Back to Journals » Risk Management and Healthcare Policy » Volume 14
Performance of Chromosomal Microarray Analysis for Detection of Copy Number Variations in Fetal Echogenic Bowel
Authors Fan X, Huang H ![]() , Lin X
, Lin X ![]() , Xue H
, Xue H ![]() , Cai M
, Cai M ![]() , Lin N, Xu L
, Lin N, Xu L ![]()
Received 30 December 2020
Accepted for publication 22 March 2021
Published 9 April 2021 Volume 2021:14 Pages 1431—1438
DOI https://doi.org/10.2147/RMHP.S299806
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Marco Carotenuto
Xiangqun Fan,1,* Hailong Huang,1,* Xiyao Lin,2 Huili Xue,1 Meiying Cai,1 Na Lin,1 Liangpu Xu1
1Fujian Maternity and Child Health Hospital, Affiliated Hospital of Fujian Medical University, Fujian Key Laboratory for Prenatal Diagnosis and Birth Defect, Fuzhou, 350001, Fujian Province, People’s Republic of China; 2School of Clinical Medicine, Fujian Medical University, Fuzhou City, Fujian Province, 350122, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liangpu Xu; Na Lin
Fujian Maternity and Child Health Hospital, Affiliated Hospital of Fujian Medical University, Fujian Key Laboratory for Prenatal Diagnosis and Birth Defect, No. 18 Daoshan Road, Gulou District, Fuzhou City, Fujian Province, 350001, People’s Republic of China
Tel +86-591-87554929
Email [email protected]; [email protected]
Background: Fetal echogenic bowel (FEB) is associated with an increased risk of poor pregnant outcomes; however, karyotyping fails to detect copy number variations (CNVs) in FEB. This study aimed to evaluate the performance of chromosomal microarray analysis (CMA) for detection of FEB.
Methods: The medical records of 147 pregnant women with FEB recruited during December 2015 to December 2018 were retrospectively reviewed, and prenatal samples were collected for karyotyping and CMA. The detection of chromosomal abnormality was compared between karyotyping and CMA.
Results: Karyotyping identified eight cases with abnormal karyotypes (5.44% prevalence), including four fetuses with pathogenic aneuploidy, three with chromosome polymorphism and one with balanced chromosome translocation. CMA identified 13 abnormal CNVs (8.84% prevalence), including 4 fetuses with pathogenic aneuploidy as detected by karyotyping and 9 additional CNVs with normal karyotypes; however, CMA failed to detect chromosome polymorphism and balanced chromosome translocation. In fetuses with isolated FEB, no cases presented pathogenic findings and CMA detected two cases with variants of uncertain significance (VOUS). In cases presenting FEB along with other ultrasound abnormalities, CMA detected three cases with pathogenic CNVs and four cases with VOUS in addition to four cases with aneuploidy. There was no significant difference in the detection of abnormal CNVs between the fetuses with echogenic bowel alone and along with other ultrasound abnormalities (10% vs 8.67%, P > 0.05). Except 9 fetuses lost to the follow-up, the other 138 fetuses with echogenic bowel were successfully followed up. Pregnancy was terminated in 5 fetuses with chromosomal abnormality, 2 with pathogenic CNVs and 1 with VOUS, and other 16 with normal karyotypes and CMA findings but showing ultrasound abnormalities or multiple malformations.
Conclusion: Isolated FEB is associated with a good prognosis, and a satisfactory pregnant outcome is expected for fetuses with echogenic bowel that are negative for chromosomal anomalies and other severe structure abnormalities. CMA shows an important value in the genetic diagnosis of FEB. As a supplement to karyotyping, CMA may improve the accuracy of prenatal diagnosis of fetal intestinal malformations in pregnant women with FEB.
Keywords: fetal echogenic bowel, chromosomal microarray analysis, karyotype analysis, copy number variation, prenatal diagnosis
Introduction
Fetal echogenic bowel (FEB) refers to an abnormality of the fetal bowel on ultrasound scans during the pregnancy, in which the bowel has brightness similar to that seen in bone.1 This disorder is estimated to occur in approximately 1% of total pregnancies.2 The majority of FEB is considered as non-specific ultrasound features, which may attribute to intestinal hypoperistalsis.3 However, this disorder is also reported to be linked with an increased risk of chromosomal abnormality, intrauterine infections, cystic fibrosis, congenital bowel abnormality, intrauterine growth restriction, and fetal mortality.4 In addition, the clinical significance of FEB is reported to vary in the echogenic intensity,5 and higher echogenic intensity predicts poorer pregnant outcomes.6
Currently, it is difficult to identify FEB as pathogenic or benign by ultrasound alone, and the identification requires the addition of medical history and laboratory examinations.7 In addition, early detection of severe fetal abnormality may facilitate the prenatal counseling and the decision to terminate the pregnancy, and a test with a higher diagnostic efficiency is required in fetuses diagnosed with abnormal ultrasound findings and a normal karyotype.8
Recently, chromosomal microarray analysis (CMA), a high-throughput assay that is effective to detect chromosomal deletions and duplications, has been widely employed in prenatal diagnosis.9–11 Unlike karyotyping that detects the abnormality of chromosomal fragments with a size of >10 Mb, CMA is sensitive to identify chromosomal microdeletion and microduplication syndromes.12 However, the detection of variants of uncertain significance (VOUS) by CMA challenges the prenatal counseling.13 The purpose of the present study was to evaluate the performance of CMA for detection of copy number variations (CNVs) in FEB.
