Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 12
Pharmacokinetic Disposition Difference Between Cyclosporine and Voclosporin Drives Their Distinct Efficacy and Safety Profiles in Clinical Studies
Authors Li Y, Palmisano M, Sun D ![]() , Zhou S
, Zhou S
Received 27 March 2020
Accepted for publication 10 June 2020
Published 1 July 2020 Volume 2020:12 Pages 83—96
DOI https://doi.org/10.2147/CPAA.S255789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Yan Li,1 Maria Palmisano,1 Duxin Sun,2 Simon Zhou1
1Translational Development and Clinical Pharmacology, Celgene Corporation, Summit, NJ 07920, USA; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, MI 48109, USA
Correspondence: Simon Zhou
Translational Development and Clinical Pharmacology, Celgene Corporation, 556 Morris Avenue, Summit, NJ 07920, USA
Tel +1 908-673-9284
Fax +1 908-673-2842
Email [email protected]
Background: Voclosporin, a more potent derivative of cyclosporine, has been studied extensively in patients with immunologic disorders such as psoriasis, organ transplantation, uvetitis and lupus nephritis. Although better tolerated and safer than cyclosporine, voclosporin is inferior to cyclosporine in treating psoriasis, non-inferior to tacrolimus in organ transplantation and efficacious in treating lupus nephritis.
Methods: The pharmacokinetic dispositions of voclosporin and cyclosporine in central and peripheral compartments were analyzed and correlated with their distinct clinical efficacy and safety profiles.
Results: Both drugs demonstrated non-linear pharmacokinetics with increasing doses, more prominently at lower doses of voclosporin than at 10-fold higher doses of cyclosporine. Repeated lower dosing of voclosporin produced preferential calcineurin inhibition in and near blood circulation, leading to relatively lower cardiovascular and renal adverse effects but inferior efficacy for psoriasis compared to cyclosporine. With 10-fold higher plasma levels and deeper tissue penetration, cyclosporine has more prevalent renal and cardiac toxicities but superior efficacy to treat psoriasis.
Conclusion: Although the two drugs are similar in structure and mechanism of action, the high potency and low dose compounded by the non-linear disposition of voclosporin resulted in more systemic versus local calcineurin inhibition than with cyclosporine. The dispositional difference between voclosporin and cyclosporine accounted for the puzzling efficacy and safety observations in different patients and was the basis for their optimal and differential use in treating diverse immunologic disorders.
Keywords: pharmacokinetic, disposition, cyclosporine and voclosporin
Introduction
Cyclosporine, a calcineurin inhibitor (CNI) that binds to intracellular cyclophilins forming a complex, which then binds to intracellular calcineurin, leading to the inhibition of its activity and subsequent T-cell activation, has been the cornerstone treatment for patients receiving organ transplants since its introduction in the 1980s. In addition, cyclosporine has been shown to be active in the treatment of psoriasis, rheumatoid arthritis and other immunologic disorders of endogenous uveitis, Sjögren’s syndrome, myasthenia gravis and Crohn’s disease. Although its clinical use in these different autoimmune diseases has been significantly reduced owing to its nephrotoxicity and the availability of newer disease-modifying anti-rheumatic drugs, cyclosporine is still an important drug.1
In an effort to find the next generation of CNIs with optimal immunosuppression and reduced nephrotoxicity, voclosporin, an analogue of cyclosporine with a methyl group addition to the aminoacid-1 residue (Figure 1), has been synthesized and studied in over 2000 patients with diverse immunologic conditions of psoriasis, organ transplantation, uveitis and lupus nephritis, with fascinating and enigmatic results. Voclosporin was designed to have enhanced action against calcineurin and greater metabolic stability than cyclosporine, which has a metabolic soft spot at the aminoacid-1 position.2 In vitro data showed that voclosporin has at least five-fold lower IC50 values on lymphocyte proliferation, T-cell cytokine production and expression of all T-cell activation surface antigens.3 Given the high in vitro potency against calcineurin, it was unexpected that voclosporin was inferior to cyclosporine (35% vs 53% in clear or almost clear static physician’s global assessment [SPGA] score at week 12) in the phase III trial for the treatment of psoriasis.2,4 This raised a question on the effectiveness of voclosporin’s engagement of calcineurin in vivo. In a subsequent phase II trial in patients with kidney transplantation, voclosporin was shown to be non-inferior to tacrolimus, consistent with its sufficient systemic calcineurin inhibition.5 Furthermore, voclosporin was shown to be active in patients with active sight-threatening, non-infectious intermediate-, anterior and intermediate-, posterior- or pan-uveitis.6 However, it did not differentiate from placebo with activity in patients with clinically quiescent sight-threatening, non-infectious intermediate-, anterior and intermediate-, posterior- or pan-uveitis. In all these patients with immunologic disorders, voclosporin was demonstrated with better tolerability and lack of nephrotoxicity. As of November 2016, voclosporin has been shown to effect complete and partial responses in a statistically significant manner in a phase II trial and has been demonstrated to be highly effective for the treatment of lupus nephritis.7,8
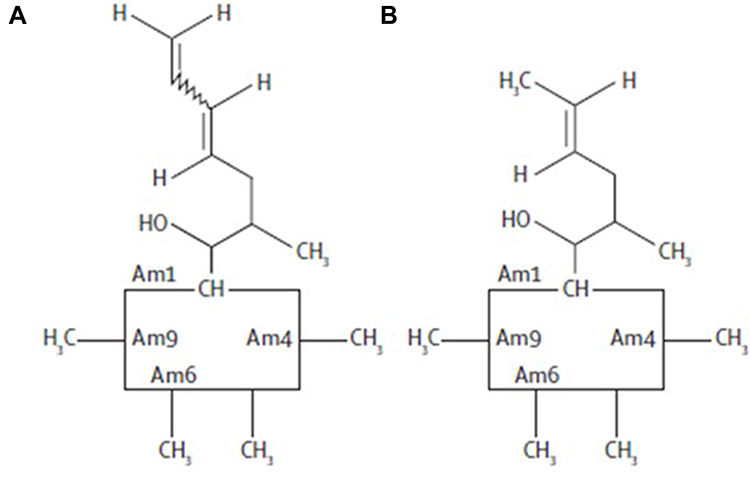 |
Figure 1 Chemical structures of (A) voclosporin and (B) cyclosporine. |
Consistent with its higher potency than cyclosporine, voclosporin has been administered at much lower doses: 20–40 mg once daily compared to 200–600 mg once daily for cyclosporine. Both cyclosporine and voclosporin demonstrated less than dose-proportional non-linear increase in exposure with increasing doses, and the non-linear exposure was more pronounced for voclosporin at low doses of 17.5–28 mg in a typical 70 kg subject compared to cyclosporine at higher dose levels from 300 to 1400 mg.9,10 As such, the aim of this analysis was to determine the difference in pharmacokinetic (PK) disposition of cyclosporine versus voclosporin from their clinical PK profiles following oral administration, and to correlate the distinct dispositions in central and peripheral compartments with their respective efficacy and adverse effect (AE) profiles in clinics.
Methods
Cyclosporine and Voclosporin Plasma Concentration–Time Data
There is a large body of PK data on cyclosporine and voclosporin, in both healthy subjects and patients with diverse immunologic disorders, in the literature. Although the pharmacokinetics in patients would be more relevant, multiple intrinsic and extrinsic factors that influence their calcineurin binding and resultant highly variable PK disposition within narrow but distinct dose ranges of both drugs make it difficult to make a meaningful comparison without individual patient-level PK data. On the other hand, both drugs have been studied extensively in healthy subjects over a wide range of doses, with good-quality reporting of detailed plasma concentration–time profiles. Thus, a consistent and in-depth comparison was possible and conducted herein based on PK data of both cyclosporine and voclosporin obtained from healthy subjects. Plasma cyclosporine and voclosporin concentration–time data in healthy subjects were obtained from six manuscripts. The PK data of voclosporin included the single ascending doses from 0.25 to 4.5 mg/kg,10 multiple doses of 0.4 mg/kg in an organ impairment study and a control cohort in a drug–drug interaction study.11,12 The PK data for cyclosporine included PK studies of single doses of cyclosporine in healthy Caucasians (350, 700 and 1400 mg)9 and drug–drug interaction studies with cyclosporine dosed at 300 and 400 mg.12,13
All of the manuscripts were either obtained in digital pdf format or scanned from paper copies into pdf formatted files. The electronic plasma concentration–time profiles were then digitized using UN-SCAN-IT Graph Digitizing Software version 6.0 (Silk Scientific, Orem, UT, USA) to obtain the plasma concentration and time data.
