Back to Journals » International Journal of Nanomedicine » Volume 13
pH-responsive charge-reversal polymer-functionalized boron nitride nanospheres for intracellular doxorubicin delivery
Authors Feng SN, Zhang HJ ![]() , Zhi CY, Gao XD
, Zhi CY, Gao XD ![]() , Nakanishi H
, Nakanishi H
Received 7 October 2017
Accepted for publication 7 December 2017
Published 31 January 2018 Volume 2018:13 Pages 641—652
DOI https://doi.org/10.2147/IJN.S153476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Shini Feng,1 Huijie Zhang,2 Chunyi Zhi,3 Xiao-Dong Gao,1 Hideki Nakanishi1
1Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi, People’s Republic of China; 2School of Pharmaceutical Sciences, Jiangnan University, Wuxi, People’s Republic of China; 3Department of Physics and Materials Science, City University of Hong Kong, Kowloon, Hong Kong SAR, People’s Republic of China
Background: Anticancer drug-delivery systems (DDSs) capable of responding to the physiological stimuli and efficiently releasing drugs inside tumor cells are highly desirable for effective cancer therapy. Herein, pH-responsive, charge-reversal poly(allylamine hydrochlorid)-citraconic anhydride (PAH-cit) functionalized boron nitride nanospheres (BNNS) were fabricated and used as a carrier for the delivery and controlled release of doxorubicin (DOX) into cancer cells.
Methods: BNNS was synthesized through a chemical vapor deposition method and then functionalized with synthesized charge-reversal PAH-cit polymer. DOX@PAH-cit–BNNS complexes were prepared via step-by-step electrostatic interactions and were fully characterized. The cellular uptake of DOX@PAH-cit–BNNS complexes and DOX release inside cancer cells were visualized by confocal laser scanning microscopy. The in vitro anticancer activity of DOX@PAH-cit–BNNS was examined using CCK-8 and live/dead viability/cytotoxicity assay.
Results: The PAH-cit–BNNS complexes were nontoxic to normal and cancer cells up to a concentration of 100 µg/mL. DOX was loaded on PAH-cit–BNNS complexes with high efficiency. In a neutral environment, the DOX@PAH-cit–BNNS was stable, whereas the loaded DOX was effectively released from these complexes at low pH condition due to amide hydrolysis of PAH-cit. Enhanced cellular uptake of DOX@PAH-cit–BNNS complexes and DOX release in the nucleus of cancer cells were revealed by confocal microscopy. Additionally, the effective delivery and release of DOX into the nucleus of cancer cells led to high therapeutic efficiency.
Conclusion: Our findings indicated that the newly developed PAH-cit–BNNS complexes are promising as an efficient pH-responsive DDS for cancer therapy.
Keywords: boron nitride nanospheres, drug delivery, doxorubicin, pH-responsive charge-reversal, controlled release
Introduction
Drug-delivery systems (DDSs) enhance drug accumulation in cancer cells through enhanced permeability and retention effects while reducing side effects on normal cells.1 Thus, they are a good approach to enhance the therapeutic efficiency of traditional anticancer drugs.2 Recently, various nanomaterial-based DDSs have been explored to improve the efficacy of cancer therapy.3 Among these nanomaterials, boron nitride (BN) has recently attracted wide attention.4–12 As structural analogs of carbon materials, BN exhibits high chemical stability and better biocompatibility than carbon materials.13–16 Therefore, it is considered promising for biomedical applications, such as drug delivery.17–23 Recently, applications of BN nanospheres (BNNS) as carriers for DNA or chemotherapy drugs have only begun to emerge.24–28 Moreover, BNNS are composed of a substantial number of boron atoms, which can serve as boron carriers for effective boron neutron capture therapy (BNCT) for cancer therapy and diagnosis.29–32 The exploration of BNNS as anticancer DDSs is a promising direction, which could provide an integrated system by combining chemotherapy with radiation therapy.33,34
As a commonly used chemotherapeutic agent, doxorubicin (DOX) can intercalate with DNA, break the DNA chain for replication, and then prevent the cell division process.35 Consequently, it only works in the nucleus of cancer cells with sufficient concentration. However, most DDSs suffer from limitations as they can neither migrate into the nucleus nor effectively release drugs in the cytoplasm.35 After delivery into cancer cells, only a small percentage of drugs can be released and eventually reach the nucleus. Thus, an ideal carrier for cancer chemotherapy should not only possess long circulation life, efficient accumulation at the tumor site, and highly efficient tumor cellular uptake but also effectively release the anticancer drug inside tumor cells in a controlled manner.36 Smart DDSs which can control their properties to maximize the efficacy and minimize the side effects by responding to internal stimuli (eg, pH,37 temperature,38 redox condition,39 enzymes40) or external stimuli (eg, magnetic field,41 ultrasound,42 light43) are ideal candidates for controlled release of anticancer drugs.
Among these stimuli, the physiological neutral pH (7.4) of normal tissue and blood, acidic extracellular environment of tumors (pH 6.0–7.0), and intracellular endosomes/lysosomes (pH 5.0–6.5) are commonly exploited in the design of pH-responsive DDSs.44 Based on the pH-responsive principle, one of the promising strategies is to develop charge-reversal nanoparticles that are able to undergo negative-to-positive charge conversion in response to environmental pH changes.44 As an anionic carboxylate-functional polyelectrolyte, poly(allylamine)-citraconic anhydride (PAH-cit) is a common pH-responsive charge-reversal polymer.45 It can be readily converted from negative to positive charge by amide hydrolysis upon exposure to mild acidic environments (Scheme 1), such as those found within late endosomes and lysosomes.46 Benefiting from this property, PAH-cit-coated nanocarriers can easily achieve long-term circulation in the blood while attaining efficient drug release in cells. Han et al fabricated a pH-responsive siRNA delivery system through polyethylenimine (PEI)/PAH-cit/gold nanoparticle-chitosan (AuNP-CS). These AuNPs were stabilized by CS to form a positively charged AuNP-CS core. Charge-reversible PAH-cit and PEI were sequentially deposited onto AuNP-CS to form a PEI/PAH-cit/AuNP-CS shell/core structure through electrostatic interaction. This DDS exhibited pH-triggered siRNA release behavior, which led to improved siRNA knockdown efficiency.47 Yin et al constructed a pH-controlled multifunctional nanosystem by using PAH-cit as a charge-reversible cross-linker for the intelligent co-delivery of DOX and siVEGF (a kind of siRNA that can inhibit the vascular endothelial growth factor signaling pathway). The results showed that PAH-cit initiated effective release of siRNA and DOX into cancer cells.48
 | Scheme 1 (A) Synthetic route of PAH-cit and its pH-responsive charge-reversal behavior. (B) Schematic illustration showing the formation of DOX@PAH-cit–BNNS and their delivery kinetics. |
In this study, we developed a novel pH-responsive DOX delivery system for cancer chemotherapy. BNNS were initially functionalized with pH-responsive charge-reversal PAH-cit polymer. Then, DOX was used as a model anticancer drug to investigate the loading and pH-responsive drug-releasing behavior of PAH-cit–BNNS complexes. Thereafter, DOX@PAH-cit–BNNS complexes were prepared via step-by-step electrostatic interactions and were fully characterized. Afterward, the controlled drug-release behavior of DOX@PAH-cit–BNNS complexes was investigated at different pH. Finally, the intracellular uptake and antitumor effects of the DOX@PAH-cit–BNNS complexes against HeLa cell lines and MCF-7 cell lines were studied.
