Back to Archived Journals » Research and Reports in Neonatology » Volume 10
Perlman Syndrome with Deletion of DIS3L2 Gene
Authors Salameh K ![]() , Viswanathan B
, Viswanathan B ![]() , Nawaz Z, Habboub L
, Nawaz Z, Habboub L ![]() , Tomerak A, Pattuvalappil R
, Tomerak A, Pattuvalappil R ![]()
Received 22 July 2020
Accepted for publication 7 October 2020
Published 23 October 2020 Volume 2020:10 Pages 89—93
DOI https://doi.org/10.2147/RRN.S270490
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Robert Schelonka
Khalil Salameh,1 Brijroy Viswanathan,1 Zafar Nawaz,2 Lina Habboub,1 Ahmed Tomerak,1 Rajesh Pattuvalappil1
1Department of Pediatrics and Neonatology, Al Wakra Hospital, Hamad Medical Corporation, Al Wakra, Qatar; 2Department of Cytogenetics and Molecular Cytogenetics Laboratory, Hamad Medical Corporation, Doha, Qatar
Correspondence: Khalil Salameh
Department of Pediatrics and Neonatology, Al Wakra Hospital, Hamad Medical Corporation, Al Wakra, Qatar
Tel +974 40114258
Email [email protected]
Abstract: Perlman syndrome is a rare genetic disorder with autosomal recessive inheritance. It is caused by deletion of the DIS3L2 gene on the long arm of chromosome 2. Though more than 30 cases have been reported in the literature with clinical features of the syndrome, very few cases have identified underlying genetic defects. The clinical features of Perlman syndrome have similarities with those of Beckwith–Wiedemann syndrome (BWS), prune belly syndrome (PBS) and Simpson–Golabi–Behmel syndrome (SGBS1). Affected patients have macrosomia, dysmorphic features, hypotonia, laxity of abdominal wall, cryptorchidism, renal anomalies and risk of development of Wilm’s tumour. Here we report a neonate with typical clinical features of Perlman syndrome, with deletion of DIS3L2 gene confirmed by array comparative genomic hybridization (aCGH).
Keywords: DIS3L2, Perlman syndrome, prune belly syndrome, Wilm’s tumour
Background
Perlman syndrome was first described in 1970 as a rare overgrowth syndrome with autosomal recessive inheritance.1,2 Affected neonates can present prenatally or at birth with macrosomia, hypotonia and dysmorphic features. Renal involvement may present with polyhydramnios and renal anomalies. Neonates with respiratory presentation show high mortality, often associated with renal involvement. The dysmorphic features and macrosomia show resemblance to BWS, PBS and SGBS1 syndromes. Mutation in the DIS3L2 gene on chromosome 2q37, causing Perlman syndrome, was reported by Astuti et al in 2012.3 Subsequently, few more cases of Perlman syndrome were reported with the deletion of the DIS3L2 gene and further detailing of its role in RNA metabolism.4,5 Perlman syndrome is associated with a predisposition to Wilm’s tumor. The earliest reported Wilm’s tumor was observed by 4 days of age and the mean age was 19 months.6 Diagnosis can be suspected clinically with typical clinical features and can be confirmed with chromosomal microarray, quantitative real-time PCR (qPCR) or next-generation sequencing.7
Case Presentation
Our patient was a male infant born at 39 weeks to a 27-year-old, Gravida 3, Para 2 mother. Parents of Pakistani origin were nonconsanguineous. Previous 2 siblings were healthy. The antenatal period was unremarkable, including the antenatal scans and GBS screening. The mother had spontaneous rupture of membranes and the newborn baby was delivered by normal vaginal method. The amniotic fluid was meconium-stained. Apgar scores were 3 and 9 at 1 and 5 minutes, respectively. The cord gases were unremarkable.
The growth centiles were weight at 85th, length at 97th and head circumference at 50th centiles. The newborn was noted to have dysmorphic features (Figures 1 and 2) such as depressed nasal bridge, hypertelorism and deep-set eyes, low-set ears, V-shaped upper lip, pectus excavatum, lax abdomen, bilateral undescended testes, generalized hypotonia, bilateral rocker bottom feet, and restricted abduction at the hips. There was apparent wrist drop but no contractures were noted. The deep tendon reflexes and neonatal reflexes were elicitable.
