Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 12
Personalized medicine in breast cancer: pharmacogenomics approaches
Authors Jeibouei S, Akbari ME, Kalbasi A, Aref AR, Ajoudanian M, Rezvani A, Zali H ![]()
Received 16 March 2018
Accepted for publication 27 March 2019
Published 27 May 2019 Volume 2019:12 Pages 59—73
DOI https://doi.org/10.2147/PGPM.S167886
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
Shabnam Jeibouei,1 Mohammad Esmael Akbari,2 Alireza Kalbasi,3 Amir Reza Aref,4,5 Mohammad Ajoudanian,6 Alireza Rezvani,7 Hakimeh Zali8
1Department of Biotechnology, School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 2Cancer Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 3Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA; 4Belfer Center for Applied Cancer Science, Dana-Farber Cancer Institute, Boston, MA, USA; 5Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA; 6Department of Tissue Engineering and Applied Sciences, School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 7Department of Hematology, Medical Oncology and Stem Cell Transplantation, Hematology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran; 8Proteomics Research Centre, Department of Tissue Engineering and Applied Sciences, School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran
Abstract: Breast cancer is the fifth cause of cancer death among women worldwide and represents a global health concern due to the lack of effective therapeutic regimens that could be applied to all disease groups. Nowadays, strategies based on pharmacogenomics constitute novel approaches that minimize toxicity while maximizing drug efficacy; this being of high importance in the oncology setting. Besides, genetic profiling of malignant tumors can lead to the development of targeted therapies to be included in effective drug regimens. Advances in molecular diagnostics have revealed that breast cancer is a multifaceted disease, characterized by inter-tumoral and intra-tumoral heterogeneity and, unlike the past, molecular classifications based on the expression of individual biomarkers have led to devising novel therapeutic strategies that improve patient survival. In this review, we report and discuss the molecular classification of breast cancer subtypes, the heterogeneity resource, and the advantages and disadvantages of current drug regimens with consideration of pharmacogenomics in response and resistance to treatment.
Keywords: breast cancer, personalized medicine, biomarker, pharmacogenomics
Introduction
Providing the most effective treatment is a task of paramount importance in personalized medicine for breast cancer.1 This approach enables categorizing cancer subtypes and, ultimately, assigning the best treatment regimen based on patient characteristics, medical history and response to therapy.2 The combination of pharmacological approaches (eg, pharmacogenetics, and pharmacodynamics) aids personalized medicine therapeutic decisions. Furthermore, pharmacogenetics and pharmacogenomics enable clinicians selecting appropriate drugs and doses to decrease adverse effects and increase efficacy. The systematic study of drug absorption, distribution, and metabolism is crucial for efficacy, and this may depend on genetic and epigenetic variations.3 In addition to the role of genetic polymorphisms, epigenetics, microbiome alterations as well as demographic characteristics are involved in the occurrence of multi-drug resistance.4–6 Historically, pharmacogenetics and pharmacogenomics refer to the effect of genetic variations on drug metabolism, and the influence of the whole genome might have in response to drugs.7 These approaches are frequently deployed in the setting of oncology as new strategies to minimize toxicity while maximizing the efficacy of target therapy and chemotherapy. They are also applied to develop new drugs based on genetic profiling and gene expression, this process being facilitated by new innovative techniques and profiling instruments. Here we overview the current molecular classification of breast cancer, the source of tumor heterogeneity and pharmacogenetics approaches in the targeted therapy and chemotherapy of the main subtypes of breast cancer.
Molecular classification
Based on molecular stratification, breast cancer is categorized into five distinct molecular classes8 1, and 2 hormone receptor positive (luminal A and luminal B), 3 human epidermal growth factor receptor-2 (HER2-positive), 4 basal-like and normal-like based on microarray and gene expression profiling described by Perou and Sorlie,9,10 and the 5 claudin-low,11 which is triple-negative breast cancer (TNBC), classified by medullary and metaplastic differentiation.12,13 The patterns of each molecular subtype differ in response to treatment.9,10 The management of the most common class – luminal A (predominantly ER+/PR+ and HER2-negative) – which accounts for 40%-60% of the disease8,14 has low pathological grade and proliferation15 and shows low response to chemotherapy16 while being sensitive to endocrine therapy,17 is focused on anti-endocrine strategies8 In luminal B (ER+/PR+ and HER2-positive), which accounts for 15% of the disease,14 and compared with luminal A,15 has higher pathological grade and proliferation, chemotherapy rather than anti-estrogen drugs is beneficial.8,18 10% of breast cancers fall into the HER2 positive category14 that displays high pathological grade.15 Although this group of tumors is chemosensitive,19 the disease prognosis has been dismal up until the introduction of targeted therapeutics.20,21 Basal-like tumors that represent 10–25% of the disease,13 and are characterized by ER−/PR− or HER2− overlap with triple-negative (TN) tumors, express either HER2 or ER or both, in 15–45% of the cases.21 Studies showed that basal-like tumors are chemo responsive,19 although they are associated with aggressive behavior and poor prognosis10,14,22–24; 3–10% of the cases classified as normal-like type.12 Those tumors that express gene expression characteristics of adipose tissue show an intermediate prognosis. In some studies, it has been proposed that these cells might represent a technical artifact during the microarrays.25 The type Claudin-low is predominantly present as TNBC that accounts for 7–14% of the tumors and displays low expression of genes claudin and E-cadherin genes26; 15% of these tumors express ER, while HER2 is overexpressed in 15% of the other cases belonging to the claudin-low type.12 Response to chemotherapy in claudin-low tumors is intermediate.12,13 Table 1 represents subtypes of breast cancer and their corresponding management strategies for cancer therapy. Despite the remarkable improvement achieved upon the use of therapeutic agents, de novo and acquired resistance remains an unsolved issue in breast cancer. The knowledge of the molecular mechanisms involved in the molecular subgroups of breast cancer might guide the development of new therapeutic strategies.27,28 Individual or multiple molecules or mutated genes could be deployed as a target of specific therapeutic strategies.