Subjects and Methods
Study Subjects
A total of 147 pregnant women with FEB detected by ultrasound at Fujian Maternity and Child Health Hospital (Fuzhou, China) during the period from December 2015 through December 2018 were recruited. The pregnant women had a mean age of 29 years (range, 20 to 42 years) and a mean gestational age of 25.7 weeks (range, 18 to 34 weeks). According to ultrasound findings, the study subjects were classified into cases with FEB alone (n = 20) and cases presenting FEB along with other ultrasound abnormalities (n = 127) (Supplementary File 1). Amniotic fluid and umbilical cord blood samples were collected from the study subjects for karyotyping and CMA.
Karyotype Analysis
Chorionic villus, amniotic fluid and umbilical cord blood samples were collected through B-mode ultrasound-guided abdominal puncture, amniocentesis and amniocentesis, respectively. All prenatal samples were routinely cultured, mounted on slides and subjected to G-banding (additional C-banding and N-banding if required). Karyotype analysis was performed on a GSL-120 Streamlines Cytogenetic Analysis System (Leica Microsystems; Mannheim, Germany). At least 40 karyotypes were counted for each case, and 5 karyotypes were randomly selected for analysis.
CMA
Approximately 10 mL of amniotic fluid was sampled and centrifuged, and the sediment was collected. Genomic DNA was extracted from amniotic fluid cells using the QIAampDNA Blood Mini Kit (Qiagen, Hilden, Germany), digested, amplified, purified, fragmented, labeled and hybridized to the array on the Affymetrix SNP Array 6.0 (Affymetrix; Santa Clara, CA, USA). The CytoScan HD array, includes the CNV probe and SNP probe, may detect CNV, mosaic (mosaic proportion >10%) and loss of heterozygosity (LOH). All data analyses were performed using the software Chromosome Analysis Suite (ChAS) version 3.2 (Affymetrix; Santa Clara, CA, USA), and the interpretation of CNV, which was classified as pathogenic, VOUS and benign, was identified using online public databases, including the database of genomic variants (DGV, http://projects.tcag.ca/variation), the DECIPHER database (htts://decinher.sanger.ac.uk/), the OMIM database (http://www.omim.org), the International Standards for Cytogenomic Arrays (ISCA) Consortium and Public Database (https://www.iscaconsortium.org/), the CAGdb database (http://www.cagdb.org/), the CHDWiki database and the NCBI database. The pathogenic CNVs detected by the SNP array were validated using fluorescence in situ hybridization (FISH) assay. Peripheral blood was sampled from the parents of the fetus with VOUS for the SNP array, and the type of CNV was identified by means of the SNP array and pedigree analysis.
Statistical Analysis
All statistical analyses were performed using the statistical software SPSS version 22.0 (SPSS, Inc.; Chicago, IL, USA). Differences of proportions were tested for statistical significance with a chi-square test, and a P value of <0.05 was considered statistically significant.
Ethical Statement
This study was approved by the Ethics Review Committee of Fujian Maternity and Child Health Hospital. All procedures were performed following the Declaration of Helsinki, as well as international and national laws, guidelines and regulations. Signed informed consent was obtained from all subjects with a detailed description of the purpose of the study.
Results
Chromosomal Karyotypes
Karyotype analysis was successfully performed in 147 prenatal samples. Karyotype analysis identified eight cases with abnormal karyotypes (5.44% prevalence), including four fetuses with aneuploidy (one case with T21, two cases with 47,XXY and one case with small supernumerary marker chromosome (47,XY,+mar)), three fetuses with chromosome polymorphism (two fetuses with 46,XX,inv(9)(p12q13) and one fetus with 46,inv(Y)(p12q13)), and one fetus with balanced chromosome translocation ((46,XY,t(10;17)(q26;p11.2)).
