Back to Journals » International Journal of Nanomedicine » Volume 13
Peptide T7-modified polypeptide with disulfide bonds for targeted delivery of plasmid DNA for gene therapy of prostate cancer
Authors Lu Y, Jiang W, Wu X, Huang S, Huang Z, Shi Y, Dai Q ![]() , Chen J, Ren F
, Chen J, Ren F ![]() , Gao S
, Gao S ![]()
Received 20 July 2018
Accepted for publication 19 September 2018
Published 30 October 2018 Volume 2018:13 Pages 6913—6927
DOI https://doi.org/10.2147/IJN.S180957
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yue Lu,1 Wenjun Jiang,1 Xin Wu,2 Saixu Huang,1 Zhiyong Huang,1 Yamin Shi,3 Qi Dai,1 Jianming Chen,2 Fuzheng Ren,1 Shen Gao4
1Shanghai Key Lab New Drug Design, School of Pharmacy, East China University of Science and Technology, Shanghai, People’s Republic of China; 2Shanghai Weier Biological Medicine Science and Technology Co. Ltd., Shanghai, People’s Republic of China; 3Department of Pharmacy, Fujian University of Traditional Chinese Medicine, Fuzhou, People’s Republic of China; 4Department of Pharmaceutics, Changhai Hospital, Second Military Medical University, Shanghai, People’s Republic of China
Background: Vectors are essential for successful gene delivery. In the present study, a tumor-targeting cationic gene vector, known as the disulfide cross-linked arginine-aspartic acid peptide modified by HAIYPRH (T7) peptide (CRD-PEG-T7), was designed for targeted delivery of plasmid DNA (pDNA) for gene therapy of prostate cancer (PCa).
Methods: The structure of CRD-PEG-T7 was determined and the cellular uptake efficacy, gene transfection efficacy, cytotoxicity, and the targeting effect of the CRD-PEG-T7–plasmid DNA complex were examined.
Results: The results demonstrated that the CRD-PEG-T7–plasmid DNA complex was nanosized and had a positively charged surface, good cellular uptake efficacy, minimal cytotoxicity, and a dual-targeting effect as compared with the CRD-PEG–plasmid DNA complex. The peptide T7-modifed new delivery system was able to target the highly expressed transferrin receptor (TfR) on tumor cells with an efficiency four-fold higher than that of the non-modified system.
Conclusion: The results above indicatd that the CRD-PEG-T7–plasmid DNA complex may prove to be a promising gene delivery system targeting bone-metastatic tumor.
Keywords: arginine peptide, aspartic acid peptide, tumor targeting, DNA delivery, bone metastasis prostate cancer
Introduction
Prostate cancer (PCa) is the second leading malignant tumor causing death among Western populations, constituting a grave threat to human health. Bone metastasis is generally regarded as an incurable symbol of cancer, occurring in approximately 80% of advanced PCa cases.1 Conventional treatment usually involves surgery to remove the prostate, but is usually associated with serious morbidities, especially in cases of bone metastatic prostate cancer (BMPCa).2 The advent of gene therapy seems to provide a promising treatment for BMPCa.3,4 Studies have reported that the risk of bone metastasis was increased in PCa cells owing to the low expression of the prostate transmembrane protein androgen induced-1 (PMEPA1).5,6 Further research revealed that PMEPA1 inhibited tumor invasion, especially bone metastasis, by blocking the TGF-β signaling pathway.7 Therefore, plasmid PMEPA1 (pPMEPA1 [DPM]) may serve as a prospective gene therapeutic option for BMPCa.