Non-Compartmental PK Analyses
Plasma concentration–time data for cyclosporine or voclosporin were analyzed using non-compartmental PK methodology with Phoenix WinNonlin version 7.3 (CERTARA, Princeton, NJ, USA). Typical PK parameters of the area under the plasma concentration–time curve (AUC), maximum observed plasma concentration (Cmax), apparent total volume of distribution during the terminal phase (V/F) and apparent total plasma clearance (CL/F) were estimated based on the standard non-parametric methodology in WinNonlin.
Compartmental PK Analyses
Compartmental model PK analyses were performed in NONMEM, version 7.2 (ICON Development Solutions, MD, USA). Concentration data were transformed by natural logarithm (ln) prior to compartmental modeling. The concentration–time profiles were visually inspected for mono-, or bi-exponential decline. Based on visual inspection, the starting point was a one-compartment structural PK model with first-order elimination and distribution (using ADVAN2, TRANS2 routine). Various alternative models were tested to identify the structural model that best described the data. A two-compartment structure PK model (using ADVAN4 and TRANS4 routine) was selected for both compounds based on the objective function value (OFV) using the log-likelihood ratio test and the goodness-of-fit criteria. The final structure PK model was parameterized in terms of:
- absorption lag time in healthy subjects (Tlag)
- first-order absorption rate constant (KA)
- apparent volume of distribution for the central compartment (V2/F)
- apparent volume of distribution for the peripheral compartment (V3/F)
- apparent intercompartmental clearance between the central compartment and the peripheral compartment in healthy subjects (Q/F)
- apparent clearance in healthy subjects (CL/F).
In addition, micro-constants describing the distribution rates between the central and peripheral compartments (k23: the rate constant from the central compartment to the peripheral compartment defined as Q/V2; k32: the rate constant from the peripheral compartment to the central compartment defined as Q/V3) and the elimination rate (kel: the elimination rate constant from the central compartment defined as CL/V2) were calculated and compared between voclosporin and cyclosporine.
Comparison of structural models was based on the OFV and goodness-of-fit criteria. A value of p<0.001, representing a decrease in OFV of >10.83, was considered statistically significant. Selection criteria during the model development process were based on goodness-of-fit plots, including the weighted residuals versus time plot, weighted residuals versus population predicted concentrations plot, population predicted concentration versus observed concentrations plot and individual predicted concentration versus observed concentrations plot, changes in the OFV, residual distributions, and parameter estimates and their relative SE values.
Efficacy and Safety Comparison Between Voclosporin and Cyclosporine
