (1 mg/kg) increased the area under the plasma concentration–time curve from time zero to infinity (AUC0–∞) and absolute bioavailability of oral nimodipine by 30.9% and 31.1%, respectively. However, the pharmacokinetics of IV nimodipine were not affected by the concomitant use of pravastatin, in contrast to those of oral nimodipine.10 The enhanced oral bioavailability of nimodipine might be mainly due to inhibition of the CYP3A-mediated metabolism of nimodipine in the small intestine and/or in the liver. Due to the concentration-dependent CYP3A4 inhibition characteristics of pravastatin, coadministration of pravastatin and nimodipine would require close monitoring for potential DDI for safe therapy of cardiovascular diseases, especially in the situation where patients need intensive lipid-lowering therapy and receive a high dose of pravastatin. Of course, the clinical relevance of this DDI should be investigated in clinical trials.
Lovastatin–nicardipine
Nicardipine has been reported to be a substrate of CYP3A4 and P-glycoprotein (P-gp) in humans,21 and lovastatin is a dual inhibitor of both CYP3A4 and P-gp.11 Therefore, lovastatin could affect the pharmacokinetics of nicardipine when they are used concomitantly for the prevention or therapy of cardiovascular diseases.
Chung et al investigated the effects of oral lovastatin on the pharmacokinetics of IV and oral nicardipine in rats. Nicardipine was given intravenously (40 mg/kg) and orally (120 mg/kg) with 0 (control), 0.3, and 10 mg/kg of oral lovastatin to rats. Lovastatin was administered 30 minutes before nicardipine dosing. The AUC0–∞ of IV nicardipine was not altered in the presence of lovastatin. However, the AUC0–∞ and absolute bioavailability of oral nicardipine were significantly increased, by 67.4% and 38.5%, respectively. This indicated that lovastatin did not significantly inhibit the hepatic CYP3A-mediated metabolism of nicardipine, but it inhibited intestinal P-gp and/or CYP3A.11
In humans, the extent of absolute oral bioavailability of nicardipine is about 0.35, following a 30 mg dose at steady state. The increased bioavailability and drug exposure caused by a lovastatin–nicardipine DDI would be of clinical relevance in consideration of the enhanced vasodilatory effect of nicardipine.
Additionally, lovastatin shows high first-pass effects. Therefore, the rate of its metabolism is most likely determined by the blood flow through the liver and not the rate of hydroxylation. Because the DHP-CCBs are vasodilators, it is possible that they could increase hepatic blood flow and thereby increase lovastatin clearance.12 It is very necessary to investigate the effect of nicardipine on lovastatin pharmacokinetics, which was not addressed in the study by Chung et al.11
Coincidentally, the studies described under circumstance 1 were rat studies. It is worthwhile to address the DDI potential of pravastatin–nimodipine and lovastatin–nicardipine combination by conducting human pharmacokinetic studies or in vitro studies using human intestinal and hepatic cells.
Circumstance 2: statin is comedicated as an object drug
Isradipine–lovastatin
A randomized, double-blind, crossover study investigated the DDI profile of lovastatin (20 mg once daily) concomitantly with isradipine (5 mg twice daily) in healthy volunteers.12 Isradipine decreased the lovastatin AUC in male subjects by 40% (P<0.001) but did not alter the maximum concentration (Cmax) or the time-to-peak concentrations (Tmax) of lovastatin. In the female subjects, isradipine treatment had no significant effect on the AUC, Cmax, or Tmax of lovastatin. On the contrary, lovastatin had no effect on the clearance of isradipine, meaning the DDI was unidirectional. Lovastatin has a high hepatic clearance. Increases in hepatic blood flow by isradipine would seem to be the most likely explanation for the increased apparent clearance and decreased AUC of lovastatin in male subjects. It is important to emphasize that this was the first report on a sex-specific interaction between a DHP-CCB and statin. In clinical practice, decreased cholesterol-lowering efficacy would possibly be expected when isradipine is comedicated with lovastatin in male patients, and their combination should be avoided.