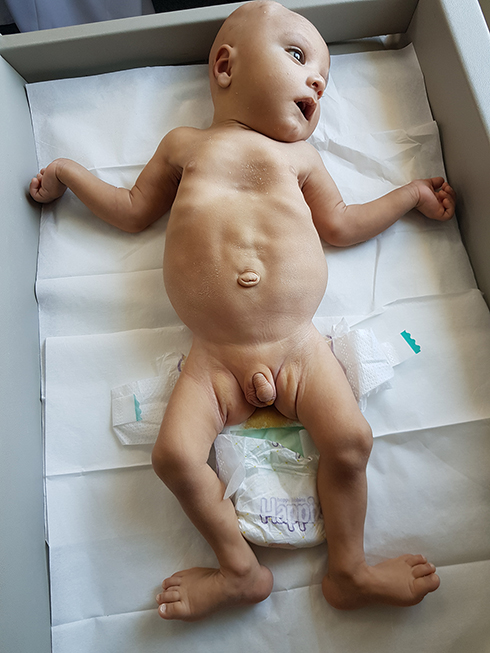 |
Figure 1 Pectus excavatum, lax abdomen, bilateral cryptorchidism and rocker bottom feet. |
 |
Figure 2 Dysmorphic features – Hypertelorism, deep-seated eyes and depressed nasal bridge. |
Investigations
Complete blood count and biochemical profile did not show any abnormality except for mild hypoglycemia. Blood gases and chest X-ray were unremarkable. Abdomen ultrasound detected both testes at the inguinal region with normal kidneys and urinary tract. Brain ultrasound did not show any abnormality. Echocardiography did not show structural anomalies of the heart. Chromosomal microarray revealed homozygous loss of ~104 kilobases of the long arm of chromosome 2 within the cytogenetic band 2q37.1, resulting in the deletion of the DIS3L2 gene (Figure 3).
 |
Figure 3 Array comparative genomic hybridization analysis revealed two-copy loss of the long arm of chromosome 2 within the cytogenetic band 2q37. The deletion was ~204 kilobases (kb) in size and caused intragenic deletion in the DIS3L2 gene. |
Differential Diagnosis
Perlman syndrome has phenotypic similarities with other overgrowth syndromes such as Beckwith–Wiedemann syndrome and Simpson–Golabi–Behmel syndrome (SGBS1). The dysmorphic facies with undescended testes and laxity of abdomen resemble the symptoms of prune belly syndrome too.
BWS occurs due to the mutation or deletion of imprinted genes within the chromosome 11p15.5 region. Affected neonates are macrosomic at birth but may have variable presentations. Hypoglycemia is reported in 30% to 50% of babies with BWS. The affected babies are also known to have abdominal wall defects, such as omphalocele, and a predisposition to develop embryological malignancies, such as Wilm’s tumor and hepatoblastoma.
Simpson–Golabi–Behmel syndrome type 1 (SGBS1- 312870) is inherited as an X-linked condition characterized by pre- and post-natal overgrowth, coarse facies, and congenital abnormalities including congenital heart defects. SGBS1 is caused by mutation or deletion in the gene encoding glypican–3 (GPC3) on chromosome Xq26.
Prune belly syndrome (PBS - 100100) occurs due to homozygous mutation in the CHRM3 gene on chromosome 1q43. The symptoms include megacystis (massively enlarged bladder) with disorganized detrusor muscle, cryptorchidism, and thin abdominal musculature with overlying lax skin.
Outcome and Follow-Up
During the neonatal period, the baby had mild tachypnoea and needed nasal cannula oxygen for its first 2 days of life. Mild asymptomatic hypoglycemia was also noted which needed 10% dextrose infusion for the first 48 hours of the baby’s life. The baby was slow to oral-feed and needed NGT (nasogastric tube) feeding for its first 5 days of life. Oral feeding improved gradually, and the baby was discharged in a stable condition after 5 days. During the post-natal follow-up after 1 month of age, the baby was not thriving. Its weight was just below the 50th centile, length at the 5th centile and head circumference at the 10th centile. A follow-up abdomen ultrasound at two months of age showed a bulky left kidney and small cortical cysts on both kidneys (Figures 4 and 5). The baby was kept under close monitoring for the risk of Wilm’s tumor and follow-up was arranged with a multidisciplinary team.
 |
Figure 4 Left kidney (bulky: 6.6 x 2.2 cm) with multiple tiny cortical cysts during follow up. |
 |
Figure 5 Right kidney (5.4 x 2.2 cm) with multiple small cortical cysts during follow up. |
Discussion
Perlman syndrome is a rare genetic disorder, and over 30 cases have been reported in the literature. Liban and Kozenitzky in 1970 and Perlman et al in 1973 first described the clinical features of Perlman syndrome.1,2 Alessandri et al summarized the clinical features of 28 patients reported in the world literature.8 The genetic diagnosis remained elusive until Astuti et al identified germline mutations in DIS3L2 on chromosome 2q37.1 and described its critical role in RNA metabolism, regulation of cell growth and division.3
Affected children are noted at birth due to the dysmorphic features and macrosomia. The characteristic facial dysmorphism includes prominent forehead, deep-set eyes, broad and flat nasal bridge, inverted V-shaped upper lip and low-set ears. These neonates are hypotonic with laxity of abdomen, cryptorchidism and renal anomalies. Prenatal suspicion and diagnosis is possible with the findings of macrosomia, polyhydramnios and renal anomalies or nephromegaly.9 High neonatal mortality has been reported and >53% die within 28 days from birth. The high mortality has been linked to renal involvement and associated pulmonary disease. The high predisposition to the development of Wilm’s tumor is widely reported and the earliest reported Wilms tumor was observed by 4 days of age with a mean age of 19 months.6 So it is recommended to monitor these cases with abdomen ultrasound every 3 months.6 Though the majority of cases have high mortality and poor neurodevelopmental outcome, there are reports of a case of Perlman syndrome that recovered from Wilm’s tumour and survived for more than 9 years with sound neurological outcome.10
Due to the similarities with overgrowth syndromes and prune belly syndrome, clinical diagnosis could be delayed.6 Detection of typical genetic abnormality by an appropriate genetic test can help in early diagnosis and therefore management.