The efficacy and safety data of voclosporin were compiled based on a phase II study in patients with renal transplantation5 and a phase III study in patients with psoriasis.14 The efficacy and safety data of cyclosporine in patients with renal transplantation or psoriasis were extracted from the product label of Sandimmune® and published reviews.15 Of note, the efficacy of voclosporin in kidney transplants cannot be directly compared to cyclosporine owing to a lack of clinical trial data with head-to-head comparison or similar comparator arms. An indirect comparison was made since voclosporin was studied relative to tacrolimus in a head-to-head comparison trial in patients with kidney transplantation in the context of cumulative data on cyclosporine and tacrolimus use in treating patients with kidney transplantation. The efficacy of voclosporin and cyclosporine in psoriasis patients was compared based on the same efficacy endpoint of PASI 75 (the psoriasis area and severity index) at 12 weeks. The safety of voclosporin and cyclosporine was compared based on the adverse events in >3% or 5% in comparable kidney transplant or psoriasis patients receiving either voclosporin or cyclosporine.
Results
Single-Dose Pharmacokinetic Disposition of Voclosporin versus Cyclosporine at Comparable Doses of 4.5 mg/kg and 300–400 mg, Respectively, in Healthy Subjects
The plasma concentration–time profiles of cyclosporine and voclosporin following oral administration of comparable doses in the range of 300–400 mg in healthy subjects are shown in Figure 2. The PK data were subjected to non-compartmental and two-compartmental analyses, and the results are summarized in Table 1.
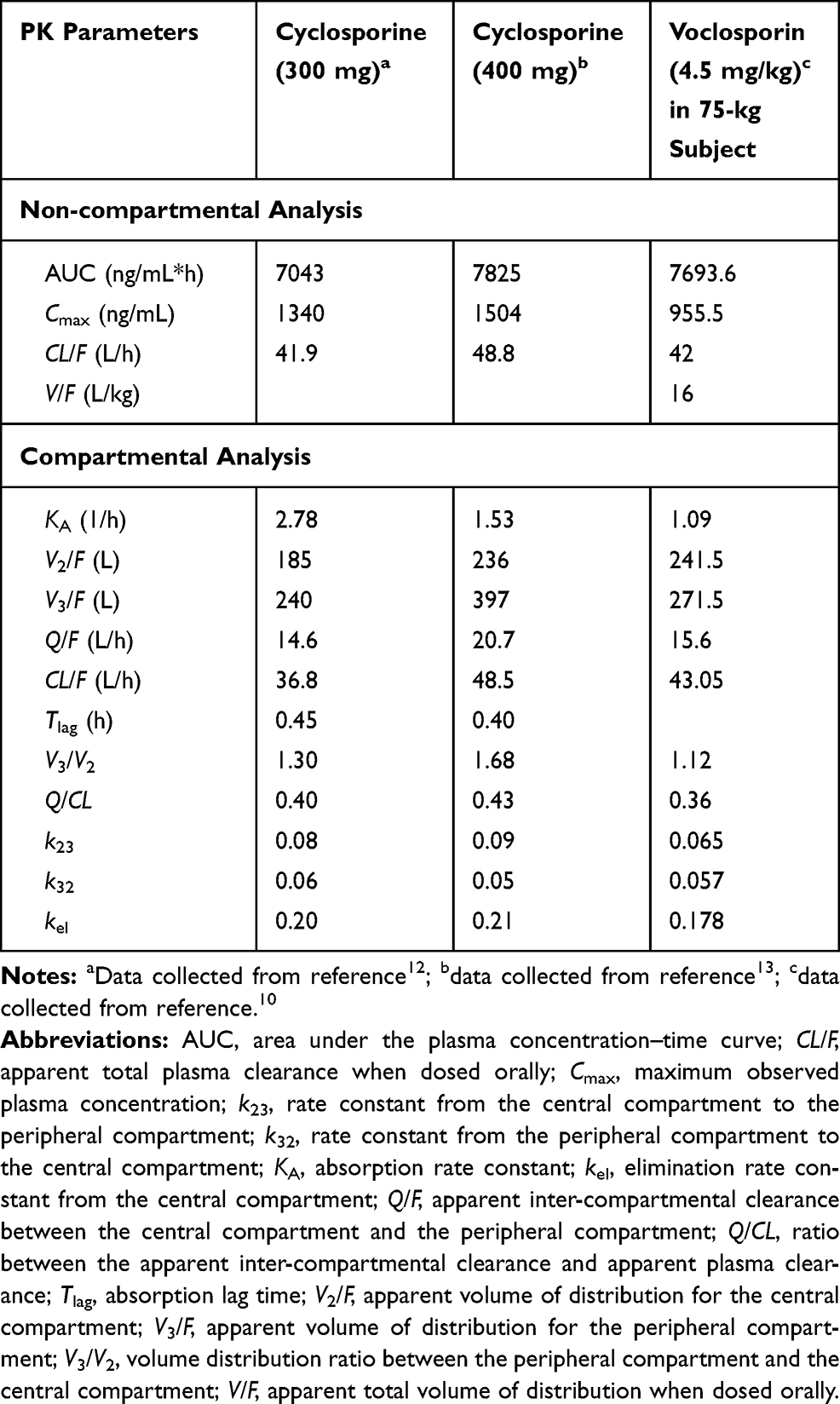 |
Table 1 Pharmacokinetic Parameter Estimates for Cyclosporine and Voclosporin at Comparable Doses (300–400 mg) |
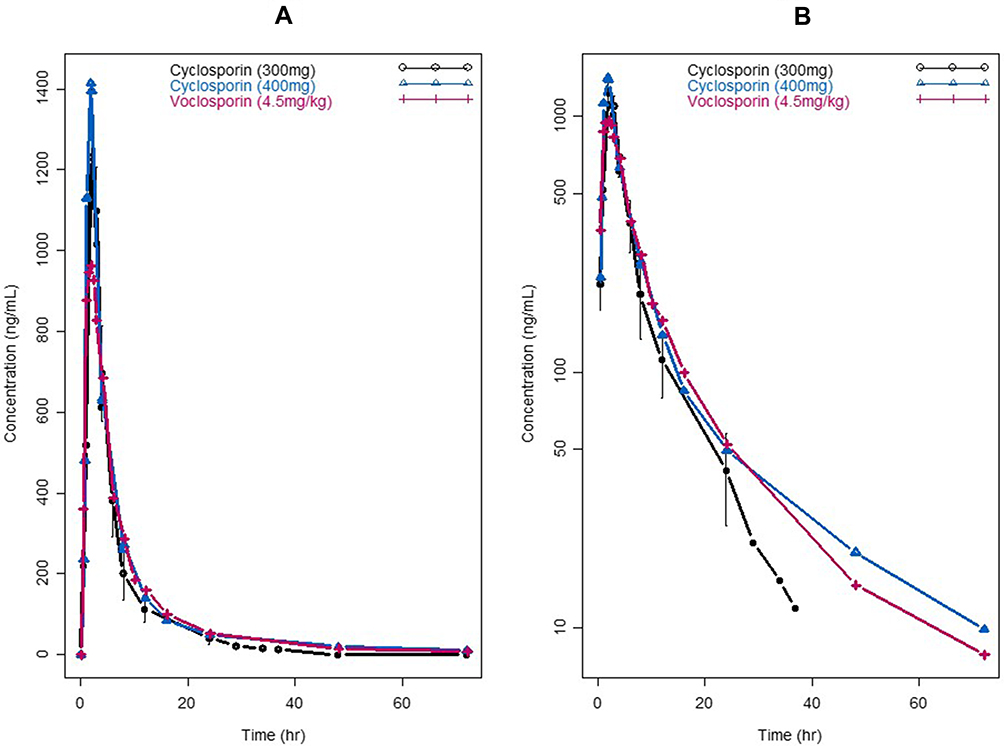 |
Figure 2 Pharmacokinetic profiles of cyclosporine (300 mg and 400 mg) versus voclosporin (4.5 mg/kg). (A) Linear scale; (B) semi-log scale. |
As shown in Figure 2 and Table 1, at similar nominal dose ranges between 300 and 400 mg, voclosporin and cyclosporine had similar plasma exposure profiles and comparable dispositional PK parameters from both non-compartmental and two-compartmental analyses. It was noted that both drugs showed a slightly larger peripheral volume of distribution than central volume of distribution, with a V3/V2 ratio of 1.12–1.68. Both drugs also showed a comparable slower distributional clearance than elimination clearance, with a Q/CL ratio of 0.36–0.43. As a result, both drugs were cleared out of the circulation much more rapidly than they were distributed into peripheral tissues (k23 of 0.065–0.09 h−1 vs kel of 0.178–0.21 h−1). In addition, once bound to targets in peripheral tissue, both drugs showed a slightly faster rate of association/penetration versus disassociation/back from tissues tothe blood circulation (k23 of 0.065–0.09 h−1 vs k32 of 0.05–0.06 h−1). Taking these results together, both drugs showed disposition preference in the central circulating system, instead of distributing into deeper tissues/organs at the nominal dose range of 300–400 mg, which is the top end of doses for voclosporin and low end of doses for cyclosporine.