Lacidipine–simvastatin
Lacidipine, a long-acting DHP-CCB, is frequently administered with simvastatin. It is necessary to evaluate the possible pharmacokinetic interactions between simvastatin and lacidipine as they share the CYP3A4 metabolic pathway. One randomized, two-way crossover study investigated the effects of lacidipine on the pharmacokinetics of simvastatin. Lacidipine (4 mg once daily, for 8 days) increased the Cmax of simvastatin (40 mg once daily) by 70% (P=0.016) and of the AUC from time zero to 24 hours (AUC0–24h) by 35% (P=0.001). Significant differences were not observed in either the Tmax or in the half-life (T1/2).13 This finding suggested that the effects of lacidipine on the pharmacokinetics of simvastatin probably occurred in the absorption phase, possibly reducing the first-pass metabolism of simvastatin.
According to the dosing recommendations in the package inserts, it is preferable to take lacidipine in the morning and simvastatin in the evening.22,23 In the study by Ziviani et al,13 simvastatin and lacidipine were given together in the morning, so the extent of any possible interaction at the given doses was maximized under a worst-case scenario as far as an interaction on a first-pass effect of the two drugs was concerned. Despite that the observed increased exposure did not exceed the therapeutic window for simvastatin, vigilance is still required of clinicians when prescribing a lacidipine–simvastatin combination.
Amlodipine–simvastatin
Nishio et al investigated the interaction between amlodipine and simvastatin in patients with hypercholesterolemia and hypertension. The enrolled patients were given 4 weeks of simvastatin (5 mg/day), followed by 4 weeks of amlodipine (5 mg/day) coadministered with simvastatin (5 mg/day). The comedicated amlodipine increased the Cmax of simvastatin from 9.6±3.7 ng/mL to 13.7±4.7 ng/mL (P<0.05) and the AUC from 34.3±16.5 ng · h/mL to 43.9±16.6 ng · h/mL (P<0.05), despite that the cholesterol-lowering effect of simvastatin was not affected.24
A nonconcurrent dosing method was investigated by Park et al, to ameliorate the degree of DDI between amlodipine and simvastatin.25 In a random fashion, 17 patients received daily doses of 20 mg simvastatin and 5 mg amlodipine for 6 weeks, either with both drugs at 7:00 pm (concurrent) or with simvastatin at 7:00 pm followed by amlodipine at 11:00 pm (nonconcurrent). The Cmax and AUC from time zero to the last measurable concentration (AUC0-t) of simvastatin acid in the nonconcurrent group were 63.2% and 66.0%, respectively, of the corresponding values in the concurrent group (Cmax: 1.2±1.0 versus 1.9±0.9 ng/mL; AUC0–t: 10.3±8.3 versus 15.6±7.5 ng · h/mL). The pharmacodynamic changes in lipid profile and blood pressure were comparable between the groups.
Very recently, Son et al used a modeling approach to quantitatively describe the pharmacokinetic interaction between simvastatin and amlodipine. The subjects were given simvastatin 40 mg only (single administration), or simvastatin 40 mg and amlodipine 10 mg concomitantly (coadministration) once daily for 9 days. Compared with the single administration of simvastatin, the coadministration resulted in a 1.8- and 1.9-fold increase, for simvastatin, and a 1.9- and 2.3-fold increase, for simvastatin acid, in the AUC and Cmax, respectively. Also, the coadministered amlodipine 10 mg increased the simvastatin bioavailability by 46% and decreased the simvastatin clearance by 13%. In order to minimize the interaction with amlodipine 10 mg, the optimal simvastatin dose should be 60% of the usual dose (ie, simvastatin 24 mg was an optimal dose in coadministration with amlodipine 10 mg).14
Benidipine–simvastatin
Benidipine is metabolized by CYP3A4 in humans. It has been found to inhibit the CYP3A4-mediated metabolism of simvastatin in a concentration-dependent manner.26 In one study, assuming a competitive inhibition mechanism, the inhibitor constant (Ki) value, based on the unbound concentrations, was calculated to be 0.846 μM for benidipine. A quantitative prediction of the in vivo DDI between simvastatin and benidipine was performed using the “well-stirred” mode.15 If simvastatin (10 mg) and benidipine (8 mg, the clinically recommended highest dose) were to be administered concomitantly, the ratio of AUC of simvastatin with and without inhibitor (AUC+I/AUC) was predicted to be 1.01. The result suggested that benidipine is unlikely to cause a DDI by inhibiting CYP3A4 activity in the liver and that the pharmacokinetic interaction between benidipine and simvastatin lacks clinical significance.