In our case, advanced growth parameters and dysmorphic features with hypoglycemia suggested Beckwith–Wiedemann syndrome, but the bilateral undescended testes, hypotonia, rocker bottom feet and lack of macroglossia suggested otherwise. The lax abdomen and cryptorchidism raised the suspicion of prune belly syndrome, but normal renal system ruled it out. The microarray finding of deletion of the DIS3L2 gene on chromosome 2q37.1 helped to confirm the clinical diagnosis of Perlman syndrome.
The limited abduction of hips and rocker bottom feet are not described in the literature but can be explained by the paucity of fetal movements due to hypotonia. Our newborn patient had poor feeding and mild hypoglycemia in the initial few days, but it improved later. The high early neonatal mortality published in the literature could be related to renal anomalies and associated lung disease. In our case, antenatal and post-natal ultrasound did not show renal anomalies. Though high neonatal mortality and poor neurodevelopmental outcomes are common, Piccione et al reported a 9-year follow-up of one case with normal psychomotor development.10 During follow-up, our case showed faltering growth, but it was too early to comment on the neurodevelopmental status.
Learning Points
For neonates with dysmorphism, features of overgrowth and clinical similarity to those with prune belly syndrome, diagnostic consideration should be given to Perlman syndrome. Specific gene studies are essential for all these syndromes, which will help in follow-up investigations, prognostication and genetic counselling.
Patient Consent for Publication
Parents of the patient have provided written informed consent for the case details and identifiable images to be published, and they have seen a copy of the manuscript. The patient was not deidentified due to facial dysmorphic features relevant to this report.
Author Contributions
We would like to sincerely acknowledge Prof. Allie Moosa for the guidance and critical evaluation of the draft preparation.
Funding
No funding was made available for this report.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Liban E, Kozenitzky IL. Metanephric hamartomas and nephroblastomatosis in siblings. Cancer. 1970;25:885–888. doi:10.1002/1097-0142(197004)25:4<885::aid-cncr2820250420>3.0.co;2-#
2. Perlman M, Levin M, Wittels B. Syndrome of fetal gigantism, renal hamartomas, and nephroblastomatosis with Wilms’ tumor. Cancer. 1975;35(4):1212–1217. doi:10.1002/1097-0142(197504)35:4<1212::aid-cncr2820350427>3.0.co;2-2
3. Astuti D, Morris MR, Cooper WN, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet. 2012;44(3):277–284. doi:10.1038/ng.1071
4. Labno A, Warkocki Z, Kulinski T, et al. Perlman syndrome nuclease DIS3L2 controls cytoplasmic non-coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Res. 2016;44:10437–10453. doi:10.1093/nar/gkw649
5. Morris MR, Astuti D, Maher ER. Perlman syndrome: overgrowth, Wilms tumor predisposition and DIS3L2. Am J Med Genet C Semin Med Genet. 2013;163C:106–113. doi:10.1002/ajmg.c.31358
6. Fahmy J, Kaminsky CK, Parisi MT. Perlman syndrome: a case report emphasizing its similarity to and distinction from Beckwith-Wiedemann and prune-belly syndromes. Pediatr Radiol. 1998;28(3):179–182. doi:10.1007/s002470050326
7. Higashimoto K, Maeda T, Okada J, et al. Homozygous deletion of dis3l2 exon 9 due to non-allelic homologous recombination between line-1s in a japanese patient with Perlman syndrome. Eur J Hum Genet. 2013;21(11):1316–1319. doi:10.1038/ejhg.2013.45
8. Alessandri J-L, Cuillier F, Ramful D, et al. Perlman syndrome: report, prenatal findings and review. Am J Med Genet A. 2008;146A(19):2532–2537. doi:10.1002/ajmg.a.32391
9. Demirel G, Oguz SS, Celik IH, et al. Rare clinical entity Perlman syndrome: is cholestasis a new finding? Congenit Anom (Kyoto). 2011;51(1):43–45. doi:10.1111/j.1741-4520.2010.00294.x
10. Piccione M, Cecconi M, Giuffre M, et al. Perlman syndrome: clinical report and nine-year follow-up. Am J Med Genet A. 2005;139A(2):131–135. doi:10.1002/ajmg.a.30994
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.