Dose-Dependent Single-Dose Disposition of Voclosporin versus Cyclosporine
The single-dose plasma concentration–time profiles of voclosporin and cyclosporine by dose level are shown in Figures 3 and 4, respectively. The PK parameters of voclosporin and cyclosporine by dose level from non-compartmental and two-compartmental analyses were summarized in Table 2.
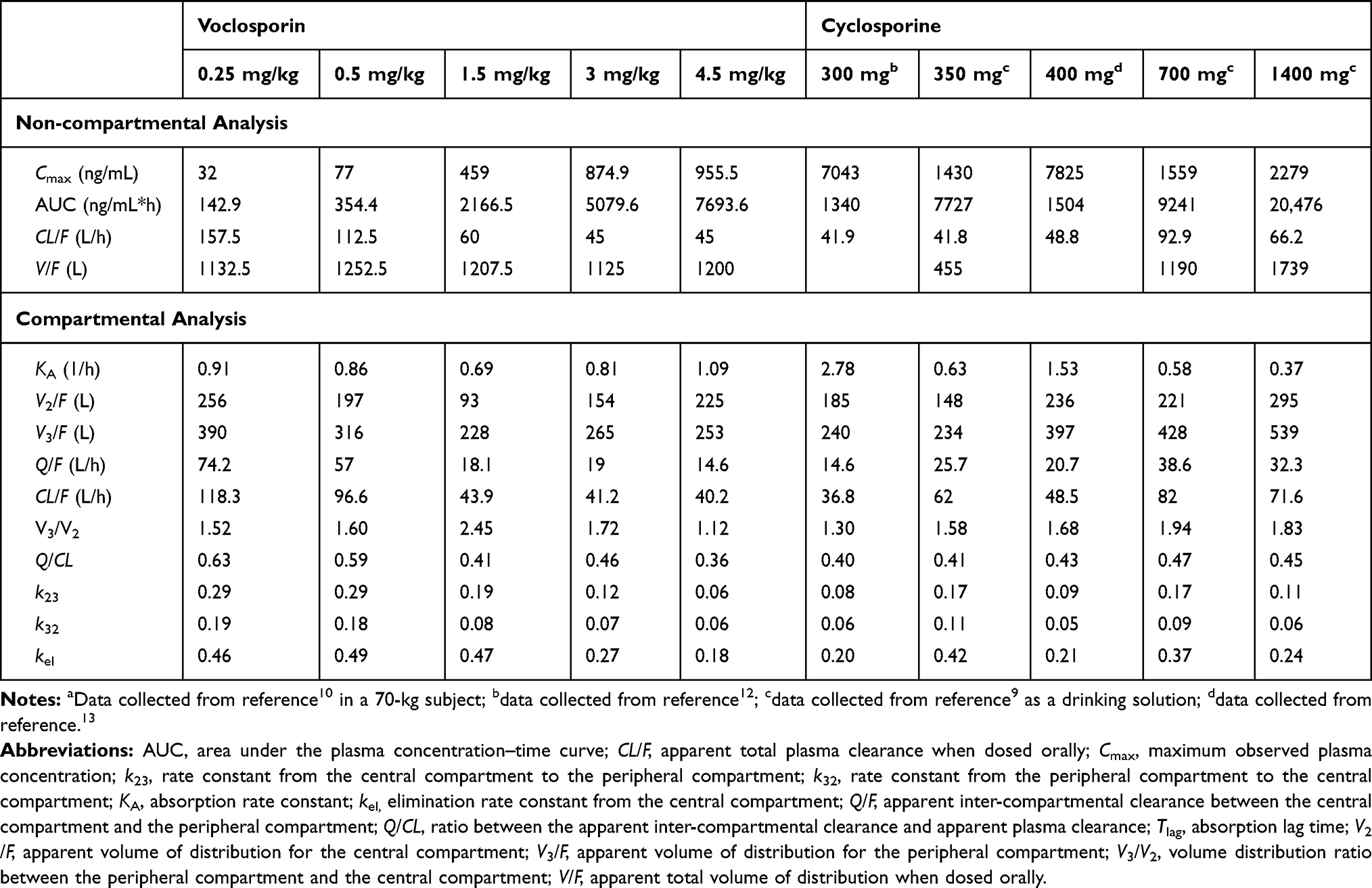 |
Table 2 Pharmacokinetic Parameters of Single Doses of Voclosporin and Cyclosporine |
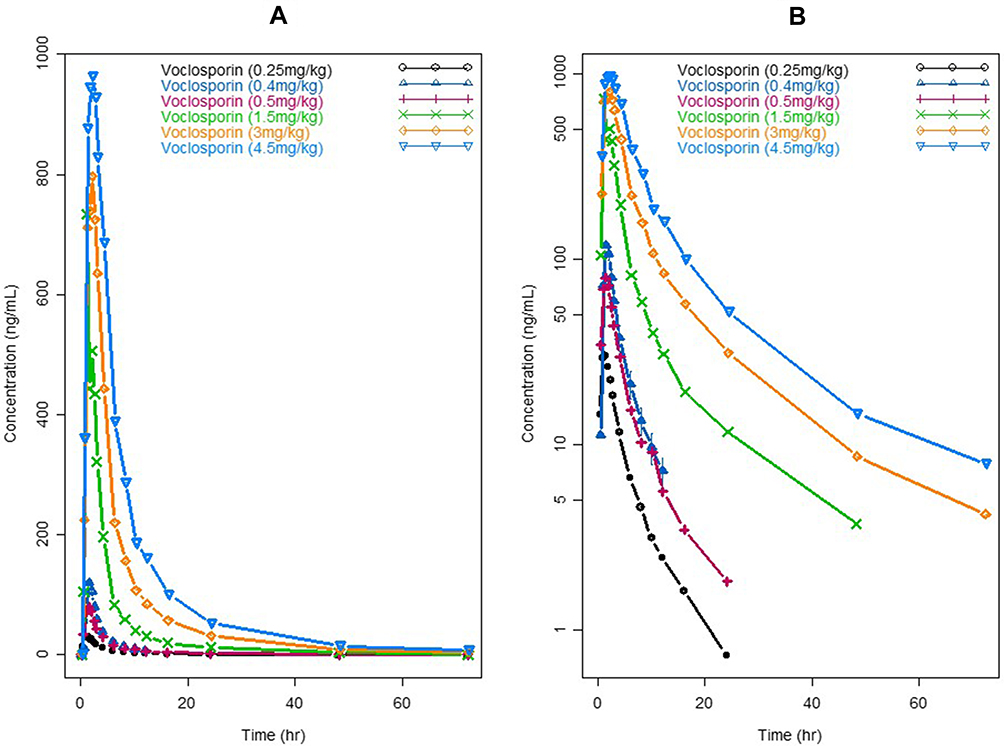 |
Figure 3 Pharmacokinetic profiles of single doses of voclosporin. (A) Linear scale; (B) semi-log scale. |
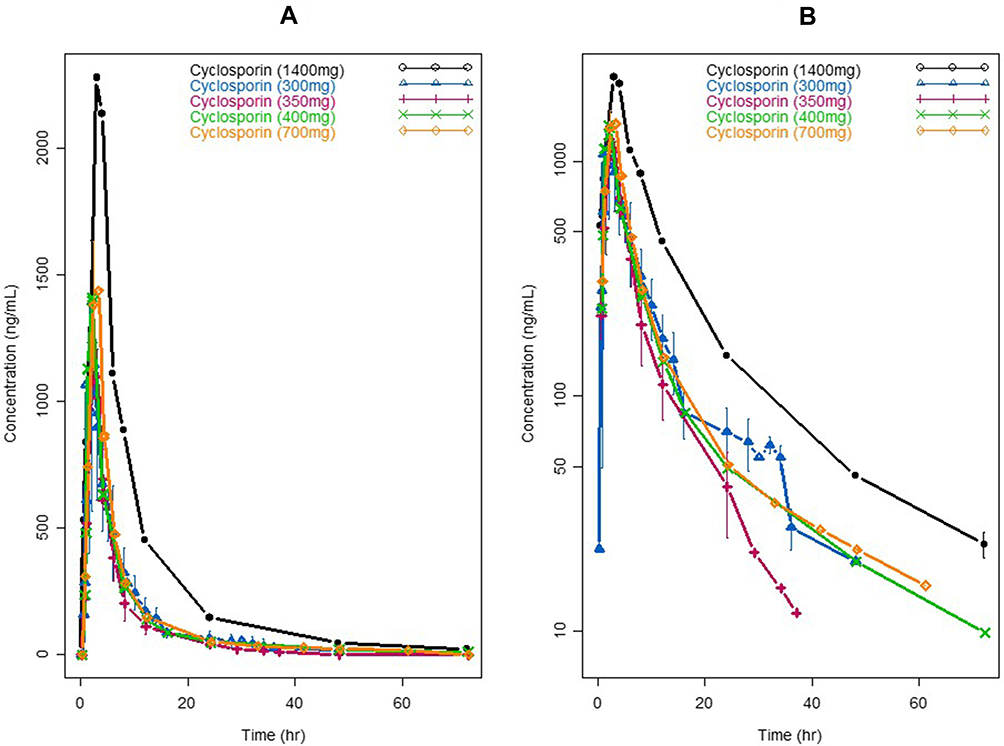 |
Figure 4 Pharmacokinetic profiles of single doses of cyclosporine. (A) Linear scale; (B) semi-log scale. |
As shown in Figures 3 and 4 and Table 2, voclosporin demonstrated strong non-linearity in its PK disposition from 0.25 to 4.5 mg/kg. While the V/F from the non-compartmental analysis, and the rate of absorption (ka), central and peripheral volumes (V2/F and V3/F) from compartmental analyses were generally comparable from low to high doses, there were a strong trend towards decreasing clearance (both distribution and elimination) with increasing doses. The distribution clearance decreased from approximately 1 L/h/kg (74.2 L/h) to 0.2 L/h/kg (14.6 L/h) while elimination clearance decreased from approximately 1.7 L/h/kg (118.3 L/h) to 0.6 L/h/kg (40.2 L/h), with the resultant Q/CL ratio decreasing from 0.63 to 0.36. The decreasing distribution clearance with increasing dose suggested target-mediated disposition with a saturation of target binding for voclosporin. In contrast, the degree of non-linearity in cyclosporine disposition from 300 mg to 1400 mg was much less and mild (Figure 4 and Table 2). Although the distribution and elimination clearance of cyclosporine appeared to increase with dose (Table 2), the increased clearance at doses above 300 mg was caused by the introduction of a new formulation. Note that the apparent clearance and volume of distribution were approximately 30–40% larger based on studies using the older cyclosporine formulation than those obtained based on studies using the new cyclosporine formulation, consistent with the improvement of around 40% in oral bioavailability by the new formulation, which has been widely adopted since its introduction. The formulation-adjusted distribution and elimination clearance (multiplication of apparent clearance of Q/F or CL/F by relative bioavailability of 40%) also appeared to decrease with increasing doses, but the changes in the overall rates of distribution and elimination were not as significant as those of voclosporin.
Repeated-Dose Disposition of Voclosporin and Cyclosporine
The repeated-dose plasma concentration–time profiles of voclosporin are shown in Figure 5. The PK parameters of voclosporin by non-compartmental and two-compartmental analyses are presented in Table 3.