Azelnidipine–simvastatin
Azelnidipine is also metabolized by CYP3A4, and it can concentration-dependently competitively inhibit the in vitro metabolism of simvastatin. Under study, the Ki value for azelnidipine, based on the unbound concentrations, was found to be 0.0181 μM. If simvastatin (10 mg) and azelnidipine (8 mg) were coadministered, the AUC+I/AUC for simvastatin was predicted to be 1.72, which is close to the observed value (1.9) in healthy volunteers. The predicted AUC+I/AUC of simvastatin coadministered with repeat administration of 16 mg azelnidipine was 2.68.15 Therefore, the DDI between azelnidipine and simvastatin may be of clinical relevance, and their combination use should be avoided.
Lercanidipine–simvastatin
Lercanidipine undergoes extensive hepatic metabolism to inactive metabolites via CYP3A4. The prescribing information for lercanidipine tablets (Apotex Pty Ltd, Macquarie Park, NSW, Australia) specifically describes the DDI between lercanidipine and simvastatin. Under study, the coadministration of a 20 mg dose of lercanidipine with 40 mg simvastatin did not change the bioavailability of lercanidipine; however, a 56% increase in bioavailability was observed for simvastatin and a 28% increase for its active metabolite β-hydroxy acid. The prescribing information recommends adoption of a nonconcurrent dosing method, with lercanidipine administered in the morning and simvastatin in the evening.16
Amlodipine–atorvastatin
These two drugs have been investigated together and found to be safe, whether in free formulations or in the combination pill formulation. Data from a DDI study in healthy subjects indicated that the coadministration of amlodipine (10 mg) tablets and atorvastatin (80 mg) tablets did not alter the pharmacokinetics of amlodipine. Comedicated amlodipine had no effect on the Cmax of atorvastatin but increased the AUC of atorvastatin by 18%, which was not clinically meaningful.18 A randomized, two-way crossover study in healthy volunteers confirmed the bioequivalence of a combination pill formulation containing amlodipine besylate/atorvastatin calcium with coadministered matching doses of amlodipine besylate and atorvastatin calcium tablets at the highest (10/80 mg) and lowest (5/10 mg) dose strengths.27 The administration of a single tablet of amlodipine/atorvastatin, compared with the coadministration of these agents as two separate tablets, improved patient adherence and reduced the likelihood that a patient would be prescribed a different statin with greater DDI potential.