It is generally recognized that vectors are essential for gene delivery. Poly-arginine (R) – belonging to the cationic cell-penetrating peptides (CPPs) – is an optional gene delivery vector due to its favorable biocompatibility.8,9 Previous studies have demonstrated that CPPs cross-linked by disulfide bonds are promising gene delivery vectors owing to their admirable gene transfection efficiency and low cytotoxicity,10–12 given that disulfide bonds can be rapidly cleaved by intracellular reductive compounds such as glutathione to release DNA.13,14 Interestingly, tumor cells bear a relatively high intracellular glutathione concentration compared to normal cells.15 In addition, polymers cross-linked by disulfide bonds enjoy a higher charge density, thus increasing their DNA-loading ability.16
A peptide sequence of aspartic acid (D) could exclusively interact with hydroxyapatite (HA) – the main component of the bone tissue17 – and, therefore, the aspartic acid peptide has already been utilized as a bone-targeted agent.18 Peptide T7 (sequence His-Ala-Ile-Tyr-Pro-Arg-His) enjoys strong affinity with transferrin receptors (TfRs), which are abundant on the surface of PCa cells.19,20 It was claimed that the binding affinity of peptide T7 to TfRs was similar to that of transferrin, and their binding sites were different to each other.21 Therefore, peptide T7 may act as an optional targeting agent for TfR-expressing tumor cells. Moreover, tumor-targeting gene delivery systems by active targeting of peptide T7 via receptor-mediated endocytosis have been reported previously.22,23
Based on prior research, we combined arginine peptides with arginine-aspartic acid peptides via cysteine to obtain arginine-aspartic acid peptides (sequence CRRRRRRRCDDDDDD, or R7D6), and then cross-linked the R7D6 monomers with cysteine through low-concentration H2O2 to form disulfide bonds. These cross-linked monomers were named CRD. Next, we employed NHS-PEG-MAL – a bifunctional PEG – to conjugate CRD and peptide T7 for the final polymer, a disulfide bond cross-linked arginine-aspartic acid peptide modified with peptide T7 that was known as CRD-PEG-T7. We hypothesized that the final