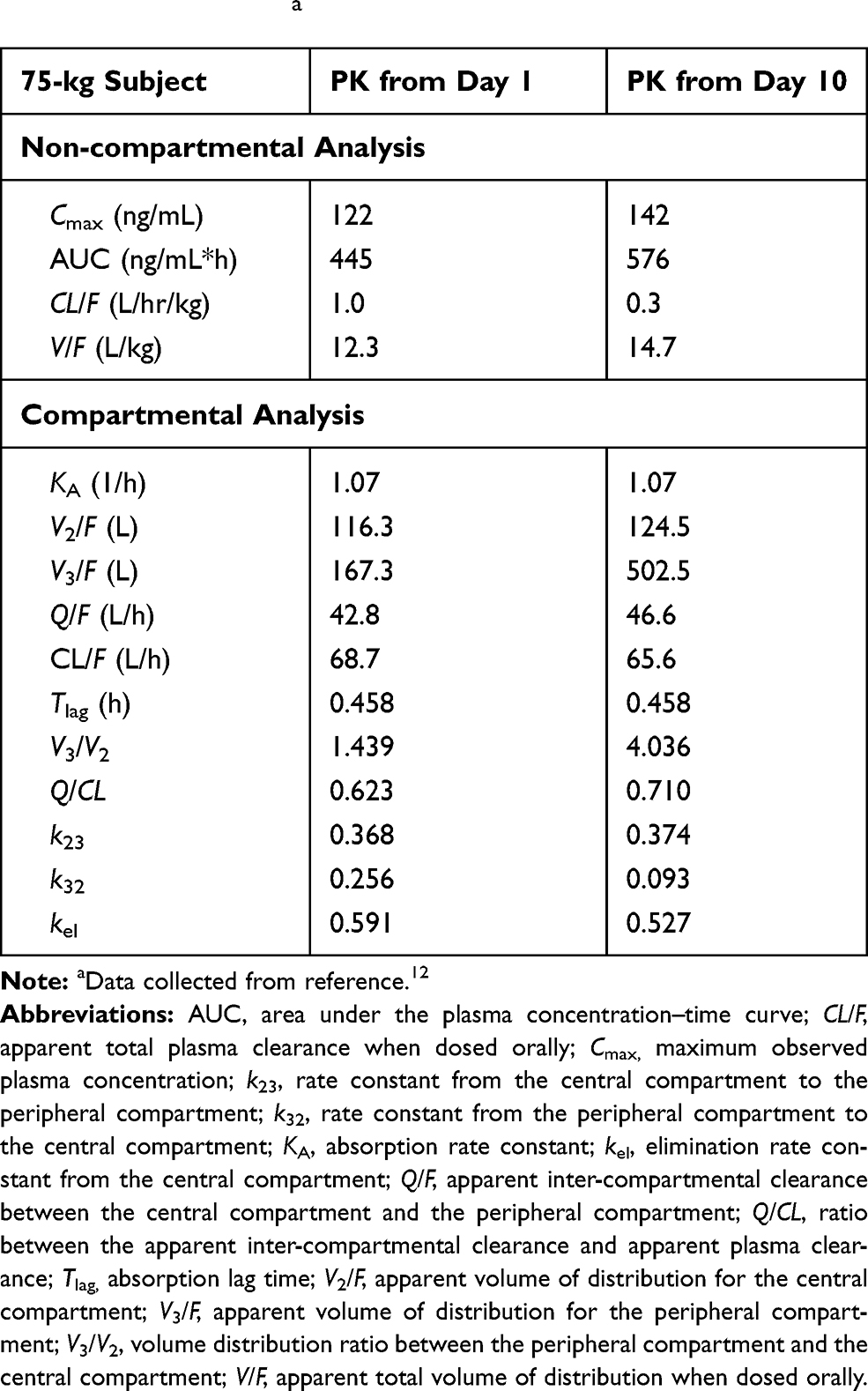 |
Table 3 Pharmacokinetic (PK) Parameters of Single and Multiple Doses of Voclosporina |
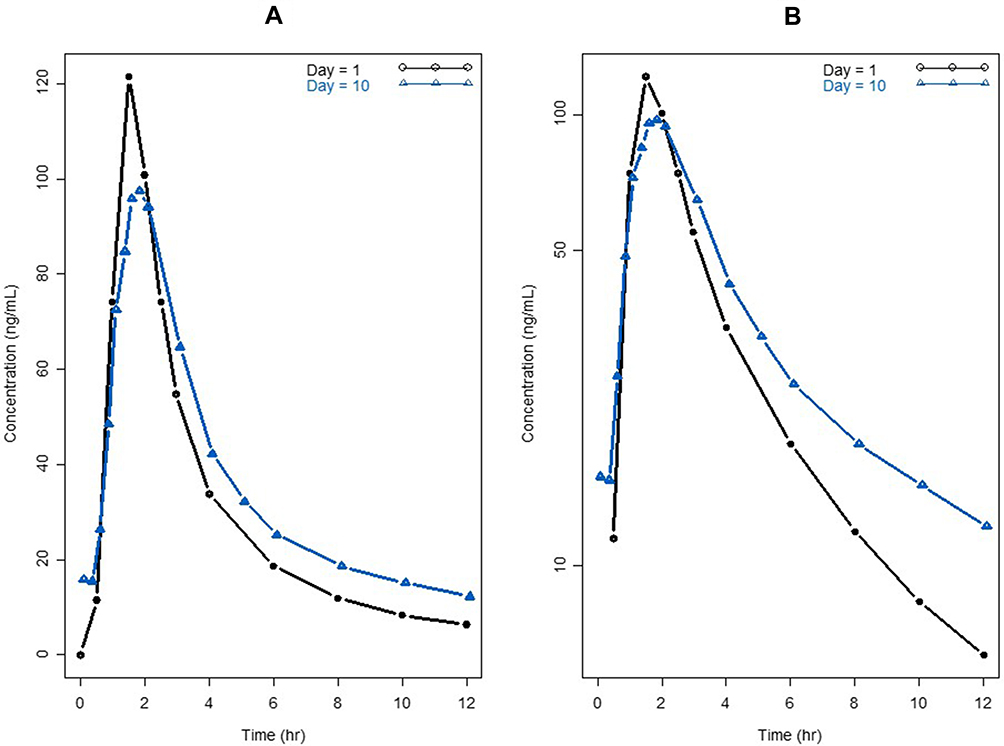 |
Figure 5 Pharmacokinetic profiles of single and multiple doses of voclosporin. (A) Linear scale; (B) semi-log scale. |
Upon repeated dosing, the peripheral volume of distribution for voclosporin increased from 167 L on day 1 to 502 L on day 10, while there was a little or no change in the central volume of distribution and elimination clearance. The resultant ratio of V3/V2 increased from 1.4 on day 1 to 4.0 at steadystate. The increase in peripheral volume of distribution suggested either a stronger peripheral binding of voclosporin and/or deeper tissue penetration of voclosporin with repeated dosing. While a similar phenomenon may happen with repeated dosing of cyclosporine, a thorough literature search did not yield any repeated-dose study of cyclosporine in healthy subjects. However, cyclosporine PK disposition has been shown to change upon repeated dosing in patients with stem cell transplant and liver transplant, consistent with either or a combination of a change in hematocrit, calcineurin expression associated with specific immunologic disorder and deeper penetration of cyclosporine with repeated dosing.16,18 The last one of these may be the reason for delayed AEs observed in patients who received long-term cyclosporine treatment.
Efficacy and Safety Comparison Between Voclosporin and Cyclosporine
A renal transplantation study of voclosporin was conducted in a phase IIb trial, PROMISE,5 a 6-month, multicenter, randomized, open-label study of three ascending concentration-controlled groups of voclosporin (low, medium and high trough concentrations of voclosporin) compared to tacrolimus (TAC) in 334 low-risk renal transplant recipients. Voclosporin was shown to be non-inferior to tacrolimus on the primary endpoint of biopsy-proven acute rejection rates (10.7%, 9.1% and 2.3%, for low, medium and high trough concentration groups of voclosporin, respectively, vs 5.8% for TAC) and on the secondary endpoint of renal function and new-onset diabetes after transplantation (1.6%, 5.7% and 17.7%, for low, medium and high trough concentration groups of voclosporin, respectively, vs 16.4% for TAC). Given that both tacrolimus and cyclosporine have been widely used in patients with kidney transplant and were shown to be similarly effective in preventing organ rejection,19 the non-inferiority of voclosporin relative to tacrolimus may reasonably be extrapolated to suggest non-inferiority of voclosporin relative to cyclosporine in the kidney transplant patients.