Materials and methods
Materials
The PAH (molecular weight [MW] ≈ 17,000) solution, citraconic anhydride, and 3-aminopropyltriethoxysilane (APTES) were obtained from Sigma-Aldrich (St Louis, MO). Rhodamine B (RhB) was purchased from Beijing XJK Biotechnology (Beijing, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY, USA). Cell Counting Kit-8 (CCK-8) was acquired from Dojindo (Kumamoto, Japan) and Alexa Fluor® 635 phalloidin and LIVE/DEAD Viability/Cytotoxicity Kit were bought from Invitrogen (Carlsbad, CA, USA).
Synthesis of PAH-cit
The charge-reversal polyelectrolyte PAH-cit was synthesized according to Liu et al’s report.45 Briefly, PAH (100 mg) was dissolved in 1.0 N NaOH and stirred overnight at room temperature. Then, 400 μL citraconic anhydride was added dropwise to the PAH solution and stirred overnight. The pH of the solution was maintained above pH 8 by 6.0 N NaOH during the reaction. The resultant mixture was dialyzed through 3.5-kDa filters (Millipore) to remove excess reagents. Finally, the product was freeze dried to yield the synthesized PAH-cit as a white amorphous powder.
Synthesis of PAH-cit–BNNS
The BNNS was prepared through a chemical vapor deposition method.49 For the preparation of the PAH-cit–BNNS complex, BNNS was firstly treated with HNO3 (65%) and stirred overnight at 70°C in order to introduce hydroxyl groups on the surface of BNNS. After that, OH-BNNS spheres were purified and dried. Next, 20 mg OH-BNNS spheres were dispersed in 10 mL ethanol containing 3 mL APTES and stirred at room temperature for 24 h to yield amino-modified BNNS (NH2-BNNS). After centrifugation and washing with water, the purified NH2-BNNS was dried in air. Subsequently, 20 mg NH2-BNNS spheres dissolved in 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) buffer (3 mL, 10 mM, pH 7.4) were mixed with 10 mg PAH-cit dispersed in a HEPES buffer (1 mL, 10 mM, pH 7.4) and stirred overnight. Finally, the synthesized PAH-cit–BNNS were freeze dried.
Characterization
Transmission electron microscopy (TEM) images were obtained on a JEM-2100 transmission electron microscope operating at an acceleration voltage of 200 kV (Jeol, Japan). Dynamic light scattering (DLS) and zeta potential measurements were conducted using a Zetasizer Nano ZS system (Malvern Instruments, Malvern, UK). Fourier transform infrared (FTIR) spectra were performed on a Nexus spectrophotometer (Nicolet, USA) at 4 cm−1 resolution with 32 scans. Ultraviolet–visible (UV–vis) absorption spectra were carried out using a Nanodrop 2000 spectrophotometer (Thermo Scientific, USA). The boron contents of samples were determined using Optima 7000DV inductively coupled plasma atomic emission spectroscopy (ICP-AES, Perkin-Elmer, USA).
DOX loading
We added 20 mg PAH-cit–BNNS complexes to 12 mL HEPES buffer (10 mM, pH 7.4) containing 5 mg DOX. After the mixture was stirred for 24 h in the dark, the resultant product was collected by centrifugation at 13,500 rpm for 10 min and washed several times by phosphate-buffered saline (PBS) to remove redundant DOX. The amount of unbound DOX in the solution was determined by UV-vis spectrophotometry. The DOX loading capacity was defined as follows: the amount of DOX loaded on 1 mg of complexes.
To investigate the effect of pH on DOX loading, 5 mL PAH-cit–BNNS suspensions at a concentration of 2 mg/mL in PBS (at different pH concentrations of 5.0, 6.5, 7.4, and 9.0) and 2.5 mg DOX solution were mixed and stirred overnight at room temperature. The DOX loading capacity at different pHs were measured by UV-vis spectrophotometry.
In vitro DOX release under different pH conditions
For the DOX release experiments, 2 mg DOX@PAH-cit–BNNS complexes were suspended in 5 mL of pH 5 acetic buffer (the endosomal pH of cancer cells), pH 6.5 phosphate buffer (the pH of cancer tissues), and pH 7.4 phosphate buffer (the physiological pH), respectively. Then, the release process was undertaken on a shaking table with constant shaking at 200 rpm at 37°C in the dark. At certain time intervals, 1 mL of the solution was taken out by micropipetting for characterization, and the same volume of fresh corresponding buffer was added to the original suspension. The concentration of DOX released was analyzed with the Nanodrop 2000 spectrophotometer at 480 nm.
Cell culture
Human embryonic kidney cells 293 (HEK 293), human cervical carcinoma cell lines, HeLa, and the human breast carcinoma cell lines, MCF-7, were all purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, People’s Republic of China) and cultured in DMEM supplemented with 10% FBS, 100 units/mL penicillin, and 100 mg/mL streptomycin. Cells were maintained at 37°C in the presence of 5% CO2 in a humidified incubator. The culture medium was replenished every 2 days.
Cytotoxicity assay
HEK 293, HeLa, and MCF-7 cells were seeded at a density of 4,000 cells per well in a 96-well plate (n=5). After 24 h, the culture medium was replaced with 100 μL fresh medium containing increased concentration of BNNS and PAH-cit–BNNS complexes, respectively. Then, cells were incubated for another 24 h at 37°C, and cell viability was qualitatively investigated with the CCK-8 assay. At the end of incubation, 10 μL CCK-8 solution was added to each well and incubated for 3 h at 37°C. Finally, cultures were observed for absorbance at 450 nm by using a microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA). Results were expressed as the percentage of viable cells over untreated control cells.
Cellular uptake and intracellular DOX release
Cellular uptake experiments were conducted using fluorescence microscopy and ICP-AES. For fluorescence microscopy observation, BNNS were first labeled with RhB to form RhB@BNNS and RhB@PAH-cit–BNNS complexes. HeLa cells were seeded at 5×104 cells per well in a 35-mm petri dish with a glass bottom and incubated in 2 mL DMEM for 24 h. Then, HeLa cells were treated with fresh medium containing RhB@BNNS or RhB@PAH-cit–BNNS at an equivalent BNNS concentration of 50 μg/mL for 4 h. Thereafter, HeLa cells were washed twice with cold PBS and immobilized with 4% (v/v) paraformaldehyde for 20 min at 37°C. Then, cells were stained by Alexa Fluor® 635 phalloidin and DAPI in the dark. Confocal fluorescence imaging of HeLa cells was visualized on a C2Si confocal laser scanning microscope (Nikon Corporation, Tokyo, Japan).