product could be a promising plasmid DNA (pDNA) delivery vector, with the capacity of enhancing cellular uptake, promoting gene transfection efficacy, and achieving the tumor-targeting purpose – both in vitro and in vivo. Therefore, we first synthesized the CRD-PEG-T7 polymer and prepared a complex through mixing the polymer with pDNA. After characterization, the cellular uptake efficacy, cytotoxicity, transfection efficiency, and targeting efficacy of these complexes were evaluated. The targeting mechanism of CRD-PEG-T7/DNA complex to BMPCa cells were illustrated in Figure 1 and its formation process, as well as the cellular uptake process, was shown in Figure 2.
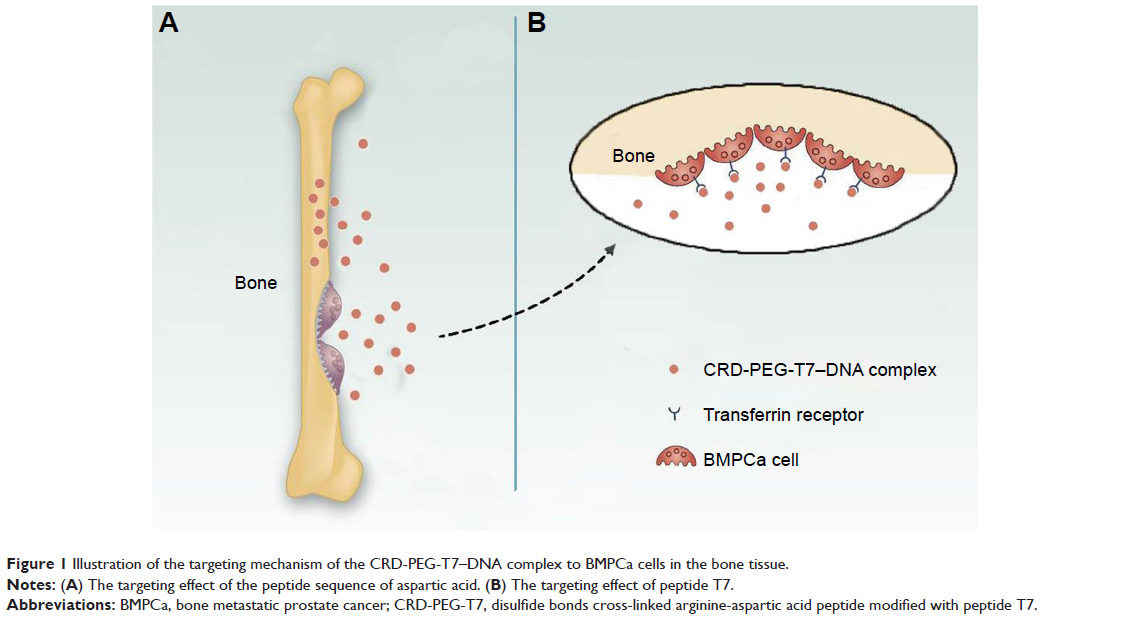 | Figure 1 Illustration of the targeting mechanism of the CRD-PEG-T7–DNA complex to BMPCa cells in the bone tissue. |
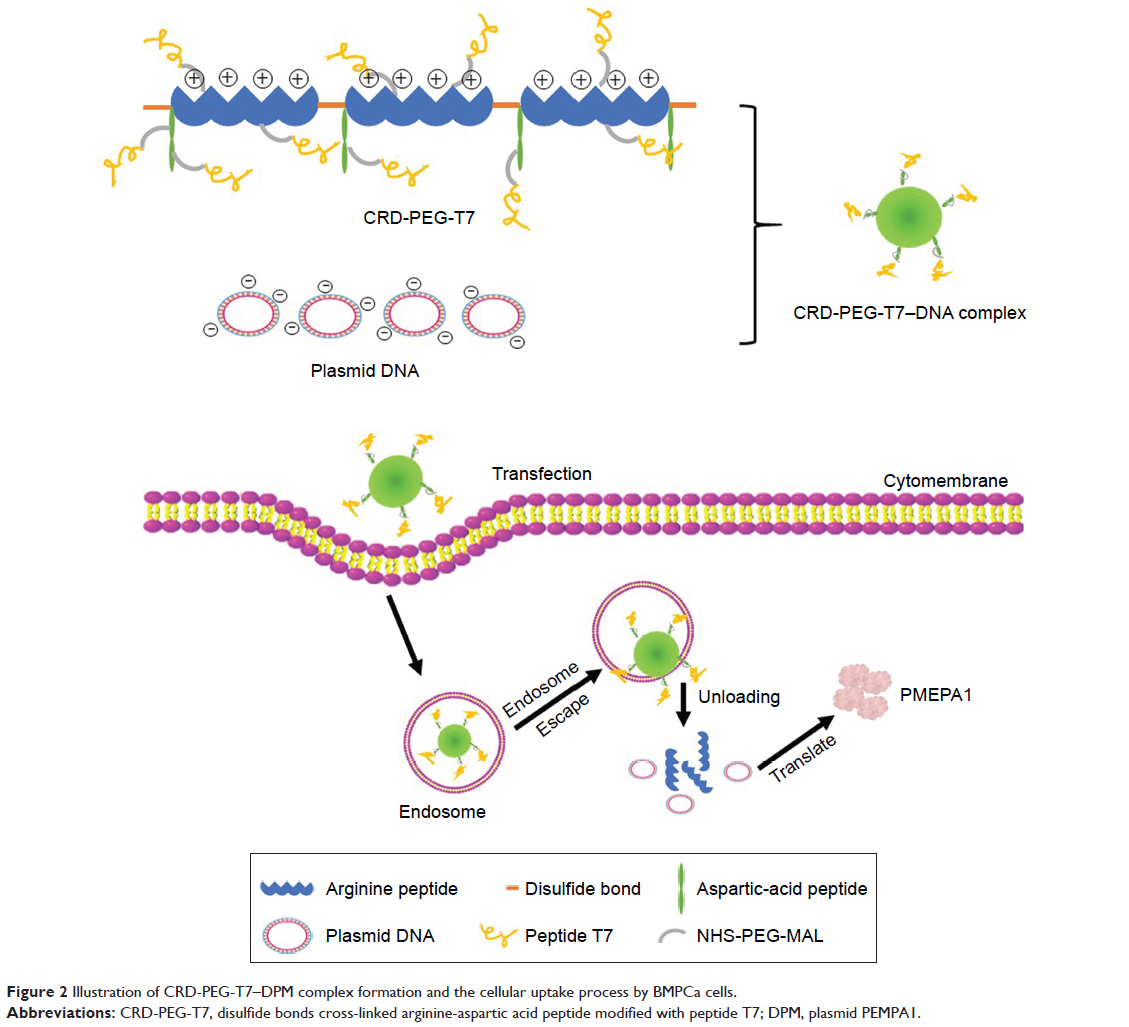 | Figure 2 Illustration of CRD-PEG-T7–DPM complex formation and the cellular uptake process by BMPCa cells. |
Material and methods
Materials
The materials used in this study included arginine-aspartic acid peptide monomer (sequence: CRRRRRRRCDDDDDD, or R7D6) and peptide T7 conjugating a cysteine on its N-terminal (sequence: CHAIYRPRH; Ontores Biotechnologies; Zhejiang, People’s Republic of China); NHS-PEG-MAL (α-malemidyl-ω-N-hydroxysuccinimidyl polyethyleneglycol, MW 3,500, Nektar Therapeutics, Huntsville, AL, USA); and DPM (General Biosystems, Anhui, People’s Republic of China); YOYO-1 (Life Technologies, CA, USA); pEGFP and pGL-3 (Innovation Biotechnology, Shanghai, People’s Republic of China); Cell Counting Kit-8 (Dojindo, Shanghai, People’s Republic of China); enhanced bicinchoninic acid protein assay kit (Pierce Biotechnology Inc., Rockford, IL, USA); cyanine 7 NHS ester (Cy-7; GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA), and PC3 and DU145 cell lines (American Type Culture Collection (ATCC), Manassas, VA, USA). The other materials were all procured from commercial providers. All animal experiments were approved by, and carried out in accordance with the guidelines of the Committee on Animals of the Second Military Medical University (Shanghai, People’s Republic of China).
