Back to Journals » International Medical Case Reports Journal » Volume 9
Peters plus anomaly in a Cameroonian child: a case report
Authors Kagmeni G ![]() , Bilong Y, Mbogos C, Bella Assumpta L
, Bilong Y, Mbogos C, Bella Assumpta L
Received 27 June 2016
Accepted for publication 9 August 2016
Published 14 October 2016 Volume 2016:9 Pages 317—319
DOI https://doi.org/10.2147/IMCRJ.S115941
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Giles Kagmeni,1,2 Yannick Bilong,2 Cedric Mbogos,2 Lucienne Bella Assumpta2
1University Teaching Hospital, Yaoundé, Cameroon; 2Eye Department, University of Yaoundé 1, Yaoundé, Cameroon
Background: Peters’ anomaly (PA) is a rare form of anterior segment dysgenesis characterized by corneal opacity with varied degree of anterior chamber affection with associated defects in the posterior layers of the cornea.
Case presentation: We report the case of a 3-month-old male infant with bilateral corneal opacity since birth who was transferred from the pediatric unit for further ophthalmic care. He was diagnosed with PA with subsequent systemic-associated malformations including craniofacial defects and skeletal defects. He was managed minimally with topical mydriatics and antiglaucomatous drugs.
Conclusion: PA is an inevitable cause of blindness in developing countries due to poor infrastructure and limited resources.
Keywords: congenital corneal opacity, polydactyly, camptodactyly, clinodactyly
Introduction
Peters’ anomaly (PA) is a rare form of anterior segment dysgenesis characterized by corneal opacity with or without irido corneal and/or corneolenticular adhesions, with associated defects in the posterior layers of the cornea.1 Embryologically, PA may result from a faulty separation of the lens from the surface ectoderm or aberrant reattachment of the lens/iris to the cornea during development of the anterior chamber between the 4th and 7th gestational weeks.2 Corneal opacity obstructs the visual axis, leading to sensory deprivation, amblyopia, and severe visual impairment.3 Two clinical types of PA have been described, Type 1 and Type 2. In Type 1, 80% of cases present bilaterally with central and paracentral corneal opacity. The cornea is typically avascular; iris strands extend from the collarette, and systemic abnormalities are not usually present. In Type 2, cases are commonly bilateral with denser corneal opacity; there is often juxtaposition of the lens, and iris strands may or may not be present.4 We report the clinical presentation and difficulties linked to the management of this rare pathology at our hospital.
Case report
We report the case of a 3-month-old male infant with bilateral corneal opacity since birth who was transferred from the pediatric unit for further ophthalmic care. Written informed consent was obtained from the patient’s parents to publish this report. Prenatal and natal history were unremarkable apart from the fact that the mother, aged 35 years at the time of childbirth, was HIV positive. There was no family history of similar ocular disorders or other congenital anomalies. Upon initial ophthalmic examination, a positive light pursuit was found. There was a homogeneous bilateral central corneal opacity without neovascularization. Ophthalmologic examination under general anesthesia revealed a deep anterior chamber with iris strands that crossed the anterior chamber from the iris. The lens was transparent on the edges and did not adhere to the cornea. The retina was difficult to evaluate because of central corneal opacity. The intraocular pressure measured with a Schiotz tonometer was respectively 18 mmHg and 20 mmHg in the right and left eye. Other malformed features found in the physical pediatric examination included: broadening of the nose base and microcephaly (Figure 1), bilateral polydactyly with camptodactyly, clinodactyly of the supernumerary finger (Figure 2), an umbilical hernia with hypochromic macules of the abdomen (Figure 3) and bilateral supernumerary toe with hypertrophy of the hallux (Figure 4). There were no genital, vertebral, or cardiac anomalies on pediatric physical examination. Laboratory examination revealed that the child was also HIV positive. The diagnosis of Peters plus anomaly Type 1 was made. Management consisted of the administration of mydriatics, one drop two times per day, 3 days a week. One drop of dorzolamide in the evening was also prescribed. Immediate penetrating keratoplasty (PKP) in both the eyes was advised. After being informed that this intervention was not available in our country, the parents did not bring the child back for further consultation, and were lost to follow-up.