The efficacy of voclosporin was studied in a phase III trial with 451 patients aged 18–65 years with plaque psoriasis involving at least 10% of the body surface area.14 The psoriasis patients were randomly assigned in equal proportions to receive placebo or voclosporin at 0.2, 0.3 or 0.4 mg/kg orally twice a day in dermatology clinics. The primary endpoint was a 75% reduction in the psoriasis area and severity index (PASI 75) score at week 12. At week 12, PASI 75 scores were achieved by 16%, 25% and 47% of patients in the voclosporin 0.2, 0.3 and 0.4 mg/kg groups, respectively, versus 4% of patients in the placebo group. By contrast, cyclosporine showed a much higher response rate in psoriasis patients, ie, at the standard doses of 2.5–3 mg/kg up to 5 mg/kg daily for 12 weeks, cyclosporine achieved PASI 75 scores of 58–89.4%.15
The types and frequency of AEs of voclosporin and cyclosporine in patients with renal transplants or psoriasis by body system are compared in Tables 4 and 5, respectively. As shown in Table 4, voclosporin had lower central nervous system (CNS), gastrointestinal and hematologic AEs, comparable to the active control group of tacrolimus and similar to those respective low AEs of cyclosporine in patients with kidney transplant. In addition, voclosporin had no cardiovascular AEs while up to 26% of transplant patients treated with cyclosporine had hypertension. Cumulative clinical experience of cyclosporine and tacrolimus use in patients with kidney transplants showed that the cholesterol and triglyceride levels in patients treated with cyclosporine were significantly higher than in the tacrolimus group, and the side effects of gingival hyperplasia and hirsutism were higher in cyclosporine use, while no such AEs were observed in voclosporin treatment.19 Given that voclosporin was demonstrated to be non-inferior to tacrolimus in overall safety in the PROMISE trial, the results further supported the between-trial safety comparison that voclosporin had fewer cardiovascular and hirsutism AEs than cyclosporine in kidney transplant patients.
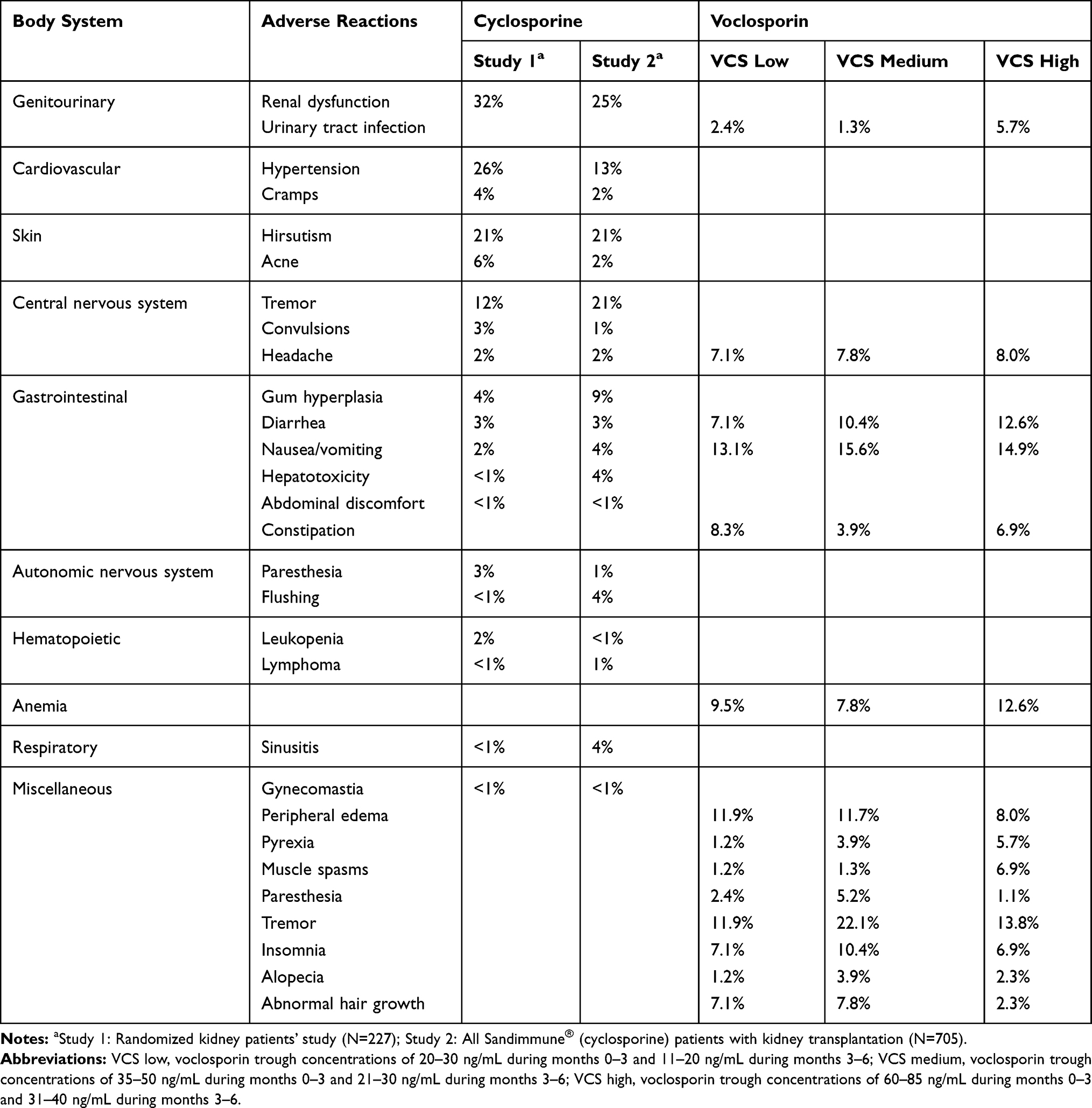 |
Table 4 Types and Frequency of Adverse Events of Voclosporin and Cyclosporine in Patients with Renal Transplants, by Body System |
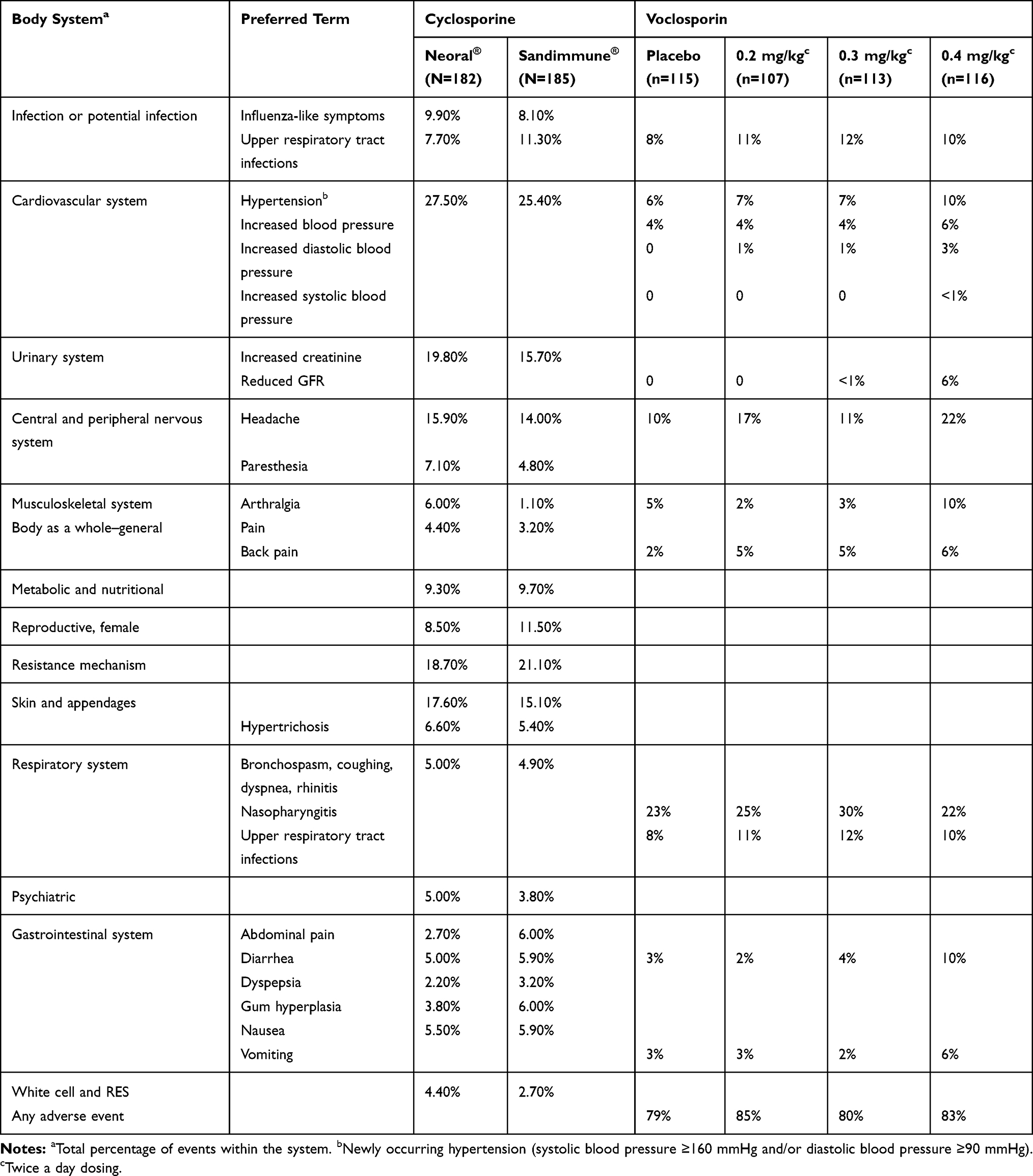 |
Table 5 Types and Frequency of Adverse Events of Voclosporin and Cyclosporine in Patients with Psoriasis, by Body System |
In patients with psoriasis, voclosporin had lower rates of hypertension of 7–10%, comparable to placebo, while cyclosporine had significantly higher rates of hypertension at 25–28% (Table 5). In addition, voclosporin had low CNS, gastrointestinal and hematologic AEs, comparable to placebo, in patients with psoriasis. By contrast, cyclosporine appeared to have higher CNS and gastrointestinal AEs in psoriasis patients.