Cells and cell culture
PC3 cells and DU145 cells were grown at 37°C under a 5% CO2 atmosphere in RPMI 1640 basic medium (GIBCO by Thermo Fisher Scientific, Bedford, MA, USA) with 10% FBS (Globo; Thermo Fisher Scientific, Waltham, MA, USA) and 1% antibiotics (penicillin and streptomycin; glibo by Life Technologies).
Synthesis and characterization of the polymers
R7D6 monomers (arginine-aspartic acid peptide, sequence CRRRRRRRCDDDDDD) was synthesized by the F-moc-solid-phase synthesis method. Then, R7D6 monomers and L-cysteine hydrochloride monohydrates (Cys) were mixed at various molar ratios (Table 1) in 10 mL distilled water, and H2O2 (1%, 0.5 mL) was added into the solution. Polymerization was conducted at room temperature for 12 hours. Next, the solution was dialyzed by a membrane (molecular weight [Mw] cutoff 3 kDa) for 12 hours in distilled water, followed by lyophilization. The products were termed as CRD1, CRD2, CRD3, and CRD4, respectively, according to their Mw. The structure and Mw of CRD was analyzed by 600 MHz nuclear magnetic resonance spectrometry (1H-NMR; Varian Inc, Palo Alto, CA, USA) and gel permeation chromatography (GPC).
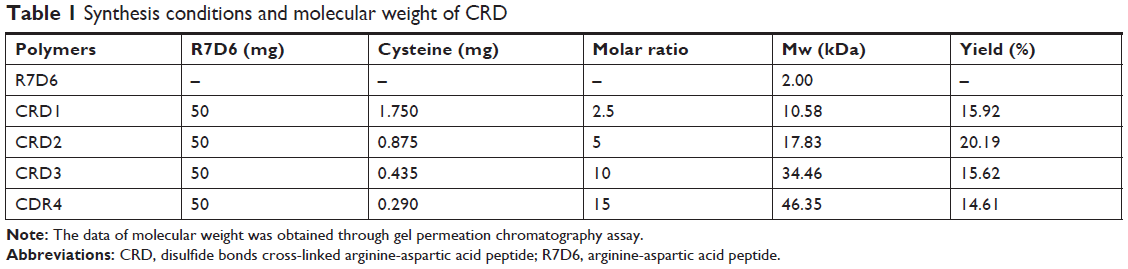 | Table 1 Synthesis conditions and molecular weight of CRD |
Subsequently, CRD1, CRD2, CRD3, and CRD4 with different cross-link degrees were reacted with the bifunctional PEG NHS-PEG-MAL (Mw 3,400) at a molar ratio of 1:10 in distilled water for 6 hours at room temperature under stirring. The amino groups of CRD could react exclusively with the NHS group of the bifunctional PEG to obtain CRD-PEG-MAL. Then, CRD-PEG-MAL was dialyzed by a dialysis membrane (Mw cutoff 3 kDa) for 6 hours in distilled water. Next, CRD-PEG-MAL was reacted with Cys-T7 at a molar ratio of 1:5 in distilled water for 6 hours under stirring to obtain CRD-PEG-T7 through the specific reaction between the MAL group and thiol group of Cys-T7. The product was purified for 6 hours in distilled water by using a dialysis membrane (Mw cutoff 3 kDa) to obtain the final product – CRD-PEG-T7. The structure of the final product CRD-PEG-T7 was analyzed by 1H-NMR.