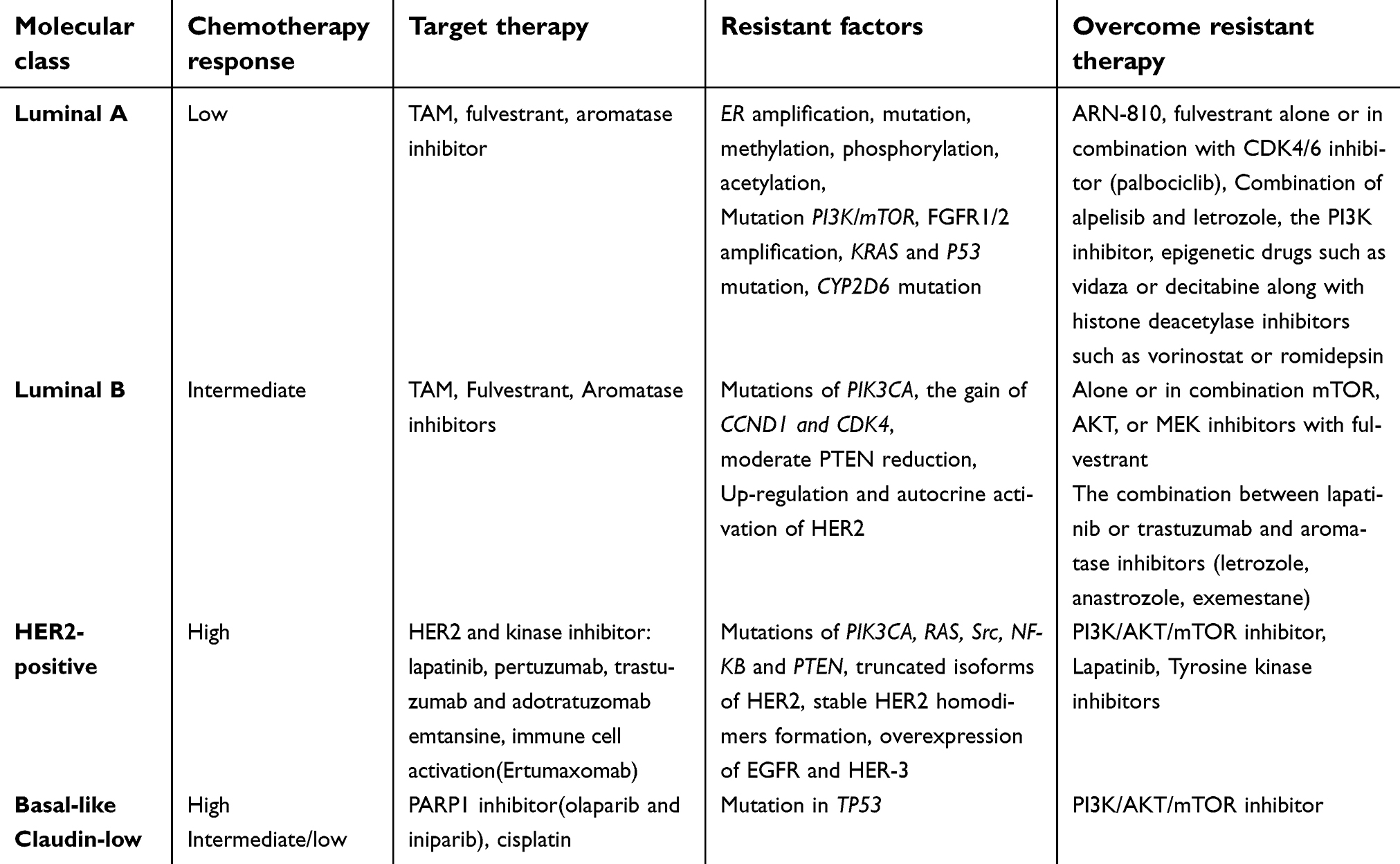 | Table 1 Breast cancer classification based on molecular profiling |
Heterogeneity resources
Heterogeneity of a subtype of breast cancer is the most frequent cause of therapy failure and unexpected outcome. In a distinct subgroup of cancer, alterations in genetic and epigenetic features of a clone of cells lead to changes in the prognosis and drug response.5,29 This heterogeneity can be intra-tumoral when involving cells within a tumor in an individual patient, or inter-tumoral when involving cells of the same subgroup of tumors in different patients. Intra-tumor heterogeneity is named “spatial” when involving a certain part of the tumor; or “temporal” when cells within the same tumor undergo changes during the course of the disease, from the primary tumor to metastasis. The heterogeneity associated with the morphological levels takes from comparing variable cell types in histopathological tests and provides grading systems.30 When considering heterogeneity at a genetic level, alterations are related to the copy number variation (CNV), overexpression and down-regulation of a gene; or from missense, nonsense and frameshift mutations. Nowadays, there is a distinct open source database (COSMIC and TCGA) that provides information related to high throughput techniques. There are different heterogeneity sources for breast cancer patients who categorize in defined groups. These include local and systemic sources.5 The COSMIC database contains data from hundreds of breast cancer tumors detected across different platforms and provides information about different analysis, mutation, CNV, methylation, over and underexpression. Table 2 shows the genetic information of different categories of breast cancer categorized based on cellular and molecular signatures. The extensive variation in top 10 genes translates to the high rate of heterogeneity not only across but also within the groups; it would, therefore, be unlikely to observe a certain molecular pattern in all patients even if they all belong to the same group. This variation may be related to spatial or temporal heterogeneity that results in different tumor responses upon exposure to the therapeutic regimens.
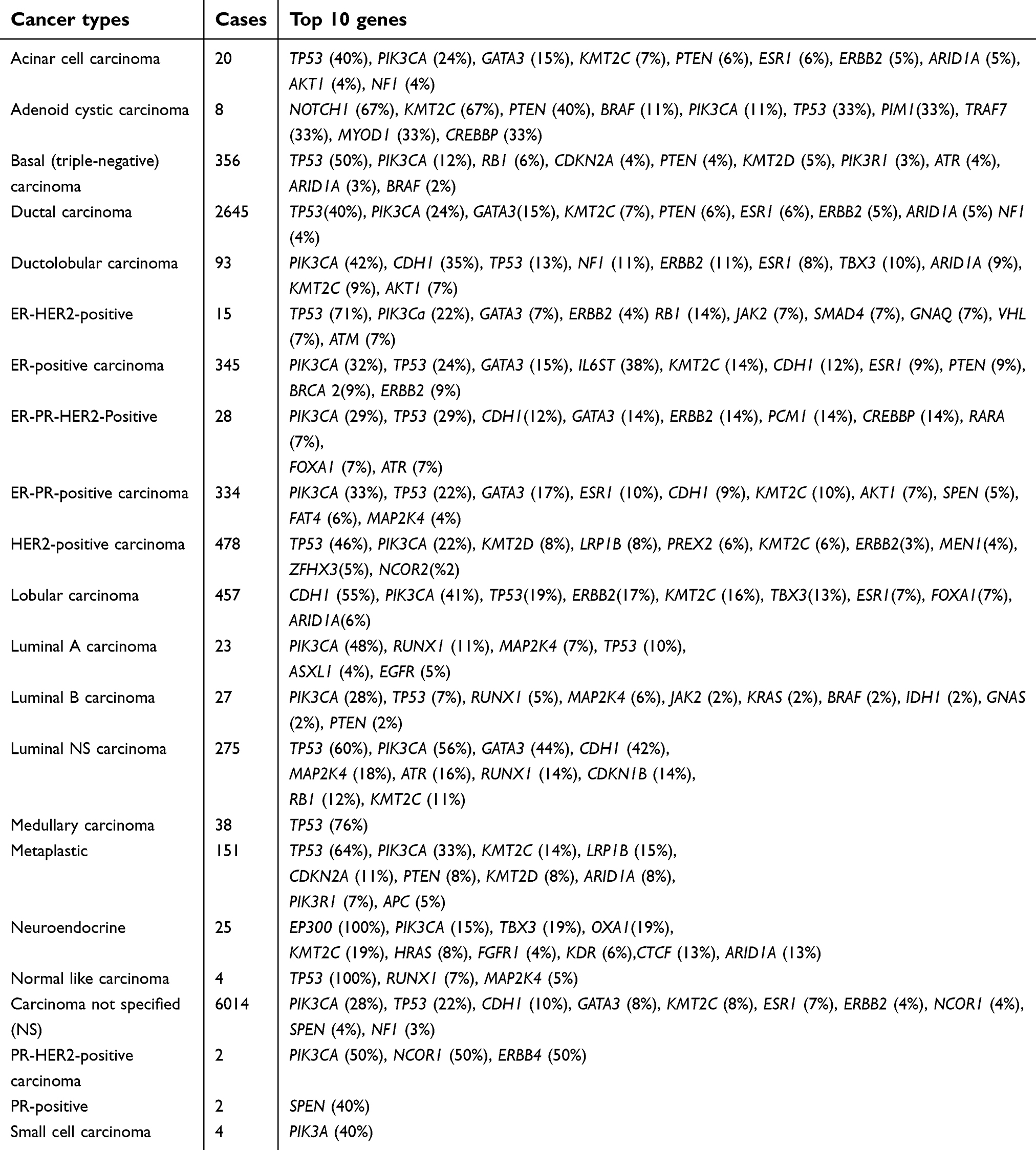 | Table 2 Breast cancer subgroups identified by genome profiling using different analysis platforms in COSMIC database |
The pharmacogenomics approach enables identifying all the genetic variations in driver genes that account for the selective growth of cells in tumor bulk. As therapeutic targets, these help the clinician to select the appropriate therapeutic regimens to overcome resistance. The use of anticancer against these driver genes requires several considerations such as the choice of drug combinations, or selected target based on the predominant subclonal population that may undergo adaptive responses or toxic effects.3 The detection of a subclonal population by accuracy methods like the ex vivo culture of tumor spheres in the tumor on-chip technique remains a critical issue.31,32 Other somatic mutations, introduced as passenger mutations, are those present in genes that help tumor survival. These mutations become acquired when cells are in the normal state or after they have undergone neoplastic transformation.5 In some cases, genes implicated in cancer development have not been found to be mutated but are frequently found to be inactivated as a result of epigenetic mechanisms.
Hereditary breast cancer is linked to genetic mutations. BRCA1, BRCA2, PALB2, TP53, CDH1, and PTEN are genes that may undergo mutations associated with cumulative breast cancer risk.33 Moreover, epigenetic mechanisms, which are known to play an essential role in the regulation of gene expression, may be involved in a hereditary form of the disease. Previous studies have shown that the tumor suppressor gene – RASSF1A– is often silenced following hypermethylation in breast cancer.34 In familial breast cancer, <5% of mutations affect the BRCA1 and BRCA2 genes; whereas sporadically arising breast cancer can be associated with 10–15% rate of BRCA1 methylation. In most subtypes of breast cancer (Figure 1), the molecular pathway35 related to cancer progression include the PI3K/AKT/mTOR and the RAS/RAF/MEK pathways. These are abnormally activated and are associated with resistance. Clinical trials are currently underway to test various PI3K inhibitors, which have been developed to target different components of the pathway. In the PI3K pathway, PTEN inactivation by epigenetic mechanisms is likely to extend the use of drug inhibitors effective in case of PTEN defective cancer cells. Another gene involved in this pathway is PPP2R2B, the negative regulator of AKT, and for which promoter methylation occurs in breast cancer.36
 | Figure 1 Signaling pathways implicated in different categories of breast cancer. Notes: Adapted with permission from Kanehisa M, Goto S. KEGG: Breast cancer - Reference pathway; 2018. Available at: https://www.genome.jp/kegg-bin/show_pathway?map05224. Accessed May 15, 2019. Copyright Kanehisa Laboratories.154 |
In some gene families, overlaps of mutation and methylation are observed, these leading to variable responses. As an example, when considering the RUNX family, RUNX1 is mutated while RUNX3 is inactivated by epigenetic mechanisms.15,37 DNA methylation can be analyzed in a blood sample using a powerful bisulfite-based PCR technique.15 The tumor microenvironment, which comprises tumor-associated macrophage, fibroblast, bone marrow-derived cell and lymphatic growth factors, chemokine, cytokines, and exosome, is also responsible for tumor heterogeneity and contributes to both growth and metastasis5 It is envisaged that in the near future genetic, genomic and immunologic consultants might help the oncologists to implement different strategies and therefore plan for the most successful therapy regimens. Pharmacogenomics will pave the way for such planning and generation of individualized treatments.