Circumstance 3: mutual interactions between DHP-CCB and statin
Lercanidipine–fluvastatin
Fluvastatin is the only statin that undergoes extensive metabolism by CYP2C9, and its pharmacokinetics mainly depend on the CYP2C9 genotype.28 Lercanidipine is a typical CYP3A4 substrate. Generally, the metabolic DDI potential between these two drugs is minimal. However, a randomized crossover, three-period clinical trial presented surprising findings.17 The study was conducted in healthy subjects treated with a single oral dose of racemic lercanidipine (20 mg) or fluvastatin (40 mg), or lercanidipine plus fluvastatin. In the monotherapy phase, the disposition of both drugs exhibited stereoselectivity. The AUC values were significantly higher for (−)-3S,5R-fluvastatin than for (+)-3R,5S-fluvastatin (358.20 versus 279.68 ng · h/mL) and for S-lercanidipine compared with R-lercanidipine (13.90 versus 11.88 ng · h/mL). The AUC values of (+)-3R,5S-fluvastatin were increased, and the stereoselectivity in the fluvastatin pharmacokinetics was abolished after the addition of lercanidipine, whereas coadministered fluvastatin significantly decreased the AUC values of lercanidipine enantiomers (S-lercanidipine: 8.06 versus 13.90 ng · h/mL and R-lercanidipine: 6.76 versus 11.88 ng · h/mL). Boralli et al speculated that fluvastatin induced intestinal P-gp and consequently, that it reduces the bioavailability of lercanidipine.17 Fluvastatin pharmacokinetics are also dependent on ABCB1 (adenosine triphosphate [ATP]-binding cassette transporter subfamily B member 1) (the gene coding P-gp) polymorphism. Lercanidipine might inhibit P-gp activity and thus elevate the fluvastatin exposure. However, the underlying mechanism for this DDI remains unclear. Such interaction between fluvastatin and lercanidipine would be clinically relevant since the therapeutic effects are 30-fold higher for (+)-3R,5S-fluvastatin and 100- to 200-fold higher for S-lercanidipine compared with their respective enantiomers. A potential clinical outcome would be anticipated, including decreased antihypertensive efficacy and increased cholesterol-lowering efficacy, as well as risk of myopathy.
Strength of pharmacokinetic interactions of DHP-CCBs
It is an interesting topic to discuss the strength of the pharmacokinetic interaction of DHP-CCBs. Although they belong to the same structural and therapeutic class, the physicochemical properties of the currently used DHP-CCBs vary significantly. Uesawa and Mohri studied the relationship between the lipophilicities of 13 DHP-CCBs and the strength of their pharmacokinetic interaction with grapefruit juice (GFJ), a strong CYP3A4 inhibitor.29 Interestingly, lipophilicity was found to be an important factor in the strength of the pharmacokinetic interactions of DHP-CCBs with concomitant intake of GFJ. The logarithm of the molecular 1-octanol-water partition coefficient (logP) values indicated significant positive correlations with the interaction strength. Lercanidipine and niguldipine, with Ghose-Crippen-Viswanadhan octanol-water partition coefficient (ALogP) being 6.42 and 6.27, respectively, were estimated to be high-risk object drugs showing a predictive increase of 300% in the AUC, with GFJ intake.
Katoh et al investigated the inhibitory effects of 13 kinds of DHP-CCBs on human CYP-isoenzyme-dependent reactions using microsomes from human B-lymphoblast cells expressing CYP and predicted the DDIs using the “well-stirred” mode.15 The investigated DHP-CCBs included amlodipine, aranidipine, barnidipine, benidipine, cilnidipine, efonidipine, felodipine, manidipine, nicardipine, nifedipine, nilvadipine, nisoldipine, and nitrendipine. In consideration of the Ki values obtained in the in vitro inhibition study and the concentration of DHP-CCBs in human liver, the researchers concluded that the CYP inhibition by DHP-CCBs, except for nicardipine, might be clinically insignificant. Since the inhibitory effects on the CYP isoforms were measured using recombinant CYP isoenzymes in the Katoh et al study,15 differences in the content of NADPH-CYP reductase and cytochrome b5 or in the lipid constitution between the expression system and human liver microsomes might affect the inhibitory effects and the reliability of the prediction.
Up to now, there has been no literature comparing the strength of the pharmacokinetic interactions of DHP-CCBs with statins. In vitro experiments have not provided evidence that amlodipine is a distinctive DHP-CCB, either as a precipitant drug or as an object drug, from the perspective of CYP3A4-mediated drug metabolism. However, currently, of all the DHP-CCBs, the FDA only requires that amlodipine not to be comedicated with more than 20 mg of simvastatin. There might be other underlying mechanisms for this strange phenomenon.