Preparation and characterization of the complexes
CRD-PEG-T7 was mixed with the model gene pGL-3 at the N/P ratio of 1, 5, 10, 15, 20, and 25 in deionized water. Basically, 0.878 μg CRD-PEG-T7 per 1 μg pDNA was equal to N/P=1. The samples were vortexed for 30 seconds and then incubated for 30 minutes at room temperature to obtain CRD-PEG-T7/pGL-3 particles. The particle size and zeta potential of CRD-PEG-T7/pGL-3 complexes were measured by Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK).
Agarose gel electrophoresis
Fresh CRD-PEG-T7/pGL-3 complexes with various N/P ratios were prepared before use. Electrophoresis was carried out at 100V in a 1% (w/v) agarose gel slab which contained GelRed for 20 minutes at room temperature. The location of pGL-3 bonds on the gel was visualized and photographed under ultraviolet light (254 nm). CRD-PEG/pGL-3 complexes were set as a control.
The ability of pGL-3 to release from the complexes was evaluated by heparin replacement test. The CRD-PEG-T7–pGL-3 complexes (N/P ratio=15) were prepared before use. Then, 10 μL heparin solution, with the concentration ranging from 50 to 1,000 μg/mL, was respectively added into each sample to replace pDNA. After 1-hour incubation, an agarose gel electrophoresis assay was conducted as described earlier.
Serum stability
The serum stability of CRD-PEG-T7–pGL-3 complexes (N/P ratio=15) was evaluated through mixing the particles with 60% FBS solution, followed by an incubation at 37°C for different time intervals. Naked pGL-3 was set as control. The mixture was then treated with 10 μL heparin solution (500 U/mL) to release pDNA. Agarose gel electrophoresis assay of the samples was conducted as described earlier.
Cellular uptake assay
Flow cytometry was used to measure the cellular uptake efficiency of the complexes. pGL-3 was labeled by YOYO-1 (YOYO-1-pGL-3) according to the manufacturing protocol before preparation of the complexes. DU145 cells were incubated in 12-well plates at a density of 2×105 cells per well for 24 hours. Then, the culture medium in each well was substituted by fresh culture medium containing CRD-PEG-T7–YOYO-1-pGL-3 complexes; following 4-hour incubation, the culture medium was discarded, and the cells were washed twice with PBS, trypsinized, and re-suspended in PBS to determine the percentage of positive YOYO-1 cells by a flow cytometer (BD, Franklin Lakes, NJ, USA). The CRD-PEG–pGL-3 complex was set as a control.
Intracellular localization experiment
A confocal laser scanning microscope (CLSM) was used to observe the intracellular distribution of the complexes. YOYO-1-pGL-3 was prepared before use. The 24-well plate used in this experiment has a cover-glass at the bottom. DU145 cells were incubated on these 24-well plates at a density of 1.0×105 cells per well for 24 hours. After removing the culture medium, fresh RPMI containing 10% FBS and CRD-PEG-T7–pGL-3 complexes was added into each well, followed by incubation for an additional 4 hours. Then, the culture medium was removes, and the cells were rinsed twice with PBS, fixed in fresh 4% paraformaldehyde solution, treated with DAPI to stain the nucleus, and, thereafter, sealed with mounting medium. The CRD-PEG–pGL-3 complex was set as a control. All samples were imaged by CLSM (Olympus Corporation, Tokyo, Japan).
Cytotoxicity assay
A Cell Counting Kit-8 (CCK-8) assay was conducted to evaluate the cytotoxicity of the polymers on PC3 and DU145 cells, which were incubated on 96-well plates at a density of 8×103 cells per well for 24 hours. When the cells achieved 70%–80% confluence, 100 μL CRD-PEG-T7–pGL-3 complex solution with various concentrations, ranging from 0 to 200 μg/mL, was added into each well. After 4-hour incubation, cells were washed twice by PBS, and incubated for an additional 20 hours in 100 μL fresh medium. Then, 100 μL fresh RPMI containing 10% CCK-8 solution was used to substitute for the culture medium, and followed by an additional 4 hours of incubation. Then, the absorbance of each well was examined through a microplate reader (Thermos, Schaumburg, IL, USA) at 450 nm. The absorbance of untreated cells was set as 100%. The CRD-PEG–pGL-3 complex was set as the control.