CMA Findings
CMA was successfully performed in 147 prenatal samples, and 13 abnormal CNVs were identified (8.84% prevalence), including 4 fetuses with pathogenic aneuploidy as detected by karyotyping. Of the fetus detected with the small supernumerary marker chromosome (47,XY,+mar) by karyotyping, CMA identified a 4.6 Mb duplication at the p12.1p11.1 region of the chromosome 3 and a 1.6 Mb duplication at the 11.1q11.2 region of the chromosome 3, which may be a novel pathogenic mutation. Moreover, CMA detected nine additional CNVs with normal karyotypes; however, CMA failed to detect chromosome polymorphism and balanced chromosome translocation due to no loss of chromosomal materials. The size of the detected abnormal CNVs was 0.708 to 19.2 Mb in fetuses with echogenic bowel, and three fetuses with pathogenic CNVs (a case with 10q11.1q11.22 microdeletion, a case with 16p13.11 microdeletion and a case with Xq28 microduplication) and six fetuses with VOUS (a case with 16p13.11 microduplication, a case with 16p13.11 microdeletion, a case with 18q21.33q22.1 microdeletion, a case with 9p21.1 microdeletion, a case with 5q33.2q33.3 microduplication, and a case with LOH at the q23.2q24.3 region) were detected (Table 1).
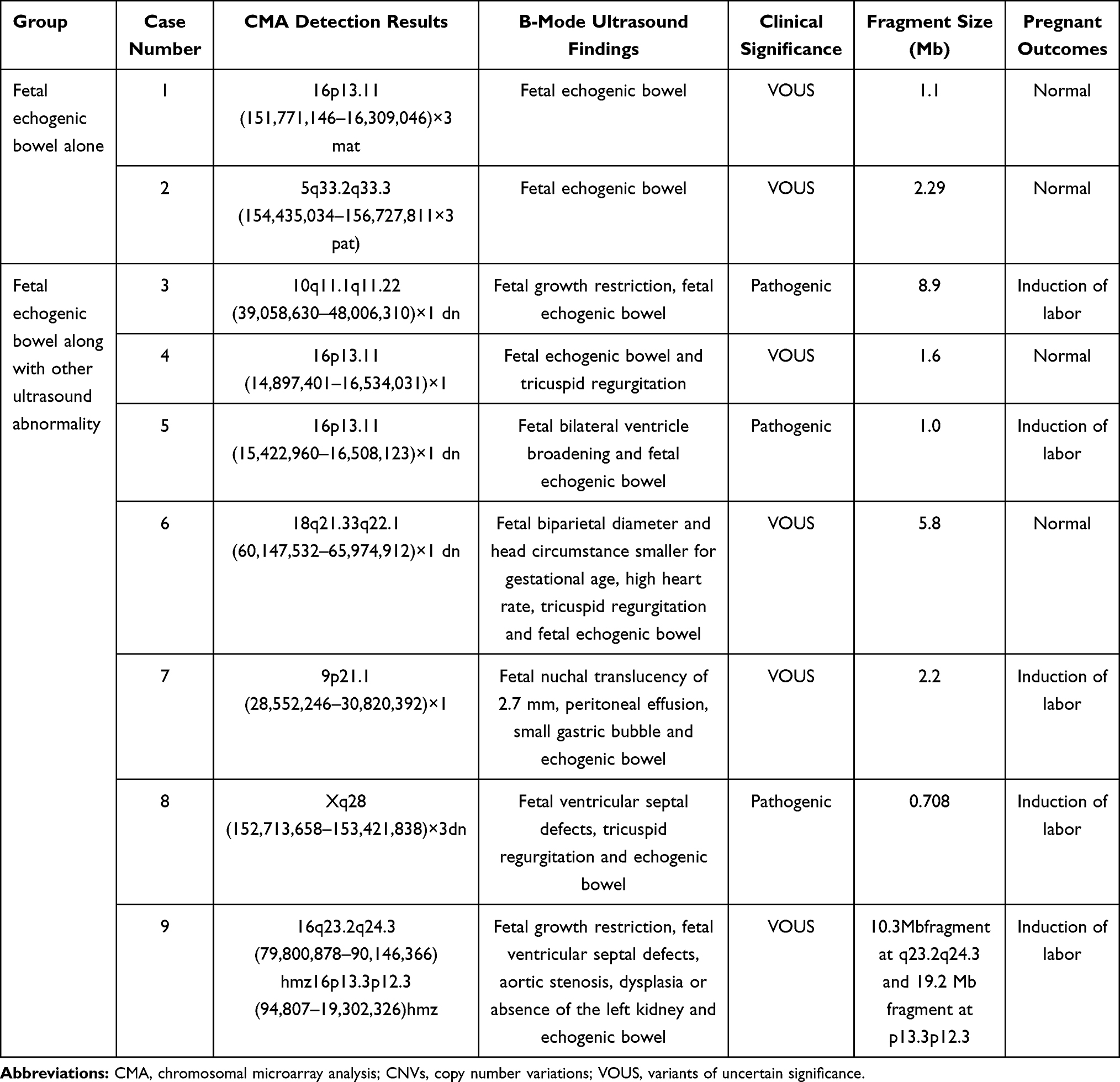 |
Table 1 Abnormal CMA Detection Results Among Fetuses Presenting Echogenic Bowel on Ultrasound with Normal Karyotypes |
In cases with isolated FEB, no case presented pathogenic findings, and two cases were detected with VOUS. In cases presenting FEB along with other ultrasound abnormalities, there were seven cases with pathogenic CNV and four cases with VOUS. However, there was no significant difference in the prevalence of abnormal CNVs between the fetuses with isolated echogenic bowel and along with other ultrasound abnormalities (10% vs 8.67%, P > 0.05) (Table 2).