 | Figure 1 Bilateral cornea opacity, broadening of the nose base and microcephaly. |
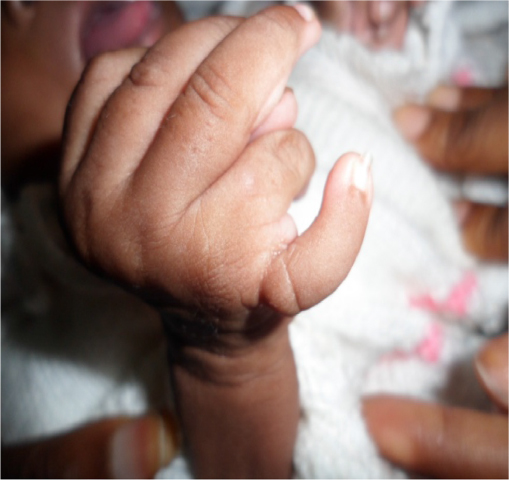 | Figure 2 Polydactyly with camptodactyly and clinodactyly of the supernumerary finger. |
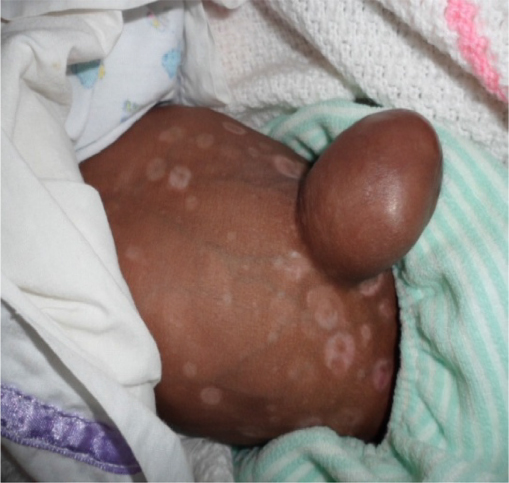 | Figure 3 Umbilical hernia with hypochromic macules of the abdomen. |
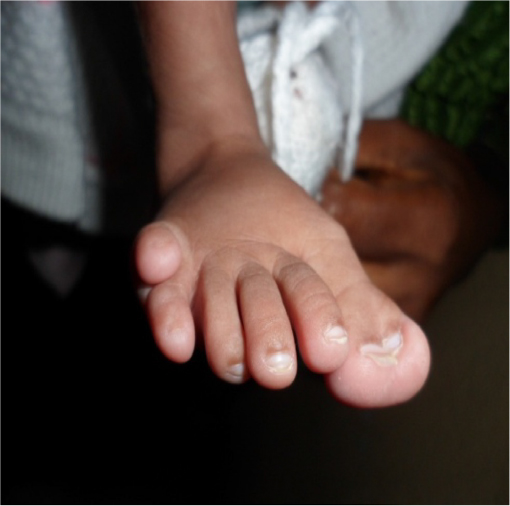 | Figure 4 Supernumerary toe with hypertrophy of the hallux. |
Discussion
First described in 1906 by Dr Alfred Peters, PA is a rare congenital dysgenesis of the anterior segment. This disease mostly appears sporadically. However, both recessive and irregular dominant inheritance has been described.4,5 Regarding causative maternal infections during pregnancy, environmental and genetic factors have been incriminated in the development of congenital eye anomalies.6 In our case, gestational maternal infection with HIV was the only associated comorbidity found. The leading clinical sign of PA is nonvascularized central or paracentral congenital corneal opacity.7 Systemic-associated malformations found in our patient included craniofacial defects and skeletal defects. Similar findings were previously reported in Kenya by Mwenda.8 Mutations in B3GLCT have been shown to explain 100% of cases of classic Peters plus syndrome.9 Although genetic diagnosis of PA is not available in our country, clinical ophthalmic findings and associated systemic abnormalities led us to classify our patient as Type 1 plus PA. This lack of genetic diagnosis is seen in the majority of reported cases of PA.1 The management of PA depends on the corneal status and accompanied eye anomalies. It should be initiated as early as possible in order to prevent sensory amblyopia and irreversible glaucoma damages. In the current cases, established medical treatment aimed to dilate the pupils and allow a child to develop peripheral vision while waiting for PKP, which, according to Parmley et al, remains the standard of care.10 In a series of 22 patients, Chang et al proved that the visual outcome of delayed PKP in PA was not inferior compared to the results of earlier PKP.11 The surgical management of PA in our setting is challenging, owing to the lack of adequate infrastructure to enable PKP, the cost of the procedure, and the lack of knowledge among patients and their guardians. Untreated, PA evolutes to blindness either by sensory deprivation or, rarely, by spontaneous corneal perforation. We cannot comment on the outcome of this child since the parents did not come back for further follow-up.
Conclusion
PA is an inevitable cause of blindness in developing countries due to poor infrastructure and limited resources. The relationship between eye anomalies and the mother’s positive serology for HIV was not established in this case.
Acknowledgment
The authors thank the father of the patient for allowing them to film and to use the photos for scientific purposes.
Author contributions
All authors contributed toward data analysis and drafting and revising the paper, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Weh E, Reis LM, Happ HC, et al. Whole exome sequence analysis of Peters anomaly. Hum Genet. 2014;133(12):1497–1511. | ||
Matsubara A, Ozeki H, Matsunaga N, et al. Histopathological examination of two cases of anterior staphyloma associated with Peters’ anomaly and persistent hyperplastic primary vitreous. Br J Ophthalmol. 2001;85(12):1421–1425. | ||
Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011;22(5):314–324. | ||
Hanson IM, Fletcher JM, Jordan T, et al. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat Genet. 1994;6(2):168–173. | ||
Ozeki H, Shirai S, Nozaki M, et al. Ocular and systemic features of Peters’ anomaly. Graefes Arch Clin Exp Ophthalmol. 2000;238(10):833–839. | ||
Lakshmi N, Maina Pk. Microphthalmia and microcornea: in congenital cytomegalovirus. Indian J Ophthalmol. 2009;57(4):323. | ||
Harissi-Dagher M, Colby K. Anterior segment dysgenesis: Peters anomaly and sclerocornea. Int Ophthalmol Clin. 2008;48(2):35–42. | ||
Mwenda AS. Peters anomaly with post axial polydactyly, bilateral camptodactyly and club foot in a Kenyan neonate: a case report. J Med Case Reports. 2012;6(1):16. | ||
Reis LM, Tyler RC, Abdul-Rahman O, et al. Mutation analysis of B3GALTL in Peters Plus syndrome. Am J Med Genet A. 2008;146A(20):2603–2610. | ||
Parmley VC, Stonecipher KG, Rowsey JJ. Peters’ anomaly: a review of 26 penetrating keratoplasties in infants. Ophthalmic Surg. 1993;24(1):31–35. | ||
Chang JW, Kim MK, Kim JH, Kim S-J, Wee WR, Yu YS. Long-term visual outcomes of penetrating keratoplasty for Peters anomaly. Graefes Arch Clin Exp Ophthalmol. 2013;251(3):953–958. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.