The quantitative measurement of cell uptake of PAH-cit–BNNS was undertaken by ICP-AES. HeLa cells were seeded at a density of 1×106 cells per well in a 10-cm petri dish and cultured in DMEM containing 10% FBS for 24 h. Then, BNNS and PAH-cit–BNNS were added with the same BNNS concentration of 50 μg/mL. Cells were further incubated for another 4 h. Then, these cells were trypsinized and centrifuged. After the cells were washed three times, 65 wt % HNO3 (0.1 mL) was added to allow dissolution of complex for 24 h. Finally, 9.9 mL ultrapure water was added to the solution. Clear acidic solutions were used to measure the mass of PAH-cit–BNNS complexes in Hela cells by detecting the boron concentration.
To investigate the intracellular drug-release behavior of DOX@PAH-cit–BNNS complexes, HeLa cells were seeded at 5×104 cells per well in a 35-mm petri dish and cultured as described earlier. These cells were treated with fresh medium containing free DOX, DOX@PAH-cit, DOX@BNNS, or DOX@PAH-cit–BNNS at an equivalent DOX concentration of 2 μg/mL for 0.5 and 4 h, respectively. After incubation, cells were immobilized and stained with DAPI. Finally, cells were observed under confocal laser scanning microscope.
In vitro antitumor effect of DOX@PAH-cit–BNNS complexes
The cytotoxicity of DOX@PAH-cit–BNNS complexes against HeLa and MCF-7 cells were firstly measured by a CCK-8 assay. Cancer cells were seeded at density of 1×104 cells per well in 96-well culture plates at 37°C in a humidified atmosphere with 5% CO2. After 24 h, the medium was replaced by fresh medium containing free DOX, DOX@PAH-cit, DOX@BNNS, or DOX@PAH-cit–BNNS complexes with increasing DOX concentration. After 48-h incubation, 10 μL CCK-8 solution was added to each well and incubated at 37°C for 3 h. Subsequently, the absorbance of solutions was monitored at 450 nm on a microplate reader. All assays were done with five parallel samples.
For the live/dead assay, HeLa cells were seeded at 5×104 cells per well in a 35-mm petri dish with a glass bottom and incubated in DMEM (2 mL) for 24 h to stain. Then, free DOX, DOX@PAH-cit, DOX@BNNS, and DOX@PAH-cit–BNNS complexes were added to cells at the same DOX concentration of 5 μg/mL for another 10 h. Subsequently, the medium was removed and 200 μL staining solution was directly added to cells. After incubation for 30 min, fluorescence microscopy images of cells were taken with filters for calcein-acetoxymethyl ester (live) and propidium iodide (dead).
Statistical analysis
Statistical analysis was conducted using Student’s t-test. Data were presented as mean ± standard deviation (SD). Differences were considered to be statistically significant *P<0.05, **P<0.01.
Results and discussion
Synthesis and characterization of PAH-cit–BNNS
The fabrication process of the DOX@PAH-cit–BNNS nanocomplex is shown in Scheme 1. BNNS was synthesized following a chemical vapor deposition method.49 PAH-cit was synthesized according to Liu et al’s report.45 Figure 1 shows TEM images of BNNS and the as-prepared PAH-cit–BNNS complexes. BNNS exhibited a spherical shape and were uniform in size (150–200 nm). After oxidation by HNO3, BNNS were reacted with APTES to obtain amino-functionalized BNNS (NH2-BNNS). Then, PAH-cit – as characterized by 1H NMR in Figure 2A – was adsorbed on the surface of NH2-BNNS through electrostatic interactions. However, compared with BNNS, no obvious morphological change could be observed after functionalization with PAH-cit (Figure 1B). The FTIR spectra verified the successful fabrication of PAH-cit–BNNS complexes (Figure 2B). BNNS had a strong asymmetric band at approximately 1,423 cm−1, corresponding to the B-N stretching vibration, along with a less intense band at 800 cm−1, which was ascribed to the B-N-B bending vibration. The peak at 1,630 cm−1 was related to the amino groups of APTES, which appeared in the spectrum of NH2-BNNS. Following PAH-cit functionalization, PAH-cit–BNNS displayed two absorption peaks at 1,550 and 1,657 cm−1, which were respectively assigned to the N-H bending and C=O stretching vibrations of PAH-cit.