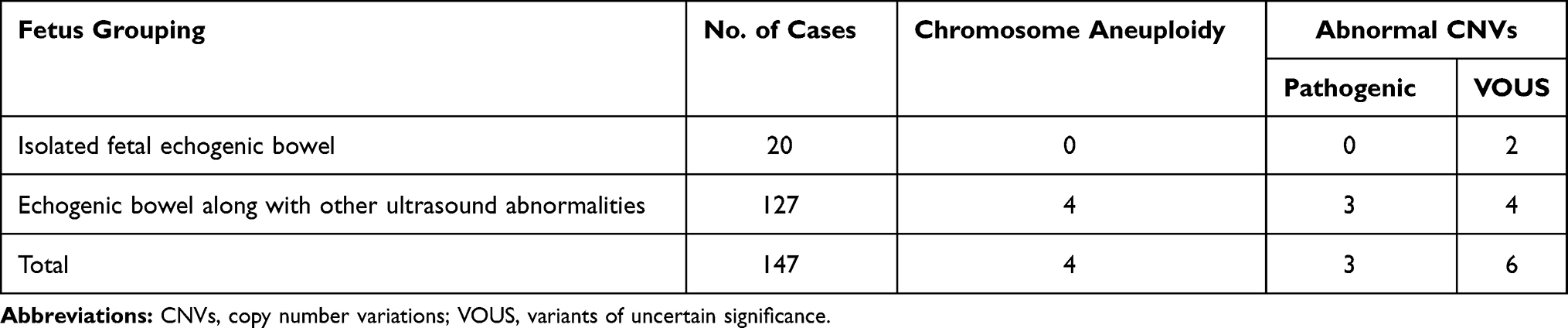 |
Table 2 Comparison of the Detection of the Prevalence of Abnormal CNVs Between Fetuses with Isolated Echogenic Bowel and Along with Other Ultrasound Abnormalities |
Follow-Up Outcomes
Except 9 fetuses lost to the follow-up, the other 138 fetuses with echogenic bowel were successfully followed up. Pregnancy was terminated in 5 fetuses with chromosomal abnormality, 2 fetuses with pathogenic CNVs, 1 fetus with VOUS and 16 fetuses with normal karyotypes and CMA findings but showing abnormal ultrasound findings or multiple malformations. In addition, the parents decided to continue the pregnancy in four fetuses with VOUS, and the postnatal follow-up showed well growth and development of the newborn (Table 3) .In second trimesters, 96% cases had normal deliveries (52/54), which higher than third trimester (87%, 73/84).