Discussion
Cyclosporine, the first calcineurin inhibitor, has been widely used in diverse populations of organ transplantation and an array of immunologic disorders. The PK disposition of cyclosporine has been shown to be highly variable among subjects,20 and significantly affected by extrinsic factors of dose and formulation, as well as intrinsic factors of patient population, such as hematocrit, CYP3A activities and baseline disease characteristics, likely associated with altered and elevated calcineurin expression under pathophysiologic conditions.21 Likewise, voclosporin, the second-generation calcineurin inhibitor, also demonstrated highly variable dispositional characteristics in a diverse population of transplantation, uveitis and psoriasis patients affected by similar extrinsic and intrinsic factors to those driving cyclosporine disposition. These vast confounding intrinsic and extrinsic factors made it very difficult to compare the disposition of voclosporin with cyclosporine in distinct target disease populations, except for the healthy normal subjects employed during the exploratory development of both drugs. In fact, the PK dispositional characteristics of voclosporin and cyclosporine in healthy subjects were consistent and reproducible among studies by different investigators conducted for a variety of objectives on different occasions (Tables 1–3). The close examination of PK dispositions of the two drugs in healthy subjects yielded profound insights into their similar but distinct pharmacology, together with their different pharmacologic activities in vitro, providing a good account of the “controversial” and “conflicting” clinical efficacy and adverse effect findings in clinical trials and the anecdotal use of both drugs in diverse populations.
While both drugs had similar chemical structure and identical mechanism of action, voclosporin was much more potent than cyclosporine, and was shown to effectively inhibit calcineurin in ex vivo blood assays with at least four-fold lower concentrations.3 For diseases with calcineurin targets close to blood and within reach by the immediate blood circulation, a direct translation of in vitro higher calcineurin inhibition potency resulting in a dose reduction for voclosporin would be expected. As such, voclosporin was demonstrated in a controlled clinical trial to be non-inferior to tacrolimus in preventing rejection of kidney transplants at much lower trough concentrations of 20–40 ng/mL compared to a trough concentration of 85 ng/mL tacrolimus, which showed comparable in vitro potency, or an equivalent of trough concentration of around 360 ng/mL cyclosporine in similar de novo renal transplant patients. The more potent calcineurin inhibition by voclosporin in blood was also confirmed by pharmacokinetic/pharmacodynamic (PK/PD) analyses.22
Because of its lower potency, the typical clinical doses of cyclosporine are from approximately180 mg in de novo patients with rheumatoid arthritis to 600 mg twice daily in patients with de novo renal transplants, given in two equally divided doses. In comparison, the typical doses of voclosporin were 0.2–0.4 mg/kg (or 14–28 mg in a 70-kg subject) in psoriasis,14 uveitis23 and lupus nephritis.7,23,24 Although both drugs showed disposition preference in the central circulating system, instead of distributing into deeper tissues and organs, the more than 10-fold difference in the daily doses between cyclosporine and voclosporin generated more than 10-fold difference in plasma drug concentrations. By mass balance, the 10-fold higher absolute molar concentration of cyclosporine was expected to exert a 10-fold stronger concentration gradient, driving cyclosporine to more organs and deeper into the tissues of these organs. Thus, it is not a surprise that cyclosporine generally has larger numbers of and sometimes more severe adverse effects than voclosporin. In the past 20 years, voclosporin has been administered to over 2000 patients with psoriasis, uveitis, renal transplantation and lupus nephritis, and proven to be safe and better tolerated than cyclosporine and similar to tacrolimus. No significant changes in mean systolic or diastolic blood pressure were reported in voclosporin-treated patients,5,6,14,23 while hypertension was a common adverse effect reported in 13% of renal transplant patients. The overall AE profiles in patients with uveitis were comparable to placebo.6,23 In the de novo renal transplantation patients,5 voclosporin appeared to offer benefit over tacrolimus and the incidence of new-onset diabetes after transplantation was significantly lower at 6 months post-transplantation in the low-voclosporin treatment group compared to the tacrolimus treatment group (1.6% vs 16.4%, respectively, p=0.031), and numerically lower with the mid-group (5.7%). Hyperlipidemia has been a major concern with the use of calcineurin inhibitors in psoriasis. In the psoriasis phase III study, no differences in mean lipid concentrations were noted between the voclosporin-treated and placebo-treated patients. By contrast, in a meta-analysis of cyclosporine treatment for psoriasis, 2% and 18% of patients given 1.25 mg/kg and 5.0 mg/kg, respectively, had a 10% increase in total cholesterol concentrations. In addition, 4% and 26% of patients given cyclosporine at 1.25 mg/kg and 5.0 mg/kg, respectively, had a 30% increase in concentrations of triglycerides.14 Most notably, at the 0.2–0.4 mg/kg dose ranges of voclosporin, no overt renal adverse effects were observed in over 2000 patients with diverse immunologic disorders. Also consistent with the low dose and lower concentration gradient, voclosporin would be expected to poorly penetrate the epidermis of skin and thus be less efficacious in treating psoriasis, an immunologic disorder occurring primarily in skin. In fact, voclosporin was shown to be statistically inferior to cyclosporine in controlled trials, with 35% of patients compared to 53% of patients in the cyclosporine control arm reaching a “clear” or “almost clear” in SPGA on week 12.2,4 Furthermore, both the percentage and higher degree of disease control improved with continual dosing beyond 12 weeks for both cyclosporine and voclosporin, in agreement with more complete calcineurin engagement with more drugs in the skin.