 | Figure 1 TEM images of BNNS (A) and PAH-cit–BNNS (B) complexes (bars =100 nm). |
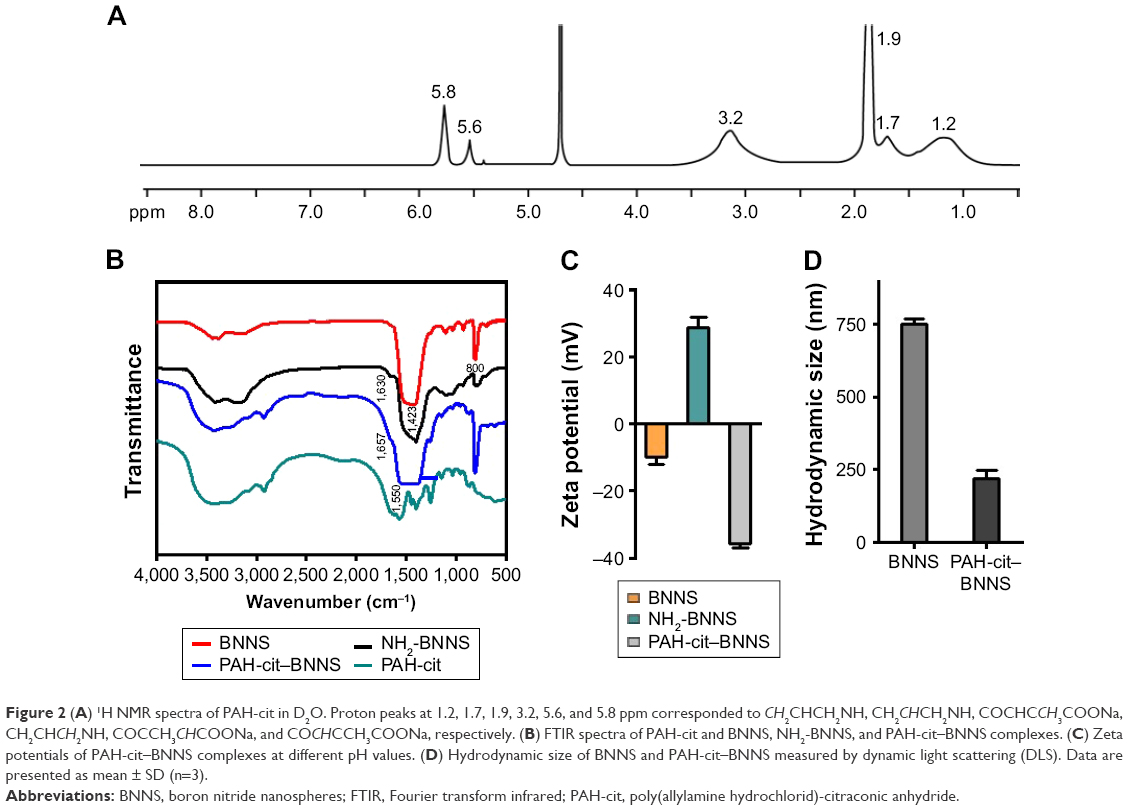 | Figure 2 (A) 1H NMR spectra of PAH-cit in D2O. Proton peaks at 1.2, 1.7, 1.9, 3.2, 5.6, and 5.8 ppm corresponded to CH2CHCH2NH, CH2CHCH2NH, COCHCCH3COONa, CH2CHCH2NH, COCCH3CHCOONa, and COCHCCH3COONa, respectively. (B) FTIR spectra of PAH-cit and BNNS, NH2-BNNS, and PAH-cit–BNNS complexes. (C) Zeta potentials of PAH-cit–BNNS complexes at different pH values. (D) Hydrodynamic size of BNNS and PAH-cit–BNNS measured by dynamic light scattering (DLS). Data are presented as mean ± SD (n=3). |
Furthermore, zeta potential measurements were used to monitor and verify the fabrication of PAH-cit–BNNS. As shown in Figure 2C, at pH 7.4, the zeta potential of BNNS was approximately −10 mV, whereas it shifted to 28.6 mV for NH2-BNNS complexes, indicating the successful amino-modification of BNNS. After the functionalization of negatively charged PAH-cit,39,46 the surface charge of these PAH-cit–BNNS complexes decreased to −35.8 mV. This result further confirmed the successful fabrication of PAH-cit–BNNS complexes. Moreover, the size distribution analysis yielded an average hydrodynamic size of 751.4 nm for BNNS and 219.7 nm for PAH-cit–BNNS (Figure 2D). This result seemed inconsistent with the TEM observation, which was mainly caused by the easy aggregation of BNNS in solutions due to its surface hydrophobicity. However, the functionalization of hydrophilic PAH-cit significantly decreased its hydrophobicity and improved its dispersity in solution.
Stability and pH-responsive charge-reversal behavior of PAH-cit–BNNS
The stability of drug carriers is important for their application in vivo. The stability of PAH-cit–BNNS complexes in PBS (pH 7.4) was examined by monitoring the variation of zeta potential and particle size. Over a period of 48 h, no obvious changes were noted in the zeta potential of PAH-cit–BNNS (Figure 3A). Moreover, the PAH-cit–BNNS suspension could be stored at room temperature for 7 days without obvious aggregation (Figure 3B). The superior stability of PAH-cit–BNNS was probably attributed to the hydrophilic nature of PAH-cit.
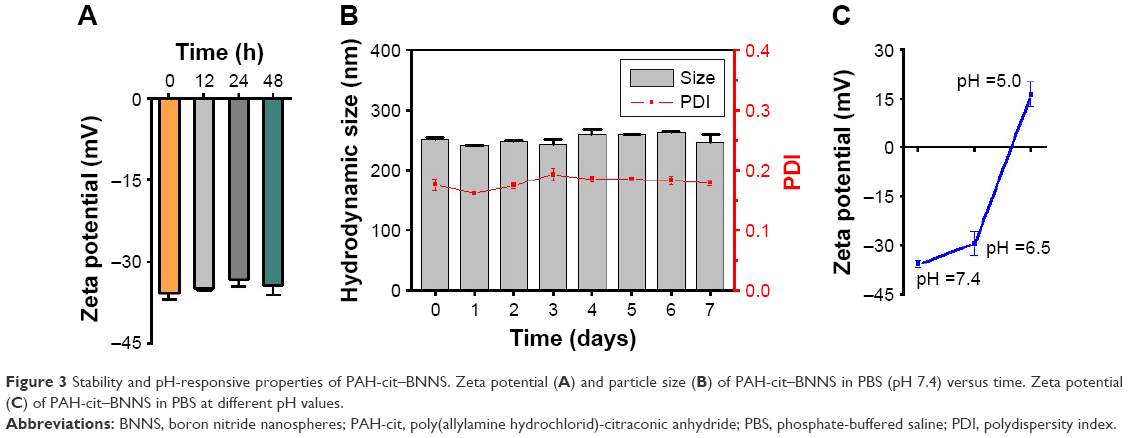 | Figure 3 Stability and pH-responsive properties of PAH-cit–BNNS. Zeta potential (A) and particle size (B) of PAH-cit–BNNS in PBS (pH 7.4) versus time. Zeta potential (C) of PAH-cit–BNNS in PBS at different pH values. |
The charge-reversal behavior of PAH-cit-BNNS complexes was further investigated through changes of zeta potential after incubation at pH 7.4, 6.5, or 5.0. As shown in Figure 3C, with the decrease in pH, the zeta potential of the PAH-cit–BNNS complexes gradually changed from negative (−35.8 mV) to positive (16.3 mV). This result demonstrated the effective pH-responsive charge-reversal behavior of PAH-cit–BNNS. However, only a slight change in charge (−29.6 mV) was found when incubated at pH 6.5, indicating their stability under the acidic extracellular environment of tumors.
In vitro cytotoxicity of PAH-cit–BNNS complexes
The cytotoxicity of PAH-cit–BNNS complexes was investigated to examine their biocompatibility. Cell proliferation was assessed by CCK-8 assay. HEK 293, HeLa, and MCF-7 cells were incubated with BNNS and PAH-cit–BNNS with a concentration range of 0–100 μg/mL for 24 h. The viability of cells treated with PBS was assumed to be 100%. As shown in Figure 4, no obvious cytotoxic effects were observed. This result clearly indicated that the synthesized PAH-cit–BNNS complexes were biocompatible and suitable for use as carriers.