Harmsze et al evaluated the effect of coadministration of P-gp-inhibiting DHP-CCBs (nifedipine and barnidipine) or non-P-gp-inhibiting DHP-CCBs (amlodipine) on clopidogrel on-treatment platelet reactivity, in patients on dual antiplatelet therapy after an elective percutaneous coronary intervention.30 Only the use of amlodipine was significantly associated with a 2.3-fold increased risk of a clopidogrel poor response. Clopidogrel is a prodrug that needs to be converted in vivo to generate its active metabolite. CYP2C19 and CYP3A4/5 are the main enzymes involved in the conversion of clopidogrel into the active compound. The inhibition of P-gp by the concomitant use of P-gp-inhibiting DHP-CCBs may cause a decreased intestinal efflux of clopidogrel, thereby increasing clopidogrel plasma concentrations and counteracting the DHP-CCB effect of impairing the metabolic activation of clopidogrel. P-gp seems unlikely to play an important role in the pharmacokinetics of simvastatin.31 Therefore, the non-P-gp-inhibiting characteristic of amlodipine compared with other DHP-CCBs seems unable to explain the DDI potential of amlodipine toward simvastatin.
Zuo et al evaluated the effect of the CYP3A5*3 allele on the DDI between tacrolimus and amlodipine in healthy subjects. Amlodipine decreased the mean apparent oral clearance of tacrolimus in subjects with the CYP3A5*1 allele (CYP3A5-expressers) by 2.2-fold (P=0.005), while it had no effect on that in the subjects with the CYP3A5*3/*3 genotype (CYP3A5-nonexpressers).32 The CYP3A system consists of the 3A4 and 3A5 isoenzymes, which share 84% amino acid sequence homology.33 We assume that amlodipine may have interactions with CYP3A5 in addition to CYP3A4, which may explain its particular characteristics in comparison with the other DHP-CCBs.
Strength of pharmacokinetic interactions of CYP3A4-metabolized statins
The comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin, when coadministered with CYP inhibitors, were investigated by Jacobson.34 Clarithromycin significantly (P<0.001) increased the AUC (and Cmax) of all three statins, most markedly simvastatin (approximately ten-fold increase in AUC) and simvastatin acid (12-fold increase), followed by atorvastatin (greater than four-fold increase) and then, pravastatin (almost two-fold increase). Clinical professionals may generally understand that pravastatin pharmacokinetics are unlikely to be altered in the presence of an CYP3A4 inhibitor, due to the fact that the statin does not undergo CYP-mediated metabolism, but they probably do not know the differences in DDI strength between simvastatin and atorvastatin.
A study by Hoch et al showed that the interaction of simvastatin with almorexant was more pronounced than with atorvastatin. More interestingly, a time-separated administration of simvastatin and almorexant significantly reduced the interaction strength, whereas relative time of administration had no effect on the magnitude of the interaction between atorvastatin and almorexant.35 The underlying mechanisms for difference in the DDI profile are as follows: 1) simvastatin has an extensive first-pass effect in the intestinal wall, whereas atorvastatin has a smaller intestinal first-pass effect; and 2) the interaction with atorvastatin is to a greater extent due to hepatic CYP3A4 inhibition, whereas the interaction of almorexant with simvastatin is mainly driven by CYP3A4 inhibition in the gut (ie, at the intestinal wall during drug absorption), where higher local concentrations of almorexant are reached than in the liver and where most of the first-pass effect of simvastatin takes place. When the drugs were ingested 2 hours apart, low residual systemic almorexant concentrations were present at the time of administration of the statin, and this significantly reduced the extent of interaction.
The coadministration of ticagrelor with atorvastatin resulted in an increase in the AUC of atorvastatin by 36% and the Cmax by 23%. However, following the coadministration of ticagrelor and simvastatin, the magnitude of the interaction was greater than that with atorvastatin, with mean increases in the simvastatin AUC and Cmax of 56% and 81%, respectively.36 Atorvastatin is thought to be less reliant than simvastatin on CYP3A4 metabolism. The calculated ratios of the contribution of CYP3A4 to the oral clearance of lovastatin, simvastatin, and atorvastatin are 1.00, 1.00, and 0.68, respectively.37 The relative lower ratio of the contribution of CYP3A4 to the oral clearance, together with smaller intestinal first-pass effect, may explain the relative difference in the impact of CYP3A inhibitors on the pharmacokinetics of atorvastatin versus the other statins metabolized by CYP3A4.