Pharmacogenetics in breast cancer subtypes
Estrogen receptor positive
The predominant type of breast cancer is associated with expression of estrogen receptor, which is used as a predictive marker in the follow up of patients (disease free). These cases benefit from hormonal therapy based on the administration of the artificial estrogen analog tamoxifen (TAM). TAM binds to estrogen receptor and disrupts the activation of the classical pathway that leads to ductal hyperplasia. Subsequent alteration of the tumor microenvironment makes the invasion state.38,39 Figure 2 represents the signaling pathways, which modulate tumor cells and tumor microenvironment components, as well as the effect that certain cancer therapeutic agents exerts on these pathways in patient with ER+. The other signaling pathways related to ER overexpression relate to nonclassical functions that facilitate genomics activity in an independent hormone manner. In this context, ER acts through growth factor signaling (FGFR, IGFR, GPCR) that activate intracellular kinase and phosphatase. Several studies determined the effect of kinases in the phosphorylation of ER protein; this phenomenon is associated with sensitivity or resistance to endocrine therapy. Baron et al provided an extensive review of all phosphorylation sites and mRNA splicing regions in ER and associated drug response. Further, the interaction between ER with other transcription factors like C-Fos/C-Jun (AP-1), Sp1 and NF-KB was found to result in tumor cell division, angiogenesis and progression to metastasis40 In addition to TAM, the ER down-regulator Fulvestrant has been approved for the treatment of ER+ breast cancer. By forming a complex with ER, Fulvestrant inhibits its dimerization, finally leading to its degradation. Moreover, Anastrozole, an aromatase inhibitor that blocks the conversion of adrenal androgen into estrogen, can be administered to a postmenopausal woman who could also benefit from Fulvestrant treatment. TAM is equally valid regardless of menopausal status.39 As indicated in Table 2, there are variations in hormone receptor subtypes, which are associated with genetic differences. Progesterone receptor gene expression is dependent on ER expression in epithelial cells so that half of the ER+ tumors are also PR+. Previous work has shown that patients who do not express PR have the worse prognosis since the start of TAM treatment when compared to those who are PR+. This group of patients benefits from the administration of Anastrozole.41 Primary endocrine resistance can occur in half of the cases and finally developed resistance in another half as acquired resistance. Several factors are contributing to resistance, and these include mutation rate, methylation, acetylation and downregulation of ERα, overexpression of ERβ as well as crosstalk between ER and growth factor and signaling pathways.40,42 ARN-810 is a selective ERα antagonist that induces proteasomal-mediated degradation of ERα. This drug is active against tumors with the ESR1 mutation, whereas conventional endocrine therapy is not sufficient.43 Mutations in the ligand binding domain of ERα are associated with ligand-independent transcriptional activity. Hot spots mutations located in the ligand-binding domain include Y537S, Y537N, Y537C, and D538G, these representing >80% of ESR1 mutations associated with acquired resistance to endocrine therapy.44–46 This type of mutations is regarded as the main resistance mechanism that is rare in primary tumors but is reported in >20% of cases of recurrence and metastatic cancer in patients treated with endocrine therapy.47–49 Since the frequency mutation of the ESR1 is higher in metastatic than in primary breast cancer, assessment of this gene mutation in plasma cfDNA could help selecting treatment strategies, eg, administration of Fulvestrant in patients with the ESR1 mutation was found to improve tumor-free survival.48,49 Although the mechanism related to the effect of Fulvestrant alone or in combination with Palbociclib in a patient with ER mutation is not known, patients with metastatic cancer benefit upon exposure to combination therapy.50 The COSMIC database reported that the ESR1 mutation Y537S affects the phosphorylation of IGF1R.51 This is associated with shorter overall survival and contributes to resistance to target therapy.42 K303R is another significant mutation that was found to reduce sensitivity to TAM through phosphorylation of AKT.52 According to the COSMIC database, most of the ER+ cases display PIK3CA mutations. In luminal B subtype with endocrine resistance generated the moderate PTEN reduction leading enhance PI3K signaling. Alone or in combination mTOR, AKT, or MEK inhibitors with Fulvestrant improved endocrine therapy.53 Luminal B are more aggressive and endocrine therapy resistant than luminal A. Moreover they are known with a different rate of PI3K pathway activation.53,54
 | Figure 2 The interaction between tumor microenvironment components, including stromal cells, and tumor cells leads to enhanced cell growth, proliferation, angiogenesis and invasion. Breast cancer epithelial cells increase tumor cell proliferation and invasion while inhibiting apoptosis through either of these following pathways: (1) classical pathway in which estrogen binds to its receptor, ER, or (2) non-classical pathway that involves post-translational modifications of ER by activation kinases, and transcription factors. Anti-cancer agents including tamoxifen (TAM), whose metabolites, 4-OH TAM and endoxifen, have higher affinity for ER when compared with TAM, exert their effects by modulating signaling pathways that regulate tumor cells.Note: Data adapted from Russell39 and Barone et al.40 |
Primary and secondary endocrine resistance can be overcome by additional agents like PI3K and CDK4/6 inhibitors. Genetic alterations were noted in sensitive endocrine patients; however, de novo resistance was related to loss of ER, loss of amplification of co-receptor or co-amplificatory molecules as well as to activation of the cyclin D pathway. In the case of acquired resistance, which develops after an initial response to endocrine therapy, the activation of the PI3Kpathway represents a common resistance mechanism.43 Previous studies have determined the relationship between ESR1 and PIK3CA mutations with clinical features. Thus, mutations of the PI3K/mTOR pathway observed in one-third of ER+ cases associated with good prognosis in early stage and with poor prognosis in the metastatic setting after start of endocrine therapy.43 A recent study that used cfDNA to detect mutations in the ESR1 and PIK3CA genes revealed higher level of heterogeneity in ESR1 than PIK3CA.55 PIK3CA mutation occurs in the early stage of tumor development;56 whereas the ESR1 mutation occurred later during endocrine treatment and was detected in metastatic lesions.57 Therefore, a mutation might result from the pressure endocrine therapy exerts on tumors. Duration of response and development of resistance are related to the rate of ESR1 mutation.57 In ER+ patients resistance to endocrine therapy deriving from aberrant activation of PI3K signaling proved to benefit from the combination of a PI3K inhibitor with the antiestrogen. Combination of Alpelisib and Letrozole were more effective in ER+/HER2 negative metastatic patients with PIK3CA mutation, patients with FGFR1/2 amplification and KRAS and P53 mutations.58 A limited number of clinical trials of luminal B HER2-enriched combination between HER2-targeted therapy (lapatinib or trastuzumab) and aromatase inhibitors provided a clinical benefit.59
Further, combinatorial treatment based on cell cycle arrest agents targeting CDK and endocrine therapy has been proven successful, especially in those cases with primary endocrine resistance. Several studies have investigated the synergistic effect of the CDK4/6 inhibitor Palbociclib and endocrine therapy in both endocrines sensitive and endocrine-resistant cases; the combined action of these drugs leads to improved prognosis. Additional agents could be deployed to target methylation by histone deacetylase; this enabling to reverse resistance.43 Hypermethylation of CPG islands within the promoter of ESR1 gene is associated with lack of response to TAM. Methylation of PITX2 has also been related to TAM-resistance. Methylation analysis of candidate genes could help in sparing patients from ineffective treatment by indicating the administration of demethylation agents such as 5-azacytidine (vidaza) or decitabine (5aza 2-deoxycytidine, dacogen) along with histone deacetylase inhibitors like vorinostat or romidepsin.15 TAM metabolites 4-OH TAM and N-desmethyl TAM (endoxifen) have a higher affinity than TAM for ER. Patients with defective alleles of CYP2D6, an enzyme involved in drug metabolism, are less responsive to TAM. In this regard, the mutation G1934A in the CYP2D6 splicing site resulting in loss of enzyme activity.3 Because the use of TAM, especially in association with chemotherapy, increases the risk of thromboembolic events, breast cancer patients should be screened before prescribing the drug.60,61 Although promising, targeted therapy might not be applicable to all patients; therefore the development of new agents based on pharmacogenomics and pharmacogenetics, resistance profile and maintenance of systemic hemostasis is needed. As highlighted above, drug resistance remains one of the most prominent clinical obstacles in breast cancer treatment. Studies have shown that 40–50% of ER+ breast cancer patients ultimately develop TAM-resistance. Thus, useful biomarkers are needed for the early diagnosis of these TAM-resistant patients.62,63