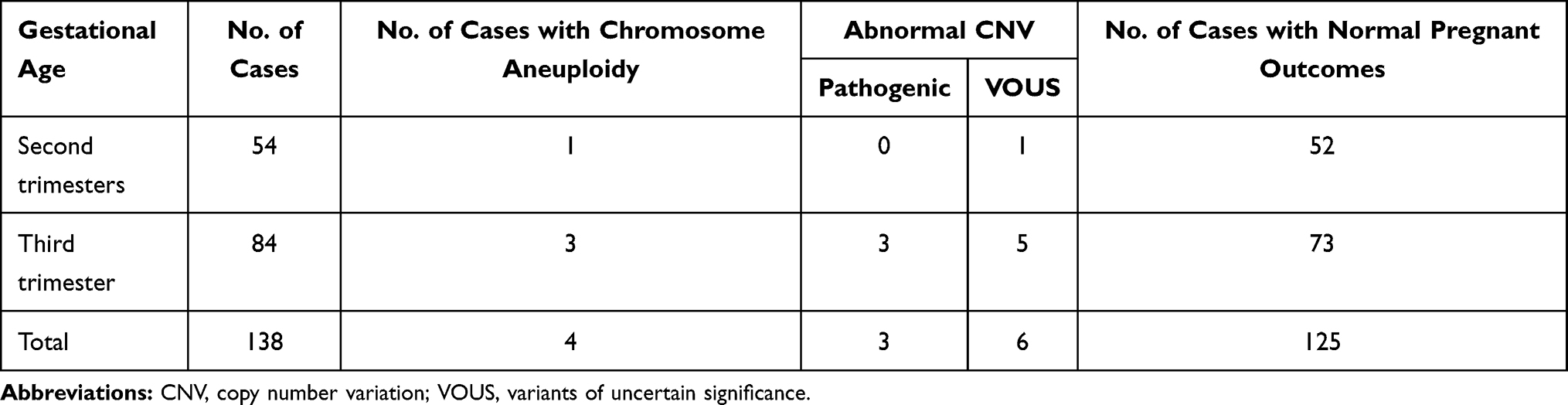 |
Table 3 Prenatal Outcomes in Cases with Fetal Echogenic Bowel Diagnosed at Different Trimesters |
Discussion
To date, the correlation between isolated FEB and chromosomal abnormality remains controversial.14,15 The incidence of gastrointestinal abnormalities was reported to be 4.76% in fetuses with antenatal echogenic bowel.16 A retrospective review of 682 cases of hyperechogenic fetal bowel collected from 22 molecular biology laboratories in France showed a 3.5% incidence rate of chromosomal anomaly in the case series (including 2.5% incidence of Down’s syndrome and 1% incidence of other severe chromosomal anomaly), 6.9% prevalence of multiple malformations, 3% prevalence of cystic fibrosis, and 2.8% prevalence of viral infections.17 Ultrasound screening of FEB is therefore of great clinical significance during the first trimester, which is considered to correlate with fetal chromosomal abnormality.5
Previous studies have shown that isolated FEB is associated with a low risk of chromosomal abnormality, while FEB along with other ultrasound soft markers is associated with a remarkable increase in the risk of chromosomal anomaly.18,19 In this study, we detected 10% and 8.67% prevalence of abnormal CNVs in fetuses with echogenic bowel alone and along with other ultrasound abnormalities (P > 0.05), which was inconsistent with previous studies.18–20 This may be attributed to the addition of CMA in our study, while karyotyping was performed in previous reports, or the small sample size of isolated fetal echogenic bowel. Further studies recruiting more fetuses with isolated echogenic bowel to compare the prevalence of chromosomal anomaly between fetuses with echogenic bowel alone and along with other ultrasound abnormalities seem justified.
In the current study, we detected three fetuses with chromosomal aneuploidy, including a case with T21 and two cases with the 47,XXY. The 47,XXY, also termed Klinefelter syndrome, is a common sex chromosomal aneuploidy and its incidence is estimated to be 1 per 1000 live male births.21 Patients with the 47,XXY are reported to present behavioral disorders, testicular abnormalities, reduced intelligence quotient relative to brothers and sisters but remaining within the normal range.22 Previous studies have shown a strong association between T21 and FEB.23,24 In this study, CMA detected additional chromosomal abnormalities as compared to karyotyping in fetuses with echogenic bowel, and we identified a 10q11.1q11.22 microdeletion with a fragment of 8.9 Mb that contained 20 OMIM genes. Loss-of-function mutations in the Ret gene (MIM# 164761) may cause autosomal dominant familial and sporadic Hirschsprung’s disease (MIM# 142623),25 and the Ret gene mutation is detected in more than 50% of patients with familial Hirschsprung’s disease.26 Infants with Hirschsprung’s disease may frequently develop symptoms of impaired bowel peristalsis within 2 months after birth, including failure of meconium passage within 48 hours of birth, constipation, vomiting, abnormal pain or distension, and diarrhea, and patients may manifest mental retardation and hypotonia.27 Besides, the Ret gene was found to show a haploinsufficiency (score of 3).28 In our study, CMA identified a pathogenic CNV in this fetus with the 10q11.1q11.22 microdeletion, and the pregnancy was terminated.