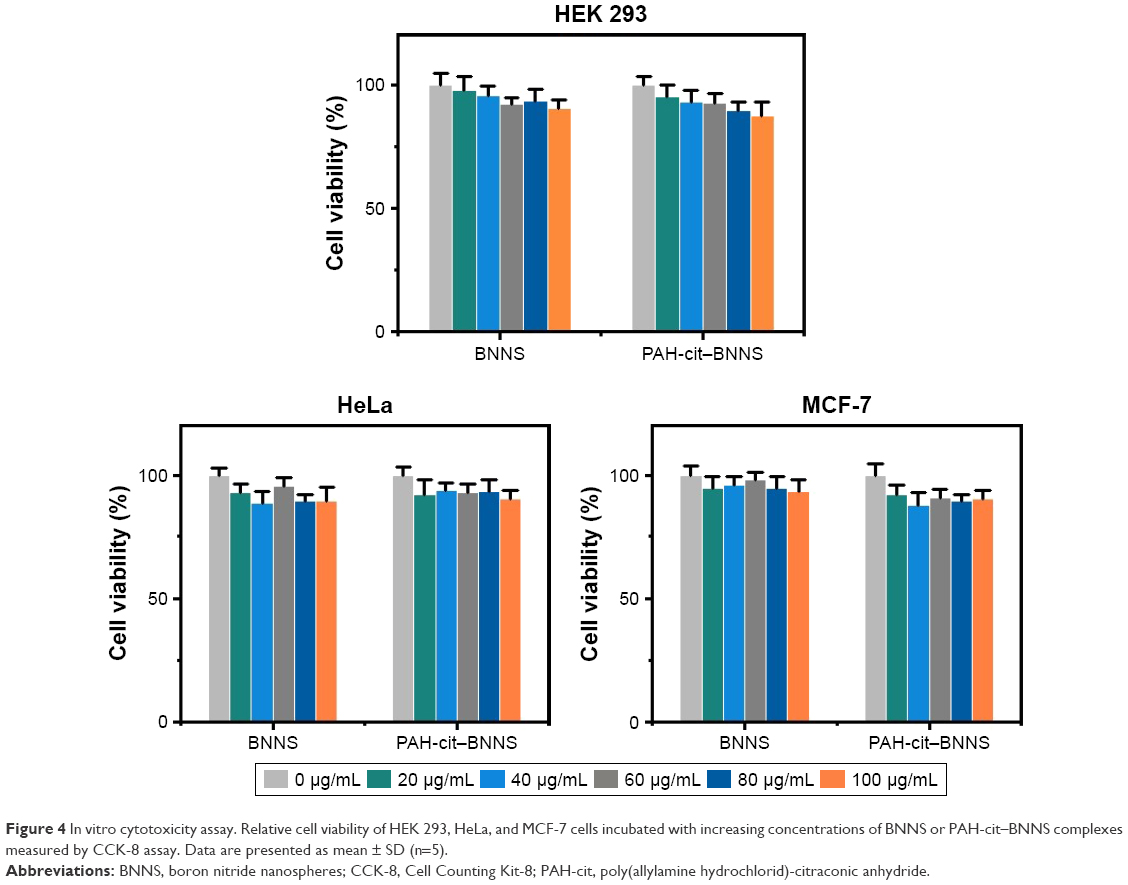 | Figure 4 In vitro cytotoxicity assay. Relative cell viability of HEK 293, HeLa, and MCF-7 cells incubated with increasing concentrations of BNNS or PAH-cit–BNNS complexes measured by CCK-8 assay. Data are presented as mean ± SD (n=5). |
DOX loading and in vitro release
The loading capacity of vehicles is a crucial parameter to evaluate the therapeutic efficacy of DDSs. The loading of DOX on PAH-cit–BNNS complexes was investigated by UV–vis spectra (Figure 5A). Fluorescence intensity of DOX decreased after incubation with PAH-cit–BNNS complexes in HEPES buffer, indicating the loading of DOX onto PAH-cit–BNNS complexes. The DOX loading capacity of BNNS and PAH-cit–BNNS complexes was calculated to be 6.9 and 72.6 μg per 1 mg of carriers, respectively (Figure 5B). The significantly increased DOX loading of PAH-cit–BNNS complexes was mainly due to their highly negative surface charge, which enhanced electrostatic attractions with positively charged DOX. Moreover, the effect of pH on DOX loading was investigated. As illustrated in Figure 5C, the loading of DOX on the PAH-cit–BNNS complexes increased with increasing pH values. At acidic pH condition, the surface charge of PAH-cit–BNNS complexes became positive, which limited the loading of positively charged DOX. On the other hand, at alkaline pH conditions, the deprotonation of −NH2 groups increased the hydrophobicity of DOX, which further improved the interactions between DOX and negatively charged PAH-cit–BNNS complexes. To investigate the in vitro DOX release, DOX@PAH-cit–BNNS complexes were dispersed in PBS at different pH values. The release of DOX from the DOX@PAH-cit–BNNS complexes was pH-responsive. As shown in Figure 5D, DOX was released slowly from DOX@PAH-cit–BNNS at neutral conditions. Only about 26.5% of DOX was released until 60 h. The amount of DOX released at pH 6.5 was slightly higher than that at pH 7.4. However, when the pH decreased to 5.0, the release efficiency of DOX was enhanced evidently. Up to 89% of total DOX was released from these complexes until the end of the assay. The increased drug release from DOX@PAH-cit–BNNS complexes under acidic condition was mainly associated with the change of electrostatic interactions between PAH-cit–BNNS and DOX. Meanwhile, the enhanced solubility of DOX at acidic condition facilitated its release from the complexes. This result demonstrated that, at lower pH, the anionic PAH-cit gradually converted into cationic PAH, and the loaded DOX could be effectively released from DOX@PAH-cit–BNNS via electrostatic repulsion.
 | Figure 5 Loading and releasing of DOX. (A) UV–vis spectra of DOX before and after incubation with PAH-cit–BNNS complexes in HEPES buffer. (B) Loading capacity of DOX on BNNS and PAH-cit–BNNS complexes. (C) UV–vis spectra of DOX before and after loading on PAH-cit–BNNS complexes in PBS with different pH values. (D) DOX-release profiles of the DOX@PAH-cit–BNNS complexes at different pH values. Data were presented as mean ± SD (n=3). |
Intracellular uptake and drug-release behavior of DOX@PAH-cit–BNNS complexes
To investigate the cellular uptake efficiency of PAH-cit–BNNS complexes, HeLa cells were incubated with 100 μg/mL RhB@BNNS and RhB@PAH-cit–BNNS complexes for 4 h. As shown in Figure 6A, strong RhB fluorescence was observed in the cytoplasm of HeLa cells incubated with RhB@PAH-cit–BNNS. By contrast, only aggregated green fluorescent dots were seen in cells treated with RhB@BNNS. The mass of complexes internalized in HeLa cells were quantitatively determined by ICP-AES. Figure 6B shows that the internalization of RhB@PAH-cit–BNNS complexes (0.493 pg/cell) was nearly five times higher than that of RhB@BNNS complexes (0.082 pg/cell) in HeLa cells. Given the significantly improved dispersity by PAH-cit coating, cancer cells easily ingested RhB@PAH-cit–BNNS complexes than the highly aggregated RhB@BNNS complexes.