HER2 positive
HER2 amplification has been identified in >14% of metastatic breast cancer that associated with increased cell proliferation, angiogenesis, invasion and reduced apoptosis.64 It has been found that in HER2- tumors there are compensatory driver genes like BRF2 and DSN1, which undergo amplification or overexpression as oncogenes that bestow a neoplastic advantage.29 HER2+ patients are sensitive to HER2 antibodies and kinase inhibitors lapatinib, pertuzumab, trastuzumab, and ado-trastuzumab and emtansine.65 Trastuzumab (Herceptin™, Genentech/Roche, South San Francisco, CA, USA) was the first humanized monoclonal antibody against the domain IV of human epidermal receptor 2 to be developed.66 This antibody was the first to receive FDA approval as a targeted drug for breast cancer.67 In some clinical studies, a combination of trastuzumab with standard chemotherapy regimens resulted in better response rates than prescription chemotherapy alone.68–70 Subsequent studies, however, showed that some patients tend to develop resistance to therapy.71 The predominant mechanisms of resistance to trastuzumab appear to be related to the HER2 signaling pathway71,72 and be associated with activating mutations in PIK3CA, RAS, Src, NF-KB and inactivating mutations in PTEN, a negative regulator.73–75
Moreover, truncated isoforms of HER2, which lack trastuzumab target epitope,76 constitute an active source of HER2 generation due to the formation of stable HER2 homodimers.77,78 Overexpression of EGFR and HER-3 – the HER2 co-receptors – and their ligands,73 interaction with adhesion molecules such as MUC1-C or MUC479,80 and incorporation into heterologous receptors (HER-3) with HER2, are additional mechanisms of resistance to trastuzumab. Targeted strategies to overcome resistance to this drug include: 1) use of pan PI3K inhibitor, specific PIK3CA inhibitors, AKT inhibitors, and mTOR inhibitors when resistance results from PIK3CA alterations; 2) Lapatinib chemotherapy to overcome the high level of p95HER2; 3) Tyrosine kinase inhibitors or IGF1R monoclonal antibodies to overcome activation of IGF1R tyrosine kinase receptor; 4) MET inhibitors to overcome MET alterations (mutation and amplification); and 5) immune checkpoints inhibitors to overcome low immune response.81 Heterogeneity in HER2+ breast cancers was associated with failure of target therapy, for example, like in the case of HER2+ and luminal molecular subtype that also expresses ER. In this regard, clinical trials highlighted the variability in the efficacy of regimens based on the combination of trastuzumab and endocrine therapy.82 As depicted in Table 2, HER2+ patients display various molecular alterations and gene expression mutations that may impact prognosis and response to treatment.
Trastuzumab-DM1 (T-DM1: Genentech/Roche, South San Francisco, CA, USA) a novel monoclonal antibody that is conjugated with maytansine - a fungal toxin,80 requires a high level of HER2 expression on the targeted cells in order to be effective. Trastuzumab and its metabolites require accumulating in the cytosol of the target cancer cell to reach an optimum concentration and therefore induce cell death.83 In this context, it appears that low intra-tumor levels of HER2 and poor internalization of the HER2-drug, which is associated with insufficient intracellular expression of DM1, represent the primary mechanisms to drug resistance.84 CYD985 is another antibody-drug conjugate administered to and well tolerated in TDM1 pretreated patients.85 Lapatinib (Tykerb™, GlaxoSmithKline, Research Triangle Park, NC, USA) is a tyrosine kinase receptor inhibitor that inhibits autophosphorylation of both EGFR and HER2.86,87 Administration of lapatinib in combination with trastuzumab represents a strategy to target both intracellular and extracellular domains of HER2.88 However, the main issue related to lapatinib is the acquired resistance due to the Presence of HER2 overexpression in tumor cells which are primarily sensitive to lapatinib apoptotic effects.89 Activation of AXL, a membrane-bound RTK, constitutes another mechanism of resistance to lapatinib.90 Pertuzumab (Omnitarg™, Genentech/Roche, South San Francisco, CA, USA), a humanized monoclonal antibody that binds to an extracellular domain of HER2, different from that bound by trastuzumab, inhibits hetero and homo-dimerization of HER291 and could partially reverse resistance to trastuzumab in metastatic breast cancer patients.92 Administration of pertuzumab in combination with lapatinib could be a promising strategy to overcome lapatinib resistance, as demonstrated in an animal model. Pertuzumab inhibited NRG1-induced signaling that is activated 24 hrs following lapatinib administration.93 Ertumaxomab (Rexoman™, Fresenius Biotech, Hamburg, Germany) is an antibody that activates and aggregates T-cells, NK-cells, dendritic cells, and macrophages through the formation of the HER2-drug-CD3 complex.93 Although the drug is still being evaluated in clinical trials, it might represent a promising targeted therapy. The presence of PIK3CA mutation might account for resistance to anti-HER2 therapy in HER2+ tumors. Neratinib has received increasing interest in the treatment of recurrent and metastatic tumors characterized by non-amplifying HER2 alterations.94 Neratinib, a reversible tyrosine kinase inhibitor, can inhibit HER-1, HER2, and HER-4.95 In a study including 1,420 women with breast cancer, neratinib significantly improved disease-free survival by 12 months.96 In the presence of HER2 mutations, afatinib, another kinase inhibitor has also been used. Afatinib efficacy has been evaluated both in vivo and in vitro.97 The duration of afatinib effects is longer compared with other reversible EGFR inhibitors.98 In a recent study, administration of afatinib with letrozole to metastatic breast cancer patients resulted in stabilization of the disease in 54% of cases, who previously progressed while they were on letrozole therapy.99
Triple negative
Triple-negative cancers include three subtypes: normal like, basal-like and non-basal like; 75% of basal-like cancers are TN, and 80% of these TN breast cancer display P53 mutations (nonsense and frameshift).100 Basal and non-basal TN also present a different type of mutation, ie, the homologous recombination deficiency (HRD) and homologous recombination repair (HRR). Homologous recombination is the repair mechanism of double-strand DNA breaks (DSB). Several clinical trials on patients with TN cancers highlighted the importance of HRD genomic signature in the prediction of therapeutic response.100–102
The COSMIC database had identified the genes mainly involved in TN variations. These genes have been extensively studied and include TP53, BRCA1, PIK3CA, RB, and PTEN. In TN tumors, there is no correlation between TP53 and BRCA1 mutations,103 although the methylation rate of BRCA1 was found to correlate with the TP53 mutation.104
Increased copy number and mutations of the PIK3CA gene, loss of PTEN and INPP4B and overexpression of EGFR were found to activate the PI3K pathway. In the basal-like tumor, 72% of cases are RB-/P16+ and display p53 overexpression, which is correlated with high proliferation. Further, mutations in BRCA1, PTEN, ERBB2 genes were correlated with increased risk of metastasis.100 Genetic and epigenetic alterations in the DNA repair system may act as initial causes of cell transformation. Drugs targeting cells with a deficit in the DSB repair are highly relevant as anti-cancer agents. These are platinum-based compounds that lead to DSB.105 The PARP inhibitor is effective against cancer cells with a deficit in DNA repair of DSBs. This inhibitor blocks the activity of the PARP enzyme, of which PARP1 is an important target indicating the presence of single-strand breaks. Therefore, the inhibition of PARP1 leads to more “unrepaired” single-strand breaks. These breaks in replication forks lead to the formation of DSB that cannot be repaired in BRCA1 or BRCA2 defective cells, thereby leading to the accumulation of DSB and subsequent cellular death.15 There are similarities between BRCA1 mutated breast cancer and basal-like breast cancer. The TN cancer due to inherited BRCA1 mutation is the same as basal-like breast cancer in the presence of acquired BRCA1 mutations or other mutations in genes within same pathways. This phenotype has been found in the sporadic case of basal-like or TNBC. PARP inhibitors initially gave promising results in TN tumors; however, subsequent trials failed to confirm these findings. Therefore, it remains unclear which subset of TN or inherited BRCA1 or BRCA2 can benefit from treatment with the PARP inhibitors olaparib and iniparib.106,107 Genetic studies of TN and basal-like cancers have reported defects in BRCA1 resulting in half of TN tumors having acquired BRCA1 methylation. Preclinical studies indicate that the use of BRCA1 methylation might present a promising biomarker of response to PARP inhibitor.15 In the case of mutation or methylation impacting BRCA1 and BRCA2, defective cells could benefit from platinum-based agents like cisplatin. By crosslinking to DNA, this drug can target the lesions that are ineffectively repaired because of DSB formation. Mutations in the TP53 gene have been associated with resistance to cisplatin treatment.108 In TN subtype, mutations in the PI3K/AKT/mTOR pathway could be overcome by target therapy in combination with chemotherapy.5