In this study, CMA detected 16p13.11 microdeletions in prenatal samples 4 and 5, and this microdeletion contained 16p13.11 recurrent deletion/duplication regions, including MYH11. A previous case–control study reported that microdeletions at the 16p13.11 region strongly correlated with epilepsy, and recurrent microdeletions at 16p13.11 conferred a pleiotropic susceptibility effect to a broad range of neuropsychiatric disorders.29 Infants with the 16p13.11 microdeletion may present a wide range of clinical manifestations, and the common clinical symptoms include mental retardation, epilepsy and microcephaly.30 This microdeletion may be inherited from parents with normal phenotypes, and a 13.1% penetrance was reported.31 These two prenatal samples were finally detected with pathogenic CNVs by CMA. In our study, microduplication was detected at 16p13.11 in prenatal sample 1. Mutation of the MYH11 gene is associated with the development of aortic aneurysm and dissection,32 and overexpression of the MYH11 gene correlates with the increased risk of aortic dissection and schizophrenia.33 In addition, the NED1 gene is involved in mental and behavioral abnormalities.34 It has been demonstrated that duplication at 16p13.11 exhibits triploinsufficiency and clinical heterogeneity.35 Furthermore, two fetuses were identified with VOUS in prenatal samples 2, 6 and 7, which contained unclear pathogenic OMIM genes. The labor was induced in fetus 7 that presented additional ultrasound abnormalities, while normal pregnant outcomes were observed in fetuses 2 and 6.
In this study, maternal uniparental disomy of chromosome 16 [UPD (16) mat] was identified in prenatal sample 9. Uniparental disomy of chromosome 16 has been reported to cause fetal developmental retardation, intrauterine growth restriction, reduced fetal movements, cardiac malformations and dysplasia of the urinary system.36 However, there is also a report showing that uniparental disomy of chromosome 16 alone does not result in intrauterine growth restriction.37 Since there is no pathogenic imprinting in chromosome 16, the exact pathogenicity of uniparental disomy of chromosome 16 remains to be investigated.38
In addition to the increase in the detection of pathogenic CNVs, CMA may detect VOUS.39 Because of penetrance and environmental factors, the individuals with the same pathogenic CNVs or VOUS may present diverse manifestations among family members, and there are also individuals with completely normal phenotypes. In addition, the detection of fetal parental samples with CMA may remain failure to identify the exact clinical significance of the VOUS.40 In this study, we detected a case with novel Xq28 microduplication. The repetitive fragment contained L1CAM and MECP2 genes that are strongly associated with mental and intelligent developments,41,42 and the major clinical manifestations of the Xq28 microduplication syndrome include autism, mental retardation, communication dysfunctions, convulsion, hypotonia, repeated infections and abnormal bladder functions.43
In addition to abnormal chromosome numbers and structure, chromosomal microdeletions or microduplications may contribute to the pathogenesis of fetal echogenic bowel.3 Our data showed that both karyotyping and CMA are effective to detect chromosomal aneuploidy; however, CMA may identify the origin, fragment size and pathogenicity of the supernumerary marker chromosome, which facilitates the precision evaluation of the prognosis in fetuses with echogenic bowel during the genetic counseling.11 Previous studies have shown that CNV, a risk factor for complicated developmental malformations,44,45 is a major contributor to fetal echogenic bowel.1
In summary, the results of the present study demonstrate that prenatal ultrasound is difficult to detect benign or pathogenic echogenic bowel, and a satisfactory pregnant outcome may be expected for fetuses with echogenic bowel that are negative for chromosomal anomalies and other severe anatomic abnormalities. Isolated FEB is associated with a good prognosis compared with those presenting multiple ultrasound abnormalities. In addition, CMA shows an important value in the genetic diagnosis of FEB, and such a tool, as a supplement to karyotyping, may improve the accuracy of prenatal diagnosis of fetal intestinal malformations in pregnant women with FEB.
Acknowledgments
We are grateful to Prof. Yuan Lin for her kind assistance during the study period. This study was funded by the grants from the Natural Science Foundation of Fujian Province (grant no. 2017J01238).
Disclosure
The authors declare no conflicts of interest.
References
1. Sepulveda W, Sebire NJ. Fetal echogenic bowel: a complex scenario. Ultrasound Obstet Gynecol. 2000;16(6):510–514. doi:10.1046/j.1469-0705.2000.00322.x
2. Maillet L, Rudigoz RC, Buffin R, Massardier J, Gaucherand P, Huissoud C. Neonatal outcome of fetal hyperechogenic bowel. Gynecol Obstet Fertil. 2014;42(6):383–386. doi:10.1016/j.gyobfe.2014.01.006
3. Kesrouani AK, Guibourdenche J, Muller F, et al. Etiology and outcome of fetal echogenic bowel. Ten years of experience. Fetal Diagn Ther. 2003;18(4):240–246. doi:10.1159/000070803
4. Findley R, Allen VM, Brock JK. Adverse perinatal conditions associated with prenatally detected fetal echogenic bowel in Nova Scotia. J Obstet Gynaecol Can. 2018;40(5):555–560. doi:10.1016/j.jogc.2017.09.017
5. D’Amico A, Buca D, Rizzo G, et al. Outcome of fetal echogenic bowel: a systematic review and meta-analysis. Prenat Diagn. 2020.