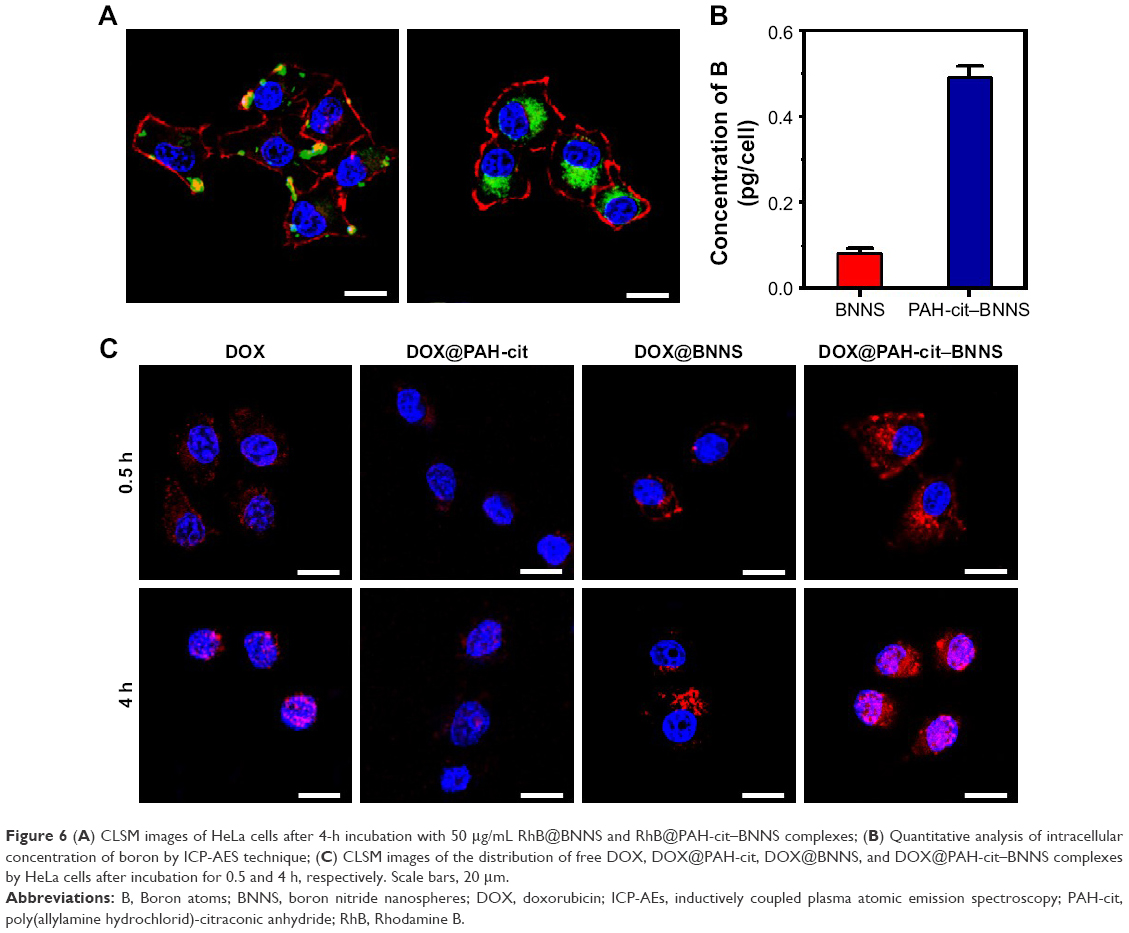 | Figure 6 (A) CLSM images of HeLa cells after 4-h incubation with 50 μg/mL RhB@BNNS and RhB@PAH-cit–BNNS complexes; (B) Quantitative analysis of intracellular concentration of boron by ICP-AES technique; (C) CLSM images of the distribution of free DOX, DOX@PAH-cit, DOX@BNNS, and DOX@PAH-cit–BNNS complexes by HeLa cells after incubation for 0.5 and 4 h, respectively. Scale bars, 20 μm. |
To confirm that the DOX@PAH-cit–BNNS complexes could efficiently transport and release DOX in cancer cells, free DOX, DOX@PAH-cit, DOX@BNNS, and DOX@PAH-cit–BNNS with equivalent DOX concentration were incubated with HeLa cells for 0.5 and 4 h. CLSM was used to visualize the intracellular fluorescence of DOX. As shown in Figure 6C, very weak fluorescence was observed in the cytoplasm of free DOX-treated HeLa cells after 0.5 h of treatment. By contrast, strong red fluorescence was seen in HeLa cells treated with DOX@PAH-cit–BNNS complexes. This result was due to the fact that free DOX was internalized inside the cells in a passive diffusion manner, and some of them were easily flushed out by P-gp. However, the DOX@PAH-cit–BNNS complexes were internalized into cells though the endocytosis pathway, which avoided the active efflux of drugs by the multidrug transporter. Similarly, cells treated with the DOX@PAH-cit complexes showed extremely weak fluorescence, indicating invalid intracellular DOX delivery (Figure 6C). This result indicated that DOX and the PAH-cit polymer might not form an ordered nanostructure. Therefore, DOX@PAH-cit complexes were similar to free DOX and exhibited low cellular uptake. Notably, a dramatic enhancement of fluorescence intensity was observed in the DOX@PAH-cit–BNNS complex-treated cells compared with that of DOX@BNNS complex-treated cells. This result might be due to the improved dispersity of DOX@PAH-cit–BNNS complexes in solution, which contributed to their uptake by cancer cells. Moreover, strong DOX fluorescence was observed in the nucleus of cells after 4-h incubation with DOX@PAH-cit–BNNS, whereas the fluorescence of DOX@BNNS still gathered in the cytoplasm of HeLa cells. This result indicated that after transportation into the cytoplasm, DOX was triggered and gradually released from DOX@PAH-cit–BNNS complexes under the acidic environment in the lysosome and diffused into the nucleus. These results suggested that the charge-reversal PAH-cit–BNNS is a potential candidate for efficient delivery and controlled release of DOX for cancer therapy.
In vitro antitumor effect of DOX@PAH-cit–BNNS complexes
We further investigated the in vitro antitumor effect of DOX@PAH-cit–BNNS complexes. HeLa and MCF-7 cells were treated with free DOX, DOX@PAH-cit, DOX@BNNS, and DOX@PAH-cit–BNNS complexes at different DOX concentrations for 48 h. The cell viability was determined by a CCK-8 assay. Free DOX and DOX@PAH-cit–BNNS complexes exhibited dose-dependent cytotoxicity to cancer cells (Figure 7). Both HeLa and MCF-7 cells showed the lowest viability after treatment with DOX@PAH-cit–BNNS complexes, much lower than that of free DOX, DOX@PAH-cit, and DOX@BNNS complexes. Considering the increased cellular uptake and efficient intracellular pH-responsive DOX release, it was reasonable that DOX@PAH-cit–BNNS complexes induced significant cell death. Moreover, free DOX exhibited lower tumor-inhibition efficacy than that of the DOX@PAH-cit–BNNS complexes with equal DOX concentration, which was caused by reduced cellular uptake and fast efflux of free DOX.