Chemotherapy
One of the cornerstones in the treatment of disease, both when dealing with primary and metastatic breast cancer, is to administer medications systematically.108 Until now numerous classes of drugs used in the treatment of breast cancer have been given systematically, and these include anthracyclines such as doxorubicin, epirubicin, and mitoxantrone that have pleiotropic effects109 and induce cell death via a number of proposed mechanisms.110–112 Resistance to anthracylines is related to the increase in expression of P-170 glycoprotein that results in increased drug efflux.113 A number of approaches have been applied to overcome tumor resistance to anthracyclines, and these are based on the use of fostriecin, merbarone, aclarubicin and bis (2,6-dioxopiperazine) as novel inhibitors of topoisomerase II,114,115 altered topoisomerase II,116 use of non-cross-resistant drugs after administration of anthracyclines117,118 and changes in the route and time of drug delivery.119 Another class of drugs, the taxanes, includes paclitaxel, docetaxel, and nab-paclitaxel that bind to microtubules with high-affinity; inhibit mitosis and therefore disrupting cell division.120 Cancer cells manifest resistance to these drugs by changes in the expression levels of beta-tubulin isotopes and by upregulating caveolin-1.121 The effects of cyclosporine A, PC833 and verapamil have been evaluated with the view of overcoming resistance to this class of chemotherapeutic agents.122 New microtubule inhibitors like ixabepilone and eribulin were developed to overcome resistance against anthracyclines and taxanes; these were approved by FDA but not by EMA.123 Recently, irinotecan pegol (NKTR-102), a long-acting topoisomerase I inhibitor, has been launched as a new agent to overcome resistance related to anthracyclines and taxanes.124,125 A study reported that CPG island hypermethylation in the promoter of the GSTP1 gene was associated with a therapeutic response to doxorubicin.15 Antimetabolites such as methotrexate (MTX) and 5-fluorouracil (5-FU) inhibit dihydrofolate reductase (DHFR)126 and thymidylate synthase enzymes127 respectively. Resistance to MTX may be conferred by a decrease in drug uptake,128,129 increase in drug efflux,130,131 reduction in polyglutamation,132 increase in the level of DHFR133 and expression of Bcl2 during apoptosis.134 Resistance to 5-FU results from alterations in enzymes involved in the metabolism of fluoropyrimidine.135 Strategies have been evaluated to overcome resistance to antimetabolite drugs. These include manipulation of drug metabolism, using high-dose medications and the development of novel antimetabolites.136 Alkylating agents and platinum-based drugs alkylate DNA through the formation of reactive intermediates formation.109 Tumors can develop resistance to these drugs through decreased alterations linked to transmembrane cellular drug accumulation,137 increase in drug inactivation in the cytosol, increase in repair of damaged DNA138 and anti-apoptotic mechanisms.139 Changes in the regulators of apoptosis have been considered as an approach to overcome resistance observed with the existing drugs.140 Vinca alkaloids that comprise vincristine, vinblastine, vinorelbine, vindesine, vinflunine, and vinpocetine have clinical uses141 and are also susceptible to multidrug resistance.142 The mechanism of drug resistance in this class of drugs results from decreased drug accumulation and retention. In this case, methods to overcome resistance have considered administration of DNA polymerase alpha inhibitors like Gemcitabine.109
New challenges in precision medicine
Despite the progress achieved in targeted therapy, numerous unsolved challenges still exist. The lack of safe and effective drugs is an important issue that might impact the effective control of the disease. Even though the molecular mechanisms underlying the anticancer effects of some common drugs like metformin – an antihyperglycemic drug – remain to be fully understood, growing evidence suggests that diabetic patients who receive metformin exhibit lower rate of cancers, including breast cancer.143 A recent study investigating the effects of metformin on cells overexpressing the tumor suppressor microRNA (miR-200c), showed that the drug inhibits proliferation, migration, and invasion of cancer cells while augmenting the levels of miR-200c.144 The Authors suggested miR-200c as a potential target for the treatment of breast cancer patients. With the view of revealing the molecular mechanisms involved in the anticancer effects of metformin and to introduce novel therapeutic approaches, a recent proteomics analysis proposed developing further generations of metformin.145
On the other hand, some factors such as diet and psycho-social status should be considered in personalized medicine. The steroid hormone – vitamin D – which affects almost every cell in the human body plays a significant role in breast cancer prevention.146
Epidemiological studies have reported the role of vitamin D and vitamin D-binding protein-related genes in the association between vitamin D and the risk of the disease. There is mounting evidence indicating that psycho-social factors such as stress, depression, anxiety, and spiritual status markedly impact the development of breast cancer and response to treatment.147–149 It is plausible that the involved molecular mechanisms are used as a step towards precision medicine. Recent studies have shown the potential role for dopamine and serotonin in this context. Experimental research on Iranian women has shown that spiritual intervention causes a decrease in dopamine receptor gene expression in peripheral blood mononuclear cells (PBMCs) of breast cancer patients compared with a control group who did not receive the intervention.150 Another study on PBMCs of breast cancer patients revealed that serotonin receptor gene expression was increased compared to the healthy group.151 The authors proposed antagonists of serotonin receptors as a new therapeutic agent.
Microbiome profiling is one of the emerging tools in the clarification of the role of environmental factors in the etiology of breast cancer. Recent work indicates there are differences between bacteria present in the normal tumor-adjacent tissue of breast cancer patients and tissue from healthy women.152
The advents of modern diagnostics and therapeutics have provided significant advances compared with traditional medicine. Nowadays, novel biomarkers, therapeutic targets and data mining constitute essential parts of science. Hence, researchers spend a long time analyzing existing databases. Analytic and modeling tools accompanied by experimental modules have been key in providing a comprehensive and meticulous view of the interactions between cellular components in biological processes. This so-called 'systems biology'153 proves pivotal in implementing personalized approaches, especially therapeutics.
Acknowledgments
The studies reported in this publication were supported by Proteomics Research Centre of Shahid Beheshti University of Medical Sciences; there was no conflict of interest in relation to the support.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wang C, Machiraju R, Huang K. Breast cancer patient stratification using a molecular regularized consensus clustering method. Methods. 2014;67(3):304–312. doi:10.1016/j.ymeth.2014.03.005
2. Chen X, Shachter RD, Kurian AW, Rubin DL. Dynamic strategy for personalized medicine: an application to metastatic breast cancer. J Biomed Inform. 2017;68:50–57. doi:10.1016/j.jbi.2017.02.012
3. Nerenz RD. Pharmacogenomics and personalized medicine in the treatment of human diseases. In: Coleman WB, Tsongalis GJ, editors. Molecular pathology.