6. Bashiri A, Burstein E, Hershkowitz R, Mazor M. Fetal echogenic bowel by ultrasound: what is the clinical significance? Harefuah. 2007;146(12):
7. Strocker AM, Snijders RJ, Carlson DE, et al. Fetal echogenic bowel: parameters to be considered in differential diagnosis. Ultrasound Obstet Gynecol. 2000;16(6):519–523. doi:10.1046/j.1469-0705.2000.00241.x
8. Ronin C, Mace P, Stenard F, et al. Antenatal prognostic factor of fetal echogenic bowel. Eur J Obstet Gynecol Reprod Biol. 2017;212:166–170. doi:10.1016/j.ejogrb.2017.01.060
9. Levy B, Wapner R. Prenatal diagnosis by chromosomal microarray analysis. Fertil Steril. 2018;109(2):201–212. doi:10.1016/j.fertnstert.2018.01.005
10. Stosic M, Levy B, Wapner R. The use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol Clin North Am. 2018;45(1):55–68. doi:10.1016/j.ogc.2017.10.002
11. Xie Y, Sun X. Chromosomal microarray analysis in prenatal diagnosis. Clin Exp Obstet Gynecol. 2017;44(2):177–179.
12. Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175–2184. doi:10.1056/NEJMoa1203382
13. Song T, Wan S, Li Y, et al. Detection of copy number variants using chromosomal microarray analysis for the prenatal diagnosis of congenital heart defects with normal karyotype. J Clin Lab Anal. 2019;33(1):e22630. doi:10.1002/jcla.22630
14. Patel Y, Boyd PA, Chamberlain P, Lakhoo K. Follow-up of children with isolated fetal echogenic bowel with particular reference to bowel-related symptoms. Prenat Diagn. 2004;24(1):35–37. doi:10.1002/pd.787
15. Ekin A, Gezer C, Taner CE, Ozeren M. The effect of associated structural malformations in the prediction of chromosomal abnormality risk of fetuses with echogenic bowel. J Matern Fetal Neonatal Med. 2016;29(1):41–45. doi:10.3109/14767058.2014.986091
16. Tan HH, Tan VC, Yeo GS. A case series of gastrointestinal abnormalities in fetuses with echogenic bowel detected during the antenatal period. Ann Acad Med Singap. 2003;32(5):649–652.
17. Imon-Bouy B, Satre V, Ferec C, et al.; French Collaborative Group. Hyperechogenic fetal bowel: a large French collaborative study of 682 cases. Am J Med Genet A. 2003;121A(3):209–213. doi:10.1002/ajmg.a.20168
18. Grijseels EW, Cohen-Overbeek TE, Adama van Scheltema PN, et al. Sonomarkers: subtle ultrasound findings in the 20-week ultrasound examination, which have a low association with some chromosomal and non-chromosomal abnormalities in the fetus. Ned Tijdschr Geneeskd. 2008;152(41):2225–2231.
19. Buiter HD, Holswilder-Olde Scholtenhuis MA, Bouman K, van Baren R, Bilardo CM, Bos AF. Outcome of infants presenting with echogenic bowel in the second trimester of pregnancy. Arch Dis Child Fetal Neonatal Ed. 2013;98(3):F256–F259. doi:10.1136/archdischild-2012-302017
20. Tang SY, Du J, Huang J, Ling HY, Meng DH. Prenatal diagnosis of fetal echogenic bowel and its clinical significance. Matern Child Health Care Chin. 2013;28(18):3015–3017.