In vitro gene transfection efficacy
A gene transfection efficacy assay of the CRD-PEG-T7–DNA complex to DU145 cells was conducted in 24-well plates. Both pEGFP and pGL-3 were used as report genes. DU145 cells were incubated in a 24-well plate at a density of 8×104 cells per well for 24 hours to reach 70%–80% confluence. Before transfection, the culture medium was replaced by fresh FBS-free medium. The cells were then incubated with the CRD-PEG-T7–pEGFP complexes with various N/P ratios for 4 hours. Thereafter, the medium was then replaced by fresh culture medium containing 10% FBS and the cell culture was expanded for an additional 24 hours. To evaluate pEGFP gene transfection efficiency, EGFP expression was observed and photographed using a fluorescence microscope (Olympus Corporation). The CRD-PEG–pEGFP complex was used as a control.
To assess pGL-3 transfection, the luciferase assay was carried out according to the manufacturer’s protocol. In addition, the luciferase activity was normalized with the amount of cellular protein that was determined by a BCA protein assay kit. The CRD-PEG–DNA complex was set as a control.
Cell migration assay
A Transwell assay was conducted to measure cell migration. DU145 cells transfected by CRD-PEG-T7–DPM complexes (N/P ratio=15) in 200 μL FBS-free RPMI culture medium were added into the upper chambers of Transwell (Matrigel-free polycarbonate membranes, pore size 8 μm) at a density of 5×103 cells per chamber; 500 μL RPMI medium with 15% FBS, which was used as an attractant for cell migration, was added into the lower chamber. After 48-hour incubation, the cells in the upper chambers were removed and the cells on the bottom of the membrane were fixed with methanol. Then, the migrated cells were stained with 1% crystal violet solution. Before photography, excess crystal violet was washed off with deionized water. Migrated cells were imaged with a light microscope (Optical Instrument Factory, Shanghai, People’s Republic of China) and counted using ImageJ software (National Institutes of Health, Bethesda, MD, USA) in five randomly selected microscopic fields.
In vivo distribution assay
A xenograft tumor model was generated for the in vivo distribution study of the CRD-PEG-T7–pDNA complex. The bone metastasis tumor model24 was established by injecting 2 mL DU145 cell suspension (1×106 cells/mL) into the tibia of 5-week-old male BALB/c nude mice weighing from 18 to 22 g (Joint Laboratories, Suzhou, People’s Republic of China). When the tumor volume reached approximately 100 mm3, 12 mice were equally randomized to four groups. Through their caudal vein, the mice in the four groups were injected with saline, naked Cy-7, the Cy-7-CRD-PEG–pGL-3 complex (N/P=15), and the Cy-7-CRD-PEG-T7–pGL-3 complex (N/P=15) at a dosage of 1 mg/kg, and then scanned at 1, 4,12, and 24 hours post-injection using an in vivo imaging system (Bio-Real Sciences, Salzberg, Austria). Mice treated with saline were set as the control. All the mice were euthanized 24 hours post-injection to isolate the tumor, heart, liver, spleen, lungs, and kidneys for imaging. The exposure time was set as 2 seconds per image. All the images were analyzed through the software Bio-Real Quick View 3000.
Statistical analysis
Data are shown as the mean ± SD (n=3). The Student’s t-test was conducted to determine statistically significant differences, represented by *P<0.05.
Results
Synthesis and characterization of the polymers
The mass-average Mw of CRD is presented in Table 1. With the decrease in the amount of cysteine, the Mw of CRD increased enormously as compared with the RD monomer (2,008.4 Da). The 1H-NMR spectra of CRD are shown in Figure 3B. The solvent peak of D2O was at 4.7 ppm. Peaks at 1.5 and 4.4 ppm were attributed to arginine protons, and peaks at 2.6 ppm were attributed to aspartic acid protons. Both 1H-NMR results and GPC results (data not shown) indicated successful synthesis of CRD.