Simvastatin is reported to have dose-proportional pharmacokinetics up to 160 mg. It is safe and well tolerated up to at least 80 mg daily. Therefore, an observed increased exposure induced by the addition of DHP-CCB usually would not exceed the therapeutic window for simvastatin. Son et al has showed that extensive metabolizers yielded a decrease in simvastatin bioavailability of 81% and a decrease in simvastatin clearance by 4.6-fold compared with poor metabolizers, when CYP3A4/5 polymorphisms were concerned.14 Where the pharmacogenetic status (CYP3A4/5 polymorphism) is concerned, elevated Cmax and/or AUC values of simvastatin in the presence of amlodipine would be observed more distinctively with extensive metabolizers and thus may be of clinical relevance for safety.
Besides the competitive inhibition of CYP3A4 and interference in flow-dependent hepatic clearance, the mechanism for the DDI between DHP-CCBs and statins might also involve P-gp and/or organic anion transporting polypeptide (OATP) 1B1-mediated drug transport inhibition, due to the varying degrees of association between statin pharmacokinetics and transporters, like P-gp, OATP1B1, and breast cancer resistance protein.38,39 Furthermore, the phenomenon that comedicated fluvastatin decreased the AUC values of lercanidipine inspires us to elucidate the underlying mechanism.
Integrated care, in a dynamic continuum, is essential to a future vision for complex patients. In order to enable even more synergy among health care professionals in the care of complex patients, pharmacists should focus on medication therapy management and play a key role in collaborative practice. Physicians and pharmacists still have many answers to provide to the scientific question concerning DDIs between DHP-CCBs and statins.
Conclusion
In this review, we addressed the pharmacokinetic DDIs between DHP-CCBs and statins, and the factors determining the degree of those DDIs. From the current data, atorvastatin seems to be a safer CYP3A4-statin for comedication with DHP-CCBs. Clinical professionals should enhance the clinical risk management of combination use of these two classes of drugs, by increasing their awareness of the potential changes in therapeutic efficacy and adverse drug reactions, by rationally prescribing alternatives, by paying attention to dose adjustment and the administration schedule, and by ensuring an appropriateness review of physician orders before dispensing. Further study is needed – the DDIs between DHP-CCBs and statins have not all been studied in humans, either from a pharmacokinetic or a clinical perspective; also, the strength of the different pharmacokinetic interactions of DHP-CCBs with statins should be addressed by systematic investigations.
Acknowledgments
This work was supported by Zhejiang Provincial Bureau of Health grant 2012KYA090, National Natural Science Foundation of China grant 81373488, and National Major Projects of China grants 2012ZX09506001-004 and 2009ZX09304-003.
Disclosure
The authors report no conflicts of interest in this work.