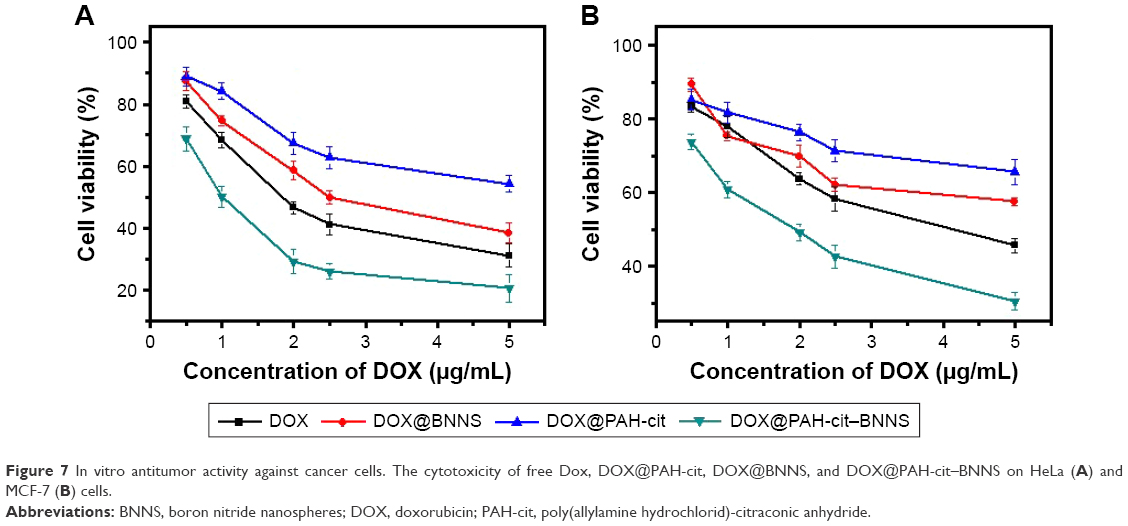 | Figure 7 In vitro antitumor activity against cancer cells. The cytotoxicity of free Dox, DOX@PAH-cit, DOX@BNNS, and DOX@PAH-cit–BNNS on HeLa (A) and MCF-7 (B) cells. |
The live/dead viability/cytotoxicity assay was conducted to further verify the superior antitumor effect of DOX@PAH-cit–BNNS. HeLa cells were incubated with free DOX, DOX@PAH-cit, DOX@BNNS, and DOX@PAH-cit–BNNS complexes with the same DOX concentration for 10 h. Then, fluorescence microscopy was used to visualize the live and dead cells. Live cells were viewed with green fluorescence, whereas dead cells were represented in red. As shown in Figure 8, DOX@PAH-cit–BNNS complexes caused the most cell death, which was consistent with the result of the CCK-8 assay. Overall, our results clearly demonstrated that the DOX@PAH-cit–BNNS complex exhibited significantly enhanced antitumor efficacy in vitro. However, the necessity for further in vivo experiment, before a final statement on their antitumor effect, is mandatory for an actual exploitation of PAH-cit–BNNS complexes in tumor therapy.
 | Figure 8 Live/dead images of HeLa cells incubated with free DOX, DOX@PAH-cit, DOX@BNNS, and DOX@PAH-cit–BNNS complexes (live cells are represented in green and dead cells in red). Scale bars denote 50 μm. |
Conclusion
We successfully prepared PAH-cit-functionalized BNNS for enhanced delivery and pH-responsive controlled release of DOX into cancer cells. PAH-cit–BNNS complexes were nontoxic to cells up to a high concentration of 100 μg/mL. DOX was loaded on PAH-cit–BNNS complexes with high efficiency. DOX@PAH-cit–BNNS complexes exhibited enhanced drug-release behaviors under acidic environment. They could be taken up by cancer cells, and then released DOX efficiently. Most importantly, DOX@PAH-cit–BNNS complexes exhibited exceptional anticancer activity in vitro. BNNS could serve as boron carriers for effective BNCT for cancer therapy. Therefore, with its pH-responsive and controlled drug-release properties, PAH-cit-functionalized BNNS will be a promising candidate for cancer therapy.
Acknowledgments
This work was supported by the Natural Science Foundation of Jiangsu Province (BK 20170202), National Natural Science of China (21778023 and 21576118), and 111 Project (111-2-06).
Disclosure
The authors report no conflicts of interest in this work.
References
Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54(5):631–651. | ||
Gu FX, Karnik R, Wang AZ, et al. Targeted nanoparticles for cancer therapy. Nano Today. 2007;2(3):14–21. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Calucci L, Ciofani G, De Marchi D, et al. Boron nitride nanotubes as T-2-weighted MRI contrast agents. J Phys Chem Lett. 2010;1(17):2561–2565. | ||
Ciofani G, Danti S, D’Alessandro D, et al. Enhancement of neurite outgrowth in neuronal-like cells following boron nitride nanotube-mediated stimulation. ACS Nano. 2010;4(10):6267–6277. | ||
Li X, Hanagata N, Wang X, et al. Multimodal luminescent-magnetic boron nitride nanotubes@NaGdF4:Eu structures for cancer therapy. Chem Commun (Camb). 2014;50(33):4371–4374. | ||
Li X, Wang X, Zhang J, et al. Hollow boron nitride nanospheres as boron reservoir for prostate cancer treatment. Nat Commun. 2017;8:13936. | ||
Li X, Zhi C, Hanagata N, Yamaguchi M, Bando Y, Golberg D. Boron nitride nanotubes functionalized with mesoporous silica for intracellular delivery of chemotherapy drugs. Chem Commun (Camb). 2013;49(66):7337–7339. | ||
Niskanen J, Zhang I, Xue Y, Golberg D, Maysinger D, Winnik FM. Boron nitride nanotubes as vehicles for intracellular delivery of fluorescent drugs and probes. Nanomedicine (Lond). 2016;11(5):447–463. | ||
Sukhorukova IV, Zhitnyak IY, Kovalskii AM, et al. Boron nitride nanoparticles with a petal-like surface as anticancer drug-delivery systems. ACS Appl Mater Interfaces. 2015;7(31):17217–17225. | ||
Weng Q, Wang B, Wang X, et al. Highly water-soluble, porous, and biocompatible boron nitrides for anticancer drug delivery. ACS Nano. 2014;8(6):6123–6130. | ||
Zhi C, Bando Y, Tang C, Xie R, Sekiguchi T, Golberg D. Perfectly dissolved boron nitride nanotubes due to polymer wrapping. J Am Chem Soc. 2005;127(46):15996–15997. | ||
Chopra NG, Luyken RJ, Cherrey K, et al. Boron nitride nanotubes. Science. 1995;269(5226):966–967. | ||
Zhi C, Bando Y, Tang C, Golberg D. Boron nitride nanotubes. Mater Sci Eng R Rep. 2010;70(3–6):92–111. | ||
Pakdel A, Bando Y, Golberg D. Nano boron nitride flatland. Chem Soc Rev. 2014;43(3):934–959. | ||
Chen X, Wu P, Rousseas M, et al. Boron nitride nanotubes are noncytotoxic and can be functionalized for interaction with proteins and cells. J Am Chem Soc. 2009;131(3):890–891. | ||
Ciofani G, Raffa V, Menciassi A, Cuschieri A. Boron nitride nanotubes: an innovative tool for nanomedicine. Nano Today. 2009;4(1):8–10. | ||
Ciofani G. Potential applications of boron nitride nanotubes as drug delivery systems. Expert Opin Drug Deliv. 2010;7(8):889–893. | ||
Ciofani G, Danti S, Genchi GG, et al. Pilot in vivo toxicological investigation of boron nitride nanotubes. Int J Nanomedicine. 2012;7:19–24. | ||
Ciofani G, Danti S, Genchi GG, Mazzolai B, Mattoli V. Boron nitride nanotubes: biocompatibility and potential spill-over in nanomedicine. Small. 2013;9(9–10):1672–1685. | ||