Although cyclosporine and voclosporin may even share similar target-mediated disposition at the same nominal doses, as demonstrated by the comparable PK parameters at common doses of 300–400 mg (Table 1 and Figure 2), the high potency and low doses of voclosporin in clinical settings yielded distinct, more pronounced non-linear disposition, pointing to engagement of calcineurin targets in tissues different from cyclosporine, with important pharmacologic manifestations. The higher distribution clearance of 1 L/h/kg at a lower voclosporin dose of 0.25 mg/kg compared to 0.2 L/h/kg at the higher dose level of 4.5 mg/kg suggested preferential engagement of certain calcineurin of high affinity and/or saturation of these calcineurin targets of high affinity. Furthermore, repeated dosing at the low doses of 0.4 mg/kg increased the peripheral volume of distribution from 167 to 500 L, pointing to a stronger association of voclosporin to certain calcineurin targets in deeper or hard-to-reach tissues/organs by the immediate blood circulation, most likely different from those that were close to the blood circulation and engaged following immediate dosing. These strong non-linear disposition behaviors associated with target-mediated dispositions at low doses of voclosporin conferred unique benefit over cyclosporine in sparing tissues from untoward toxicity. The absence of renal damage by voclosporin at the low doses of 0.2–0.4 mg/kg in the large and diverse populations of psoriasis, uveitis and renal transplantation offer a unique window for its use in treating lupus nephritis. While calcineurin inhibition by cyclosporine has been shown to modulate the excessive immune responses that cause organ injury, ie, lupus nephritis, it has proven difficult to tip the delicate balance with a less potent inhibitor that requires a much larger amount of mass to easily cross the threshold to cause harm in renal and other cells. The fact that the notorious and irreversible renal damage by cyclosporine is also positively correlated with the higher doses and longer duration of cyclosporine use attests to the delicate balance of systemic immune modulation versus local tissue exposure and untoward effects and toxicity. In comparison, the high potency and preferential engagement of calcineurin targets in and near the central circulation makes it a good option to treat moderate systemic immune responses without causing direct organ damage, eg, renal toxicity. To this end, the positive phase II results with voclosporin in treating lupus nephritis are a good prognostic signal for its well-balanced pharmacology for the treatment of immunologic disorders with strong central and systemic components while sparing the notorious renal toxicity of cyclosporine. During the preparation of the manuscript, voclosporin has been demonstrated to be highly effective in treating lupus nephritis in phase III clinical studies; a positive benefit–risk profile was observed in the AURORA trial, confirming the treatment effect seen in the AURA-LV trial when comparing voclosporin 23.7 mg twice daily in combination with background standard of care versus standard of care alone.8
In conclusion, the 10-fold difference in doses and non-linear target-mediated disposition between voclosporin and cyclosporinetogether with their 10-fold difference in potency of calcineurin inhibition, is the key to unlocking the enigmatic similarities and differences of the two drugs in past clinical experience. The proper analyses of drug disposition of voclosporin versus cyclosporine offered profound insights into their differentiated pharmacology and provided sound clinical pharmacologic and therapeutic rationales for their differentiated use to treat distinct immunologic disorders with varied involvement of systemic versus local components of calcineurin expression in different tissues.
Disclosure
Yan Li, Maria Palmisano and Simon Zhou are employees of Bristol Myers Squibb (heritage Celgene) Corporation. The authors report no other possible conflicts of interest with this research work.
References
1. Azzi JR, Sayegh MH, Mallat SG. Calcineurin inhibitors: 40 years later, can’t live without. J Immunol. 2013;191(12):5785–5791.
2. Naidoo P, Rambiritch V. Voclosporin (ISA247) for plaque psoriasis. Lancet. 2008;372(9642):888–889. doi:10.1016/S0140-6736(08)61391-4
3. Birsan T, Dambrin C, Freitag DG, Yatscoff RW, Morris RE. The novel calcineurin inhibitor ISA247: a more potent immunosuppressant than cyclosporine in vitro. Transpl Int. 2005;17(12):767–771. doi:10.1111/j.1432-2277.2004.tb00509.x
4. Davis J Voclosporin’s mixed results in Phase III psoriasis trial. 2009. Available from: https://scrippharmaintelligenceinformacom/SC001881/Voclosporins-mixed-results-in-Phase-III-psoriasis-trial.
5. Busque S, Cantarovich M, Mulgaonkar S, et al. The PROMISE study: a phase 2b multicenter study of voclosporin (ISA247) versus tacrolimus in de novo kidney transplantation. Am J Transplant. 2011;11(12):2675–2684. doi:10.1111/j.1600-6143.2011.03763.x
6. EMA. Voclosporin for the treatment of non-infectious uveitis. 2013. Available from: http://wwwemaeuropaeu/docs/en_GB/document_library/Orphan_designation/2013/01/WC500137776pdf.
7. Wire B Aurinia Announces That Voclosporin Achieves Primary and All Pre-Specified Secondary Endpoints in Its Phase IIb AURA-LV Study for Lupus Nephritis (LN). 2016. Available from: http://wwwbusinesswirecom/news/home/20160929006376/en/Aurinia-Announces-Voclosporin-Achieves-Primary-Pre-Specified-Secondary.
8. Pharmaceuticals A. Aurinia Announces Positive AURORA Phase 3 Trial Results Demonstrating Voclosporin Superiority Over Standard of Care in Lupus Nephritis. 2019.
9. Reymond JP, Steimer JL, Niederberger W. On the dose dependency of cyclosporin A absorption and disposition in healthy volunteers. J Pharmacokinet Biopharm. 1988;16(4):331–353. doi:10.1007/BF01062550
10. Mayo PR, Huizinga RB, Ling SY, Freitag DG, Aspeslet LJ, Foster RT. Voclosporin food effect and single oral ascending dose pharmacokinetic and pharmacodynamic studies in healthy human subjects. J Clin Pharmacol. 2013;53(8):819–826. doi:10.1002/jcph.114
11. Ling SY, Huizinga RB, Mayo PR, Freitag DG, Aspeslet LJ, Foster RT. Pharmacokinetics of voclosporin in renal impairment and hepatic impairment. J Clin Pharmacol. 2013;53(12):1303–1312. doi:10.1002/jcph.166
12. Groll AH, Desai A, Han D, et al. Pharmacokinetic assessment of drug-drug interactions of isavuconazole with the immunosuppressants cyclosporine, mycophenolic acid, prednisolone, sirolimus, and tacrolimus in healthy adults. Clin Pharmacol Drug Dev. 2017;6(1):76–85. doi:10.1002/cpdd.284
13. Bifano M, Adamczyk R, Hwang C, Kandoussi H, Marion A, Bertz RJ. An open-label investigation into drug-drug interactions between multiple doses of daclatasvir and single-dose cyclosporine or tacrolimus in healthy subjects. Clin Drug Investig. 2015;35(5):281–289. doi:10.1007/s40261-015-0279-5
14. Papp K, Bissonnette R, Rosoph L, et al. Efficacy of ISA247 in plaque psoriasis: a randomised, multicentre, double-blind, placebo-controlled phase III study. Lancet. 2008;371(9621):1337–1342. doi:10.1016/S0140-6736(08)60593-0
15. Colombo MD, Cassano N, Bellia G, Vena GA. Cyclosporine regimens in plaque psoriasis: an overview with special emphasis on dose, duration, and old and new treatment approaches. ScientificWorldJournal. 2013;2013:805705. doi:10.1155/2013/805705
16. Burckart GJ, Venkataramanan R, Ptachcinski RJ, et al. Cyclosporine absorption following orthotopic liver transplantation. J Clin Pharmacol. 1986;26(8):647–651. doi:10.1002/j.1552-4604.1986.tb02966.x
17. Frassetto L, Baluom M, Jacobsen W, et al. Cyclosporine pharmacokinetics and dosing modifications in human immunodeficiency virus-infected liver and kidney transplant recipients. Transplantation. 2005;80(1):13–17. doi:10.1097/01.TP.0000165111.09687.4E
18. Jacobson PA, Ng J, Green KGE, Rogosheske J, Brundage R. Posttransplant day significantly influences pharmacokinetics of cyclosporine after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2003;9(5):304–311. doi:10.1016/S1083-8791(03)00076-4
19. Ekmekcioglu O, Turkan S, Yildiz S, Gunes ZE. Comparison of tacrolimus with a cyclosporine microemulsion for immunosuppressive therapy in kidney transplantation. Turk J Urol. 2013;39(1):16–21. doi:10.5152/tud.2013.004
20. Ptachcinski RJ, Venkataramanan R, Burckart GJ. Clinical pharmacokinetics of cyclosporin. Clin Pharmacokinet. 1986;11(2):107–132. doi:10.2165/00003088-198611020-00002
21. Tod M, Nkoud-Mongo C, Gueyffier F. Impact of genetic polymorphism on drug-drug interactions mediated by cytochromes: a general approach. AAPS J. 2013;15(4):1242–1252. doi:10.1208/s12248-013-9530-2
22. Anglade E, Aspeslet LJ, Weiss SL. A new agent for the treatment of noninfectious uveitis: rationale and design of three LUMINATE (Lux Uveitis Multicenter Investigation of a new approach to treatment) trials of steroid-sparing voclosporin. Clin Ophthalmol. 2008;2(4):693–702. doi:10.2147/OPTH.S2452
23. EMA. Assessment report (Luveniq). 2011. Available from: http://wwwemaeuropaeu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2012/02/WC500122082pdf.
24. Worcester S. AURA-LV study: rapid remission with voclosporin for lupus nephritis. 2016. Available from: http://wwwmdedgecom/rheumatologynews/article/118557/lupus-connective-tissue-diseases/aura-lv-study-rapid-remission.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.