4. Li H, Jia W. Cometabolism of microbes and host: implications for drug metabolism and drug‐induced toxicity. Clin Pharmacol Ther. 2013;94(5):574–581. doi:10.1038/clpt.2013.157
5. Nandy A, Gangopadhyay S, Mukhopadhyay A. Individualizing breast cancer treatment—the dawn of personalized medicine. Exp Cell Res. 2014;320(1):1–11. doi:10.1016/j.yexcr.2013.09.002
6. Alomar MJ. Factors affecting the development of adverse drug reactions. Saudi Pharm J. 2014;22(2):83–94. doi:10.1016/j.jsps.2013.02.003
7. Dickmann LJ, Ware JA. Pharmacogenomics in the age of personalized medicine. Drug Discov Today Technol. 2016;21:11–16. doi:10.1016/j.ddtec.2016.11.003
8. Eroles P, Bosch A, Pérez-Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev. 2012;38(6):698–707. doi:10.1016/j.ctrv.2011.11.005
9. Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747. doi:10.1038/35020557
10. Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci. 2001;98(19):10869–10874. doi:10.1073/pnas.191367098
11. Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8(5):R76. doi:10.1186/gb-2007-8-5-r81
12. Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5(1):5–23. doi:10.1016/j.molonc.2010.11.003
13. Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2011;16(Supplement 1):61–70. doi:10.1634/theoncologist.2011-S1-61
14. Sørlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc National Acad Sci. 2003;100(14):8418–8423. doi:10.1073/pnas.0932692100
15. Stefansson OA, Esteller M. Epigenetic modifications in breast cancer and their role in personalized medicine. Am J Pathol. 2013;183(4):1052–1063. doi:10.1016/j.ajpath.2013.04.033
16. Gnant M, Harbeck N, St. Gallen TC. summary of the consensus discussion. Breast Care. 2011;6(2):136–141.
17. Del Mastro L, De Placido S, Bruzzi P, et al. Fluorouracil and dose-dense chemotherapy in adjuvant treatment of patients with early-stage breast cancer: an open-label, 2×2 factorial, randomised phase 3 trial. Lancet. 2015;385(9980):1863–1872. doi:10.1016/S0140-6736(14)62048-1
18. Creighton CJ. The molecular profile of luminal B breast cancer. Biologics. 2012;6:289.
19. Rouzier R, Perou CM, Symmans WF, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11(16):5678–5685. doi:10.1158/1078-0432.CCR-04-2421
20. Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61. doi:10.1038/nature11412
21. Colozza M, de Azambuja E, Cardoso F, Bernard C, Piccart MJ. Breast cancer: achievements in adjuvant systemic therapies in the pre-genomic era. Oncologist. 2006;11(2):111–125. doi:10.1634/theoncologist.11-2-111
22. Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the carolina breast cancer study. JAMA. 2006;295(21):2492–2502. doi:10.1001/jama.295.21.2492
23. Rakha EA, El-Rehim DA, Paish C, et al. Basal phenotype identifies a poor prognostic subgroup of breast cancer of clinical importance. Eur J Cancer. 2006;42(18):3149–3156. doi:10.1016/j.ejca.2006.08.015
24. Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10(16):5367–5374. doi:10.1158/1078-0432.CCR-04-0220
25. Weigelt B, Mackay A, A‘Hern R, et al. Breast cancer molecular profiling with single sample predictors: a retrospective analysis. Lancet Oncol. 2010;11(4):339–349. doi:10.1016/S1470-2045(10)70008-5
26. Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi:10.1186/bcr2722
27. Cadoo KA, Traina TA, King TA. Advances in molecular and clinical subtyping of breast cancer and their implications for therapy. Surg Oncol Clin. 2013;22(4):823–840. doi:10.1016/j.soc.2013.06.006
28. Fang L, Barekati Z, Zhang B, Liu Z, Zhong X. Targeted therapy in breast cancer: what’s new. Swiss Med Wkly. 2011;141:w13231.
29. Ng CK, Martelotto LG, Gauthier A, et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 2015;16(1):107. doi:10.1186/s13059-015-0667-4
30. Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12(7):381. doi:10.1038/nrclinonc.2015.73
31. Tsai H-F, Trubelja A, Shen AQ, Bao G. Tumour-on-a-chip: microfluidic models of tumour morphology, growth and microenvironment. J R Soc Interface. 2017;14(131):20170137. doi:10.1098/rsif.2017.0137
32. Ahn J, Sei Y, Jeon N, Kim Y. Tumor microenvironment on a chip: the progress and future perspective. Bioengineering. 2017;4(3):64. doi:10.3390/bioengineering4020044
33. Peters ML, Garber JE, Tung N. Managing hereditary breast cancer risk in women with and without ovarian cancer. Gynecol Oncol. 2017;146(1):205–214. doi:10.1016/j.ygyno.2017.04.013
34. Kristiansen S, Nielsen D, Sölétormos G. Detection and monitoring of hypermethylated RASSF1A in serum from patients with metastatic breast cancer. Clin Epigen. 2016;8(1):35. doi:10.1186/s13148-016-0199-0
35. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
36. Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP 2A: exploiting the therapeutic potential of this phosphatase. FEBS J. 2016;283(6):1004–1024. doi:10.1111/febs.13573
37. Lau QC, Raja E, Salto-Tellez M, et al. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Res. 2006;66(13):6512–6520. doi:10.1158/0008-5472.CAN-06-0369
38. Pietras RJ, Márquez-Garbán DC. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin Cancer Res. 2007;13(16):4672–4676. doi:10.1158/1078-0432.CCR-07-1373
39. Russell CA. Personalized medicine for breast cancer: it is a new day! Am J Surg. 2014;207(3):321–325. doi:10.1016/j.amjsurg.2013.10.016
40. Barone I, Brusco L, Fuqua SA. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin Cancer Res. 2010;15;16(10):1078–1432. CCR-09-1753.
41. De Abreu FB, Wells WA, Tsongalis GJ. The emerging role of the molecular diagnostics laboratory in breast cancer personalized medicine. Am J Pathol. 2013;183(4):1075–1083. doi:10.1016/j.ajpath.2013.07.002
42. Yanagawa T, Kagara N, Miyake T, et al. Detection of ESR1 mutations in plasma and tumors from metastatic breast cancer patients using next-generation sequencing. Breast Cancer Res Treat. 2017;163(2):231–240. doi:10.1007/s10549-017-4190-z
43. Mayer IA. Advanced hormone-sensitive breast cancer: overcoming resistance. J Natl Compr Canc Netw. 2015;13(5S):655–657.
44. Downey C, Simpkins S, White J, et al. The prognostic significance of tumour–stroma ratio in oestrogen receptor-positive breast cancer. Br J Cancer. 2014;110(7):1744. doi:10.1038/bjc.2014.69
45. Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, et al. Clinical significance of monitoring ESR1 mutations in circulating cell-free DNA in estrogen receptor positive breast cancer patients. Oncotarget. 2016;7(22):32504. doi:10.18632/oncotarget.8839
46. Tabarestani S, Motallebi M, Akbari ME. Are estrogen receptor genomic aberrations predictive of hormone therapy response in breast cancer? Iran J Cancer Prev. 2016;9:4. doi:10.17795/ijcp
47. Segal CV, Dowsett M. Estrogen receptor mutations in breast cancer—new focus on an old target. Clin Cancer Res. 2014;20(7):1724–1726. doi:10.1158/1078-0432.CCR-14-0067
48. Fribbens C, O‘Leary B, Kilburn L, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol. 2016;34:2961–2968.
49. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425–439. doi:10.1016/S1470-2045(15)00613-0
50. Lauring J, Wolff AC. Evolving role of the estrogen receptor as a predictive biomarker: ESR1 mutational status and endocrine resistance in breast cancer. J Clin Oncol. 2016;34(25):2950–2952. doi:10.1200/JCO.2016.68.4720
51. Gelsomino L, Gu G, Rechoum Y, et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res Treat. 2016;157(2):253–265. doi:10.1007/s10549-016-3829-5
52. Fuqua SA, Gu G, Rechoum Y. Estrogen receptor (ER) α mutations in breast cancer: hidden in plain sight. Breast Cancer Res Treat. 2014;144(1):11–19. doi:10.1007/s10549-014-2847-4
53. Fu X, Creighton CJ, Biswal NC, et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. 2014;16(5):430. doi:10.1186/s13058-014-0492-9
54. Rimawi MF, Wiechmann LS, Wang Y-C, et al. Reduced dose and intermittent treatment with lapatinib and trastuzumab for potent blockade of the HER pathway in HER2/neu-overexpressing breast tumor xenografts. Clin Cancer Res. 2011;17(6):1351–1361. doi:10.1158/1078-0432.CCR-10-1905
55. Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, et al. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget. 2017;8(32):52142. doi:10.18632/oncotarget.18479
56. Van Loo P, Wedge D, Nik-Zainal S, Stratton M, Futreal P, Campbell P. 5 proffered paper: the life history of 21 breast cancers. Eur J Cancer. 2012;48:S2. doi:10.1016/S0959-8049(12)70709-8
57. Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, et al. Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Transl Res. 2015;166(6):540–53. e2. doi:10.1016/j.trsl.2015.09.003
58. Perez EA. Treatment strategies for advanced hormone receptor-positive and human epidermal growth factor 2-negative breast cancer: the role of treatment order. Drug Resist Update. 2016;24:13–22. doi:10.1016/j.drup.2015.11.001
59. Zanardi E, Bregni G, De Braud F, Di Cosimo S, editors. Better together: targeted combination therapies in breast cancer. Semin Oncol. 2015. Elsevier. doi:10.1053/j.seminoncol.2015.09.029
60. Garber JE, Halabi S, Tolaney SM, et al. Factor V Leiden mutation and thromboembolism risk in women receiving adjuvant tamoxifen for breast cancer. J Natl Cancer Inst. 2010;102(13):942–949. doi:10.1093/jnci/djq211
61. Onitilo AA, McCarty CA, Wilke RA, et al. Estrogen receptor genotype is associated with risk of venous thromboembolism during tamoxifen therapy. Breast Cancer Res Treat. 2009;115(3):643–650. doi:10.1007/s10549-008-0264-2
62. Conway K, Parrish E, Edmiston SN, et al. The estrogen receptor-α A908G (K303R) mutation occurs at a low frequency in invasive breast tumors: results from a population-based study. Breast Cancer Res. 2005;7(6):R871. doi:10.1186/bcr949
63. Roodi N, Bailey LR, Kao W-Y, et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. Jnci. 1995;87(6):446–451. doi:10.1093/jnci/87.6.446
64. Weinreb I, Piscuoglio S, Martelotto LG, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014;46(11):1166. doi:10.1038/ng.2895
65. Ross JS, Gay LM, Wang K, et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: an emerging opportunity for anti‐HER2 targeted therapies. Cancer. 2016;122(17):2654–2662. doi:10.1002/cncr.30102
66. Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc National Acad Sci. 1992;89(10):4285–4289. doi:10.1073/pnas.89.10.4285
67. Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11(2):263–275. doi:10.1586/era.10.226
68. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi:10.1056/NEJM200103153441101
69. Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–726. doi:10.1200/JCO.2002.20.3.719
70. Jahanzeb M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer. 2008;8(4):324–333. doi:10.3816/CBC.2008.n.037
71. Nahta R, Esteva F. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26(25):3637. doi:10.1038/sj.onc.1210379
72. Bedard PL, de Azambuja E, Cardoso F. Beyond trastuzumab: overcoming resistance to targeted HER-2 therapy in breast cancer. Curr Cancer Drug Targets. 2009;9(2):148–162.