References
Corsonello A, Pedone C, Corica F, Incalzi RA. Polypharmacy in elderly patients at discharge from the acute care hospital. Ther Clin Risk Manag. 2007;3(1):197–203. | |
Koh Y, Kutty FB, Li SC. Drug-related problems in hospitalized patients on polypharmacy: the influence of age and gender. Ther Clin Risk Manag. 2005;1(1):39–48. | |
Köberlein J, Gottschall M, Czarnecki K, Thomas A, Bergmann A, Voigt K. General practitioners’ views on polypharmacy and its consequences for patient health care. BMC Fam Pract. 2013;14(1):119. | |
Li W, Zeng S, Yu LS, Zhou Q. Pharmacokinetic drug interaction profile of omeprazole with adverse consequences and clinical risk management. Ther Clin Risk Manag. 2013;9:259–271. | |
Joint Commission International. Joint Commission International Accreditation Standards for Hospitals; Standards Lists Version. 4th ed. Oak Brook, IL: Joint Commission International; 2010. Available from: http://www.jointcommissioninternational.org/common/pdfs/jcia/IAS400_Standards_Lists_Only.pdf. Accessed March 20, 2013. | |
Leung A, Schaefer EW, Tempelhof MW, Stone NJ. Emphasizing statin safety in the hospitalized patient: a review. Am J Med. 2012;125(9):845–853. | |
Henneman A, Thornby KA. Risk of hypotension with concomitant use of calcium-channel blockers and macrolide antibiotics. Am J Health Syst Pharm. 2012;69(12):1038–1043. | |
fda.gov [homepage on the Internet]. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury; 2011 [updated December 15, 2011; cited October 4, 2013] Available from: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed November 17, 2013. | |
Wiwanitkit S, Wiwanitkit V. Inappropriate concomitant use of amlodipine and simvastatin: A report on its incidence in a primary care unit. Indian J Endocrinol Metab. 2011;15 Suppl 4:S409. | |
Lee CK, Choi JS, Choi DH. Effects of pravastatin on the pharmacokinetic parameters of nimodipine after oral and intravenous administration in rats: possible role of CYP3A4 inhibition by pravastatin. Indian J Pharmacol. 2012;44(5):624–628. | |
Chung JW, Yang SH, Choi JS. Effects of lovastatin on the pharmacokinetics of nicardipine in rats. Biopharm Drug Dispos. 2010;31(7):436–441. | |
Zhou LX, Finley DK, Hassell AE, Holtzman JL. Pharmacokinetic interaction between isradipine and lovastatin in normal, female and male volunteers. J Pharmacol Exp Ther. 1995;273(1):121–127. | |
Ziviani L, Da Ros L, Squassante L, Milleri S, Cugola M, Iavarone LE. The effects of lacidipine on the steady/state plasma concentrations of simvastatin in healthy subjects. Br J Clin Pharmacol. 2001;51(2):147–152. | |
Son H, Lee D, Lim LA, Jang SB, Roh H, Park K. Development of a Pharmacokinetic Interaction Model for Co-administration of Simvastatin and Amlodipine. Drug Metab Pharmacokinet. Epub August 20, 2013. | |
Katoh M, Nakajima M, Shimada N, Yamazaki H, Yokoi T. Inhibition of human cytochrome P450 enzymes by 1,4-dihydropyridine calcium antagonists: prediction of in vivo drug-drug interactions. Eur J Clin Pharmacol. 2000;55(11–12):843–852. | |
APO-Lercanidipine (lercanidipine hydrochloride) tablets [product information]. North Ryde: Apotex Pty Ltd; 2010. | |
Boralli VB, Coelho EB, Sampaio SA, Marques MP, Lanchote VL. Enantioselectivity in the pharmacokinetic interaction between fluvastatin and lercanidipine in healthy volunteers. J Clin Pharmacol. 2009; 49(2):205–211. | |
CADUET® (amlodipine besylate/atorvastatin calcium) tablets [patient information]. New York, NY: Pfizer Inc; 2012. | |
Nimotop® (nimodipine) Prescribing Information. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/018869s014lbl.pdf. Accessed October 4, 2013. | |
Liu XQ, Ren YL, Qian ZY, Wang GJ. Enzyme kinetics and inhibition of nimodipine metabolism in human liver microsomes. Acta Pharmacol Sin. 2000;21:690–694. | |