21. Crawford D, Dearmun A. Klinefelter syndrome. Nurs Child Young People. 2017;29(6):19. doi:10.7748/ncyp.29.6.19.s21
22. Bird RJ, Hurren BJ. Anatomical and clinical aspects of Klinefelter’s syndrome. Clin Anat. 2016;29(5):606–619. doi:10.1002/ca.22695
23. Berlin BM, Norton ME, Sugarman EA, Tsipis JE, Allitto BA. Cystic fibrosis and chromosome abnormalities associated with echogenic fetal bowel. Obstet Gynecol. 1999;94(1):135–138. doi:10.1016/s0029-7844(99)00286-0
24. Dagklis T, Plasencia W, Maiz N, Duarte L, Nicolaides KH. Choroid plexus cyst, intracardiac echogenic focus, hyperechogenic bowel and hydronephrosis in screening for trisomy 21 at 11 + 0 to 13 + 6 weeks. Ultrasound Obstet Gynecol. 2008;31(2):132–135. doi:10.1002/uog.5224
25. Langer JC. Hirschsprung disease. Curr Opin Pediatr. 2013;25(3):368–374. doi:10.1097/MOP.0b013e328360c2a0
26. Wang XJ, Camilleri M. Hirschsprung disease: insights on genes, penetrance, and prenatal diagnosis. Neurogastroenterol Motil. 2019;31(11):e13732. doi:10.1111/nmo.13732
27. Wester T, Granström AL. Hirschsprung disease-bowel function beyond childhood. Semin Pediatr Surg. 2017;26(5):322–327. doi:10.1053/j.sempedsurg.2017.09.008
28. Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al.; Hirschsprung Disease Consortium. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45(1):1–14. doi:10.1136/jmg.2007.053959
29. de Kovel CG, Trucks H, Helbig I, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133(Pt 1):23–32. doi:10.1093/brain/awp262
30. Smith AE, Jnah A, Newberry D. Chromosome 16p13.11 microdeletion syndrome in a newborn: a Case Study. Neonatal Netw. 2018;37(5):303–309. doi:10.1891/0730-0832.37.5.303
31. Tan L, Bi B, Zhao P, et al. Severe congenital microcephaly with 16p13.11 microdeletion combined with NDE1 mutation, a case report and literature review. BMC Med Genet. 2017;18(1):141. doi:10.1186/s12881-017-0501-9
32. Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation. 2016;133(24):2516–2528. doi:10.1161/CIRCULATIONAHA.116.009762
33. Takeda N, Morita H, Fujita D, et al. A deleterious MYH11 mutation causing familial thoracic aortic dissection. Hum Genome Var. 2015;2(1):15028. doi:10.1038/hgv.2015.28
34. Bradshaw NJ. Cloning of the promoter of NDE1, a gene implicated in psychiatric and neurodevelopmental disorders through copy number variation. Neuroscience. 2016;324:262–270. doi:10.1016/j.neuroscience.2016.03.018
35. Allach El Khattabi L, Heide S, Caberg JH, et al. 16p13.11 microduplication in 45 new patients: refined clinical significance and genotype-phenotype correlations. J Med Genet. 2020;57(5):301–307. doi:10.1136/jmedgenet-2018-105389
36. Scheuvens R, Begemann M, Soellner L, et al. Maternal uniparental disomy of chromosome 16 [upd(16)mat]: clinical features are rather caused by (hidden) trisomy 16 mosaicism than by upd(16)mat itself. Clin Genet. 2017;92(1):45–51. doi:10.1111/cge.12958
37. Wang H, Luo C, Liu Y, et al. UPD16 itself is not a cause of intrauterine growth restriction. Fetal Pediatr Pathol. 2018;37(6):452–464. doi:10.1080/15513815.2018.1538275
38. Yong PJ, Marion SA, Barrett IJ, Kalousek DK, Robinson WP. Evidence for imprinting on chromosome 16: the effect of uniparental disomy on the outcome of mosaic trisomy 16 pregnancies. Am J Med Genet. 2002;112(2):123–132. doi:10.1002/ajmg.10702
39. Desai P, Haber H, Bulafka J, et al. Impacts of variants of uncertain significance on parental perceptions of children after prenatal chromosome microarray testing. Prenat Diagn. 2018;38(10):740–747. doi:10.1002/pd.5323
40. Dugoff L, Norton ME, Kuller JA; Society for Maternal-Fetal Medicine (SMFM). The use of chromosomal microarray for prenatal diagnosis. Am J Obstet Gynecol. 2016;215(4):B2–B9. doi:10.1016/j.ajog.2016.07.016
41. Swarna M, Sujatha M, Rani PU, Reddy PP. Detection of L1 CAM mutation in a male child with mental retardation. Indian J Clin Biochem. 2004;19(2):163–167. doi:10.1007/BF02894278
42. Gonzales ML, LaSalle JM. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr Psychiatry Rep. 2010;12(2):127–134. doi:10.1007/s11920-010-0097-7
43. Kosma K, Mitrakos A, Sofokleous C, et al. A female patient with Xq28 microduplication presenting with myotubular myopathy, confirmed with a custom-designed X-array. Neuropediatrics. 2019;50(1):61–63. doi:10.1055/s-0038-1676513
44. Wang Y, Hu P, Xu Z. Copy number variations and fetal ventriculomegaly. Curr Opin Obstet Gynecol. 2018;30(2):104–110. doi:10.1097/GCO.0000000000000439
45. Hu P, Wang Y, Sun R, et al. Copy number variations with isolated fetal ventriculomegaly. Curr Mol Med. 2017;17(2):133–139. doi:10.2174/1566524017666170303125529
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.