73. Rimawi MF, Schiff R, Osborne CK. Targeting HER2 for the treatment of breast cancer. Annu Rev Med. 2015;66:111–128.
74. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9(1):16. doi:10.1038/nrclinonc.2012.154
75. Yamaguchi H, Chang S, Hsu J, Hung M. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene. 2014;33(9):1073. doi:10.1038/onc.2013.74
76. Arribas J, Baselga J, Pedersen K, Parra-Palau JL. p95HER2 and breast cancer. Cancer Res. 2011;71(5):1515–1519. doi:10.1158/0008-5472.CAN-10-3795
77. Castiglioni F, Tagliabue E, Campiglio M, Pupa S, Balsari A, Menard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006;13(1):221–232. doi:10.1677/erc.1.01047
78. Mitra D, Brumlik MJ, Okamgba SU, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–2162. MCT-09-0295.
79. Funes M, Miller JK, Lai C, Carraway KL, Sweeney C. The mucin Muc4 potentiates neuregulin signaling by increasing the cell-surface populations of ErbB2 and ErbB3. J Biol Chem. 2006;281(28):19310–19319. doi:10.1074/jbc.M603225200
80. Price‐Schiavi SA, Jepson S, Li P, et al. Rat Muc4 (sialomucin complex) reduces binding of anti‐ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer. 2002;99(6):783–791. doi:10.1002/ijc.10410
81. de Melo Gagliato D, Jardim DLF, Marchesi MSP, Hortobagyi GN. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget. 2016;7(39):64431.
82. Ferrari A, Vincent-Salomon A, Pivot X, et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat Commun. 2016;7:12222. doi:10.1038/ncomms12222
83. Kovtun YV, Goldmacher VS. Cell killing by antibody–drug conjugates. Cancer Lett. 2007;255(2):232–240. doi:10.1016/j.canlet.2007.04.010
84. Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014;16(2):209. doi:10.1186/s13058-014-0492-9
85. Van Herpen C, Banerji U, Mommers E, et al. 333 Phase I dose-escalation trial with the DNA-alkylating anti-HER2 antibody-drug conjugate SYD985. Eur J Cancer. 2015;51:S65. doi:10.1016/S0959-8049(16)30197-6
86. Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 2008;30(8):1426–1447. doi:10.1016/j.clinthera.2008.08.008
87. Tevaarwerk AJ, Kolesar JM. Lapatinib: A small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer. Clin Ther. 2009;31:2332–2348. doi:10.1016/j.clinthera.2009.11.029
88. Konecny GE, Pegram MD, Venkatesan N, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–1639. doi:10.1158/0008-5472.CAN-05-1182
89. Vicario R, Peg V, Morancho B, et al. Patterns of HER2 gene amplification and response to anti-HER2 therapies. PLoS One. 2015;10(6):e0129876. doi:10.1371/journal.pone.0129876
90. Hafizi S, Dahlbäck B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev. 2006;17(4):295–304. doi:10.1016/j.cytogfr.2006.04.004
91. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, De Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5(4):317–328.
92. Agus DB, Gordon MS, Taylor C, et al. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J clin oncol. 2005;23(11):2534–2543. doi:10.1200/JCO.2005.03.184
93. Leung W-Y, Roxanis I, Sheldon H, et al. Combining lapatinib and pertuzumab to overcome lapatinib resistance due to NRG1-mediated signalling in HER2-amplified breast cancer. Oncotarget. 2015;6(8):5678. doi:10.18632/oncotarget.3296
94. Hyman D, Piha-Paul S, Rodón J, et al. editors. Neratinib for ERBB2 mutant, HER2 non-amplified, metastatic breast cancer: preliminary analysis from a multicenter, open-label, multi-histology phase II basket trial. Cancer Res. 2016. AMER ASSOC CANCER RESEARCH 615 CHESTNUT ST, 17TH FLOOR, PHILADELPHIA, PA …. doi:10.1158/1538-7445.SABCS15-PD5-05
95. Subramaniam D, He A R, Hwang J, et al. Irreversible multitargeted ErbB family inhibitors for therapy of lung and breast cancer. Curr Cancer Drug Targets. 2014;14(9):775–793. doi:10.2174/1568009614666141111104643
96. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(3):367–377. doi:10.1016/S1470-2045(15)00551-3
97. Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702. doi:10.1038/onc.2008.109
98. Hurvitz SA, Shatsky R, Harbeck N. Afatinib in the treatment of breast cancer. Expert Opin Investig Drugs. 2014;23(7):1039–1047. doi:10.1517/13543784.2014.924505
99. Gunzer K, Joly F, Ferrero J-M, et al. A phase II study of afatinib, an irreversible ErbB family blocker, added to letrozole in patients with estrogen receptor-positive hormone-refractory metastatic breast cancer progressing on letrozole. Springerplus. 2016;5(1):45. doi:10.1186/s40064-015-1601-7
100. Judes G, Rifaï K, Daures M, et al. High-throughput «Omics» technologies: new tools for the study of triple-negative breast cancer. Cancer Lett. 2016;382(1):77–85. doi:10.1016/j.canlet.2016.03.001
101. Telli ML, Hellyer J, Audeh W, et al. Homologous recombination deficiency (HRD) status predicts response to standard neoadjuvant chemotherapy in patients with triple-negative or BRCA1/2 mutation-associated breast cancer. Breast Cancer Res Treat. 2018;168(3):625–630. doi:10.1007/s10549-017-4624-7
102. Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395. doi:10.1038/nature10933
103. Foedermayr M, Sebesta M, Rudas M, et al. BRCA-1 methylation and TP53 mutation in triple-negative breast cancer patients without pathological complete response to taxane-based neoadjuvant chemotherapy. Cancer Chemother Pharmacol. 2014;73(4):771–778. doi:10.1007/s00280-014-2404-1
104. Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006;8(4):R38. doi:10.1186/bcr1522
105. Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864. doi:10.1038/sj.onc.1209874
106. O‘Shaughnessy J, Schwartzberg L, Danso M, et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC). J Clin Oncol. 2011;29(15_suppl):1007. doi:10.1200/jco.2011.29.15_suppl.1007
107. Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH. Failure of iniparib to inhibit poly (ADP-Ribose) polymerase in vitro. Clin Cancer Res. 2012;15;18(6):1655–1662.
108. Reles A, Wen WH, Schmider A, et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001;7(10):2984–2997.
109. Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Breast Cancer Chemosensitivity. 2007;608:1–22.
110. Gewirtz D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–741. doi:10.1016/S0006-2952(98)00307-4
111. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229. doi:10.1124/pr.56.2.6
112. Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism—a strategy for overcoming drug resistance. J Natl Cancer Inst. 2001;93(5):347–357.
113. Chen G, J-P J, Fleming WH, Durán GE, Sikic BI. Prevalence of multidrug resistance related to activation of the mdr1 gene in human sarcoma mutants derived by single-step doxorubicin selection. Cancer Res. 1994;54(18):4980–4987.