Katoh M, Nakajima M, Yamazaki H, Yokoi T. Inhibitory potencies of 1,4-dihydropyridine calcium antagonists to P-glycoprotein-mediated transport: comparison with the effects on CYP3A4. Pharm Res. 2000;17:1189–1197. | |
Motens Tablets 4mg. Prescribing Information. Available from: http://www.medicines.org.uk/emc/medicine/298/SPC. Accessed October 5, 2013. | |
ZOCOR (simvastatin) Tablets. Product Information. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/019766s077s082lbl.pdf. Accessed October 5, 2013. | |
Nishio S, Watanabe H, Kosuge K, Uchida S, Hayashi H, Ohashi K. Interaction between amlodipine and simvastatin in patients with hypercholesterolemia and hypertension. Hypertens Res. 2005; 28(3):223–227. | |
Park CG, Lee H, Choi JW, Lee SJ, Kim SH, Lim HE. Non-concurrent dosing attenuates the pharmacokinetic interaction between amlodipine and simvastatin. Int J Clin Pharmacol Ther. 2010;48(8):497–503. | |
Sugiyama Y, Mimura N, Kuwabara T, Kobayashi H, Ushiki J, Fuse E. Effect of benidipine on simvastatin metabolism in human liver microsomes. Drug Metab Pharmacokinet. 2007;22(3):199–205. | |
Chung M, Calcagni A, Glue P, Bramson C. Bioavailability of amlodipine besylate/atorvastatin calcium combination tablet. J Clin Pharmacol. 2006;46(9):1030–1037. | |
Zhou Q, Ruan ZR, Yuan H, Zeng S. CYP2C9*3(1075A>C), MDR1 G2677T/A and MDR1 C3435T are determinants of inter-subject variability in fluvastatin pharmacokinetics in healthy Chinese volunteers. Arzneimittelforschung. 2012;62(11):519–524. | |
Uesawa Y, Mohri K. Relationship between lipophilicities of 1,4-dihydropyridine derivatives and pharmacokinetic interaction strengths with grapefruit juice. Yakugaku Zasshi. 2008;128(1):117–122. | |
Harmsze AM, Robijns K, van Werkum JW, et al. The use of amlodipine, but not of P-glycoprotein inhibiting calcium channel blockers is associated with clopidogrel poor-response. Thromb Haemost. 2010;103(5):920–925. | |
Zhou Q, Ruan ZR, Jiang B, Yuan H, Zeng S. Simvastatin pharmacokinetics in healthy Chinese subjects and its relations with CYP2C9, CYP3A5, ABCB1, ABCG2 and SLCO1B1 polymorphisms. Pharmazie. 2013;68(2):124–128. | |
Zuo XC, Zhou YN, Zhang BK, et al. Effect of CYP3A5*3 Polymorphism on Pharmacokinetic Drug Interaction between Tacrolimus and Amlodipine. Drug Metab Pharmacokinet. 2013;28(5):398–405. | |
Walsky RL, Obach RS, Hyland R, et al. Selective mechanism-based inactivation of CYP3A4 by CYP3cide (PF-04981517) and its utility as an in vitro tool for delineating the relative roles of CYP3A4 versus CYP3A5 in the metabolism of drugs. Drug Metab Dispos. 2012;40:1686–1697. | |
Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol. 2004;94(9):1140–1146. | |
Hoch M, Hoever P, Theodor R, Dingemanse J. Almorexant effects on CYP3A4 activity studied by its simultaneous and time-separated administration with simvastatin and atorvastatin. Eur J Clin Pharmacol. 2013;69(6):1235–1245. | |
Teng R, Mitchell PD, Butler KA. Pharmacokinetic interaction studies of co-administration of ticagrelor and atorvastatin or simvastatin in healthy volunteers. Eur J Clin Pharmacol. 2013;69(3):477–487. | |
Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681–696. | |
Zhou Q, Chen QX, Ruan ZR, Yuan H, Xu HM, Zeng S. CYP2C9*3 (1075A>C), ABCB1 and SLCO1B1 genetic polymorphisms and gender are determinants of inter-subject variability in pitavastatin pharmacokinetics. Pharmazie. 2013;68(3):187–194. | |
Zhou Q, Ruan ZR, Yuan H, Xu DH, Zeng S. ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421c>A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Pharmazie. 2013;68(2):129–134. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.