Ciofani G, Raffa V, Menciassi A, Cuschieri A. Cytocompatibility, interactions, and uptake of polyethyleneimine-coated boron nitride nanotubes by living cells: confirmation of their potential for biomedical applications. Biotechnol Bioeng. 2008;101(4):850–858. | ||
Horváth L, Magrez A, Golberg D, et al. In vitro investigation of the cellular toxicity of boron nitride nanotubes. ACS Nano. 2011;5(5):3800–3810. | ||
Salvetti A, Rossi L, Iacopetti P, et al. In vivo biocompatibility of boron nitride nanotubes: effects on stem cell biology and tissue regeneration in planarians. Nanomedicine (Lond). 2015;10(12):1911–1922. | ||
Feng S, Zhang H, Yan T, et al. Folate-conjugated boron nitride nanospheres for targeted delivery of anticancer drugs. Int J Nanomedicine. 2016;11:4573–4582. | ||
Zhang H, Chen S, Zhi C, Yamazaki T, Hanagata N. Chitosan-coated boron nitride nanospheres enhance delivery of CpG oligodeoxynucleotides and induction of cytokines. Int J Nanomedicine. 2013;8:1783–1793. | ||
Zhang H, Feng S, Yan T, Zhi C, Gao XD, Hanagata N. Polyethyleneimine-functionalized boron nitride nanospheres as efficient carriers for enhancing the immunostimulatory effect of CpG oligodeoxynucleotides. Int J Nanomedicine. 2015;10:5343–5353. | ||
Zhang H, Yamazaki T, Zhi C, Hanagata N. Identification of a boron nitride nanosphere-binding peptide for the intracellular delivery of CpG oligodeoxynucleotides. Nanoscale. 2012;4(20):6343–6350. | ||
Zhi C, Meng W, Yamazaki T, et al. BN nanospheres as CpG ODN carriers for activation of toll-like receptor 9. J Mater Chem. 2011;21(14):5219–5222. | ||
Barth RF, Coderre JA, Vicente MG, Blue TE. Boron neutron capture therapy of cancer: current status and future prospects. Clin Cancer Res. 2005;11(11):3987–4002. | ||
Barth RF, Soloway AH, Fairchild RG, Brugger RM. Boron neutron capture therapy for cancer. Realities and prospects. Cancer. 1992;70(12):2995–3007. | ||
Ciofani G, Raffa V, Menciassi A, Cuschieri A. Folate functionalized boron nitride nanotubes and their selective uptake by glioblastoma multiforme cells: implications for their use as boron carriers in clinical boron neutron capture therapy. Nanoscale Res Lett. 2008;4(2):113–121. | ||
Yanagië H, Ogata A, Sugiyama H, Eriguchi M, Takamoto S, Takahashi H. Application of drug delivery system to boron neutron capture therapy for cancer. Expert Opin Drug Deliv. 2008;5(4):427–443. | ||
Kuthala N, Vankayala R, Li YN, Chiang CS, Hwang KC. Engineering novel targeted boron-10-enriched theranostic nanomedicine to combat against murine brain tumors via MR imaging-guided boron neutron capture therapy. Adv Mater. 2017;29(31). | ||
Nakamura H, Koganei H, Miyoshi T, Sakurai Y, Ono K, Suzuki M. Antitumor effect of boron nitride nanotubes in combination with thermal neutron irradiation on BNCT. Bioorg Med Chem Lett. 2015;25(2):172–174. | ||
Guo X, Shi C, Yang G, Wang J, Cai Z, Zhou S. Dual-responsive polymer micelles for target-cell-specific anticancer drug delivery. Chem Mater. 2014;26(15):4405–4418. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. | ||
Guo S, Huang Y, Jiang Q, et al. Enhanced gene delivery and siRNA silencing by gold nanoparticles coated with charge-reversal polyelectrolyte. ACS Nano. 2010;4(9):5505–5511. | ||
Chen KJ, Liang HF, Chen HL, et al. A thermoresponsive bubble-generating liposomal system for triggering localized extracellular drug delivery. ACS Nano. 2013;7(1):438–446. | ||
Han L, Tang C, Yin C. Dual-targeting and pH/redox-responsive multi-layered nanocomplexes for smart co-delivery of doxorubicin and siRNA. Biomaterials. 2015;60:42–52. | ||
Xiong MH, Bao Y, Yang XZ, Wang YC, Sun B, Wang J. Lipase-sensitive polymeric triple-layered nanogel for “on-demand” drug delivery. J Am Chem Soc. 2012;134(9):4355–4362. | ||
Tian Y, Jiang X, Chen X, Shao Z, Yang W. Doxorubicin-loaded magnetic silk fibroin nanoparticles for targeted therapy of multidrug-resistant cancer. Adv Mater. 2014;26(43):7393–7398. | ||
Schroeder A, Honen R, Turjeman K, Gabizon A, Kost J, Barenholz Y. Ultrasound triggered release of cisplatin from liposomes in murine tumors. J Control Release. 2009;137(1):63–68. | ||
Yu J, Ju Y, Zhao L, et al. Multistimuli-regulated photochemothermal cancer therapy remotely controlled via Fe5C2 nanoparticles. ACS Nano. 2016;10(1):159–169. | ||
Han SS, Li ZY, Zhu JY, et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small. 2015;11(21):2543–2554. | ||
Liu X, Zhang J, Lynn DM. Polyelectrolyte multilayers fabricated from ‘charge-shifting’ anionic polymers: a new approach to controlled film disruption and the release of cationic agents from surfaces. Soft Matter. 2008;4(8):1688–1695. | ||
Zhang P, Wu T, Kong JL. In situ monitoring of intracellular controlled drug release from mesoporous silica nanoparticles coated with pH-responsive charge-reversal polymer. ACS Appl Mater Interfaces. 2014;6(20):17446–17453. | ||
Han L, Zhao J, Zhang X, et al. Enhanced siRNA delivery and silencing gold-chitosan nanosystem with surface charge-reversal polymer assembly and good biocompatibility. ACS Nano. 2012;6(8):7340–7351. | ||
Han L, Tang C, Yin C. Enhanced antitumor efficacies of multifunctional nanocomplexes through knocking down the barriers for siRNA delivery. Biomaterials. 2015;44:111–121. | ||
Tang CC, Bando Y, Huang Y, Zhi CY, Golberg D. Synthetic routes and formation mechanisms of spherical boron nitride nanoparticles. Adv Funct Mater. 2008;18(22):3653–3661. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.