114. Larsen AK, Skladanowski A. Cellular resistance to topoisomerase-targeted drugs: from drug uptake to cell death. Biochim Biophys Acta. 1998;1400(1–3):257–274. doi:10.1016/S0167-4781(98)00140-7
115. Withoff S, De SJ, De EV, Mulder N. Human DNA topoisomerase II: biochemistry and role in chemotherapy resistance. Anticancer Res. 1996;16(4A):1867–1880.
116. Finlay GJ, Baguley BC, Snow K, Judd W. Multiple patterns of resistance of human leukemia cell sublines to amsacrine analogues. Jnci. 1990;82(8):662–667. doi:10.1093/jnci/82.8.662
117. Seidman AD, Reichman BS, Crown J, et al. Paclitaxel as second and subsequent therapy for metastatic breast cancer: activity independent of prior anthracycline response. J Clin Oncol. 1995;13(5):1152–1159. doi:10.1200/JCO.1995.13.5.1152
118. Wilson WH, Berg SL, Bryant G, et al. Paclitaxel in doxorubicin-refractory or mitoxantrone-refractory breast cancer: a phase I/II trial of 96 hr infusion. J Clin Oncol. 1994;12(8):1621–1629. doi:10.1200/JCO.1994.12.8.1621
119. Anderson H, Hopwood P, Prendiville J, Radford JA, Thatcher N, Ashcroft L. A randomised study of bolus vs continuous pump infusion of ifosfamide and doxorubicin with oral etoposide for small cell lung cancer. Br J Cancer. 1993;67(6):1385. doi:10.1038/bjc.1993.256
120. Bristol-Myers Squibb. Taxol® (paclitaxel) [Prescribing information]. New York: Bristol-Myers Squibb; 2011.
121. Greenberger L, Williams SS, Horwitz SB. Biosynthesis of heterogeneous forms of multidrug resistance-associated glycoproteins. J Biol Chem. 1987;262(28):13685–13689.
122. Tolcher A, Cowan K, Solomon D, et al. Phase I crossover study of paclitaxel with r-verapamil in patients with metastatic breast cancer. J clin oncol. 1996;14(4):1173–1184. doi:10.1200/JCO.1996.14.4.1173
123. Twelves C, Jove M, Gombos A, Awada A. Cytotoxic chemotherapy: still the mainstay of clinical practice for all subtypes metastatic breast cancer. Crit Rev Oncol Hematol. 2016;100:74–87. doi:10.1016/j.critrevonc.2016.01.021
124. Jameson GS, Hamm JT, Weiss GJ, et al. A multicenter, phase I, dose-escalation study to assess the safety, tolerability, and pharmacokinetics of etirinotecan pegol in patients with refractory solid tumors. Clin Cancer Res. 2013;19(1):268–278. doi:10.1158/1078-0432.CCR-12-1201
125. Hoch U, Staschen C-M, Johnson RK, Eldon MA. Nonclinical pharmacokinetics and activity of etirinotecan pegol (NKTR-102), a long-acting topoisomerase 1 inhibitor, in multiple cancer models. Cancer Chemother Pharmacol. 2014;74(6):1125–1137. doi:10.1007/s00280-014-2577-7
126. Huennekens F. The methotrexate story: a paradigm for development of cancer chemotherapeutic agents. Adv Enzyme Regul. 1994;34:397–419.
127. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330. doi:10.1038/nrc1074
128. Grant SC, Kris MG, Young CW, Sirotnak FM. Edatrexate, an antifolate with antitumor activity: a review. Cancer Invest. 1993;11(1):36–45.
129. Sirotnak F, Moccio D, Kelleher L, Goutas L. Relative frequency and kinetic properties of transport-defective phenotypes among methotrexate-resistant L1210 clonal cell lines derived in vivo. Cancer Research. 1981;41(11 Pt 1):4447–4452.
130. Kool M, Van Der Linden M, de Haas M, et al. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc National Acad Sci. 1999;96(12):6914–6919. doi:10.1073/pnas.96.12.6914
131. Hooijberg J, Broxterman H, Scheffer G, et al. Potent interaction of flavopiridol with MRP1. Br J Cancer. 1999;81(2):269. doi:10.1038/sj.bjc.6690687
132. Cowan KH, Jolivet J. A methotrexate-resistant human breast cancer cell line with multiple defects, including diminished formation of methotrexate polyglutamates. J Biol Chem. 1984;259(17):10793–10800.
133. Volk EL, Rohde K, Rhee M, et al. Methotrexate cross-resistance in a mitoxantrone-selected multidrug-resistant MCF7 breast cancer cell line is attributable to enhanced energy-dependent drug efflux. Cancer Res. 2000;60(13):3514–3521.
134. Ohmori T, Podack E, Nishio K, et al. Apoptosis of lung cancer cells caused by some anti-cancer agents (MMC, CPT-11, ADM) is inhibited by bcl-2. Biochem Biophys Res Commun. 1993;192(1):30–36.
135. Priest DG, Ledford BE, Doig MT. Increased thymidylate synthetase in 5-fluorodeoxyuridine resistant cultured hepatoma cells. Biochem Pharmacol. 1980;29(11):1549–1553.
136. Spears CP. Clinical resistance to antimetabolites. Hematol Oncol Clin North Am. 1995;9(2):397–414.
137. Klatt O, Stehlin JS, McBride C, Griffin A. The effect of nitrogen mustard treatment on the deoxyribonucleic acid of sensitive and resistant Ehrlich tumor cells. Cancer Res. 1969;29(2):286–290.
138. Ichiro N, Kimitoshi K, Junko K, et al. Analysis of structural features of dihydropyridine analogs needed to reverse multidrug resistance and to inhibit photoaffinity labeling of P-glycoprotein. Biochem Pharmacol. 1989;38(3):519–527.
139. Zamble DB, Lippard SJ. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem Sci. 1995;20(10):435–439.
140. Perez R. Cellular and molecular determinants of cisplatin resistance. Eur J Cancer. 1998;34(10):1535–1542.
141. Moudi M, Go R, Yien CYS, Nazre M. Vinca alkaloids. Int J Prev Med. 2013;4(11):1231.
142. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi:10.1016/j.addr.2012.09.037
143. Coyle C, Cafferty F, Vale C, Langley R. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol. 2016;27(12):2184–2195. doi:10.1093/annonc/mdw410
144. Zhang J, Li G, Chen Y, et al. Metformin Inhibits tumorigenesis and tumor growth of breast cancer cells by upregulating miR-200c but downregulating AKT2 expression. J Cancer. 2017;8(10):1849. doi:10.7150/jca.19858
145. Al-Zaidan L, Ruz E, Abu R, Malki AM. Screening novel molecular targets of metformin in breast cancer by proteomic approach. Front Public Health. 2017;5:277. doi:10.3389/fpubh.2017.00081
146. Obaidi J, Musallam E, Al-Ghzawi HM, Azzeghaiby SN, Alzoghaibi IN. Vitamin D and its relationship with breast cancer: an evidence based practice paper. Glob J Health Sci. 2015;7(1):261.
147. Gall TL, Kristjansson E, Charbonneau C, Florack P. A longitudinal study on the role of spirituality in response to the diagnosis and treatment of breast cancer. J Behav Med. 2009;32(2):174–186. doi:10.1007/s10865-008-9182-3
148. Chida Y, Hamer M, Wardle J, Steptoe A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat Rev Clin Onco. 2008;5(8):466. doi:10.1038/ncponc1134
149. MacArthur AC, Le ND, Abanto ZU, Gallagher RP. Occupational female breast and reproductive cancer mortality in British Columbia, Canada, 1950–94. Occup Med (Chic Ill). 2007;57(4):246–253. doi:10.1093/occmed/kqm002
150. Akbari ME, Kashani FL, Ahangari G, et al. The effects of spiritual intervention and changes in dopamine receptor gene expression in breast cancer patients. Breast Cancer. 2016;23(6):893–900. doi:10.1007/s12282-015-0658-z
151. Hejazi SH, Ahangari G, Pornour M, et al. Evaluation of gene expression changes of serotonin receptors, 5-HT3AR and 5-HT2AR as main stress factors in breast cancer patients. Asian Pac J Cancer Prev. 2013;15(11):4455–4458. doi:10.7314/APJCP.2014.15.11.4455
152. Urbaniak C, Gloor GB, Brackstone M, Scott L, Tangney M, Reid G. The microbiota of breast tissue and its association with tumours. Appl Environ Microbiol. 2016;AEM:01235–16.
153. Toga AW, Foster I, Kesselman C, et al. Big biomedical data as the key resource for discovery science. JAMIA. 2015;22(6):1126–1131. doi:10.1093/jamia/ocv077
154. Kanehisa M, Goto S. KEGG: Breast cancer - Reference pathway; 2018. Available at:
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.