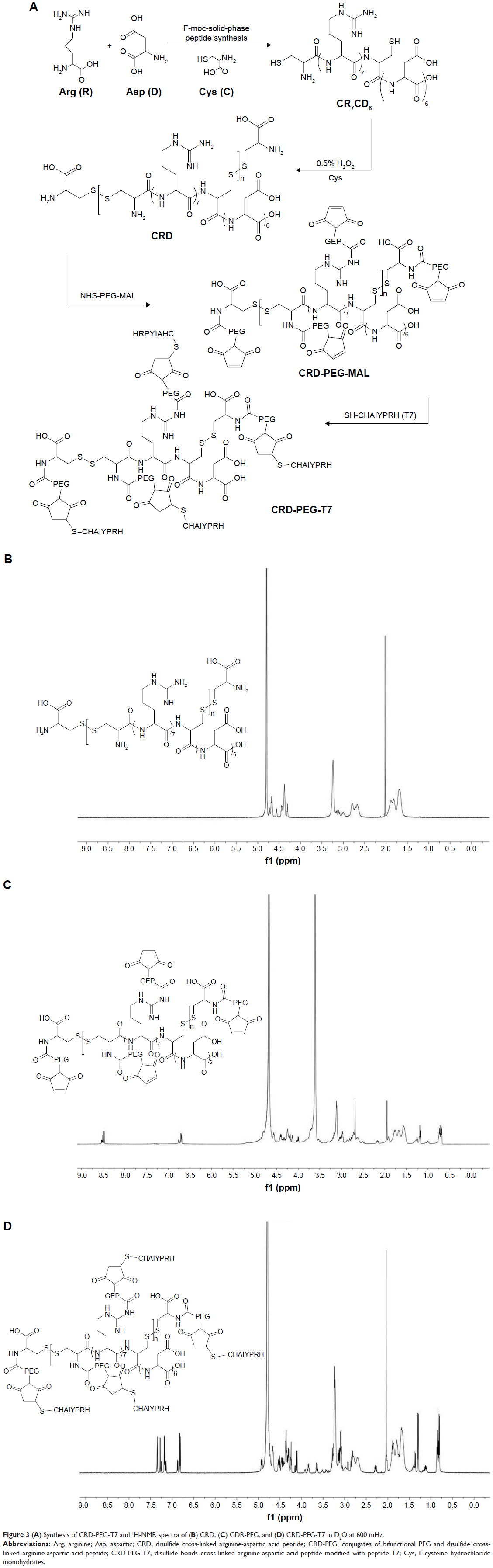 | Figure 3 (A) Synthesis of CRD-PEG-T7 and 1H-NMR spectra of (B) CRD, (C) CDR-PEG, and (D) CRD-PEG-T7 in D2O at 600 mHz. |
As shown in Figure 3D, peptide T7 was successfully conjugated to polypeptide via NHS-PEG-MAL. The solvent peak was at 4.7 ppm. The 1H-NMR spectra of CRD-PEG (Figure 3C) showed the distinctive peak of the MAL group at 8.5 ppm, which disappeared in the spectra of CRD-PEG-T7 (Figure 3D), whereas the PEG skeleton still had a sharp peak at 3.3 ppm. Peaks at 7.3 ppm were attributed to protons of peptide T7, indicating that peptide T7 was conjugated to CRD-PEG and the final gene delivery vector CRD-PEG-T7 was successfully synthesized.
Characterization of the polymer–pDNA complex
As shown in Figure 4, the particle size and zeta potential were greatly influenced by the N/P ratio. Figure 4A shows that when the N/P ratio was higher than 5, all particles carried a positive surface charge and the zeta potential increased as the N/P ratio grew. As shown in Figure 4B, smaller particles were formed when the N/P ratio reached 15. Besides that, the cross-linking degree of the carrier affected the particle size and the zeta potential of the complexes with the same N/P ratio. Among all of the particles with an N/P ratio of 15, the CRD-PEG-T7-2–pGL-3 complex reached an appropriate size of 158.8±10.3 nm, which was significantly smaller than the CRD-PEG-T7-3–pGL-3 complex (227.0±32.0 nm) and CRD-PEG-T7-4–pGL-3 complex (257.6±30.1 nm). The zeta potential of the CRD-PEG-T7-2–pGL-3 complex (N/P=15) was 15.8±0.6 mV. The nanoscale size and positive potential surface of the CRD-PEG-T7-2–pGL-3 complex were expected to exhibit great gene transfection efficiency. Thus, the CRD-PEG-T7-2–pGL-3 complex (N/P=15) was selected for further study.
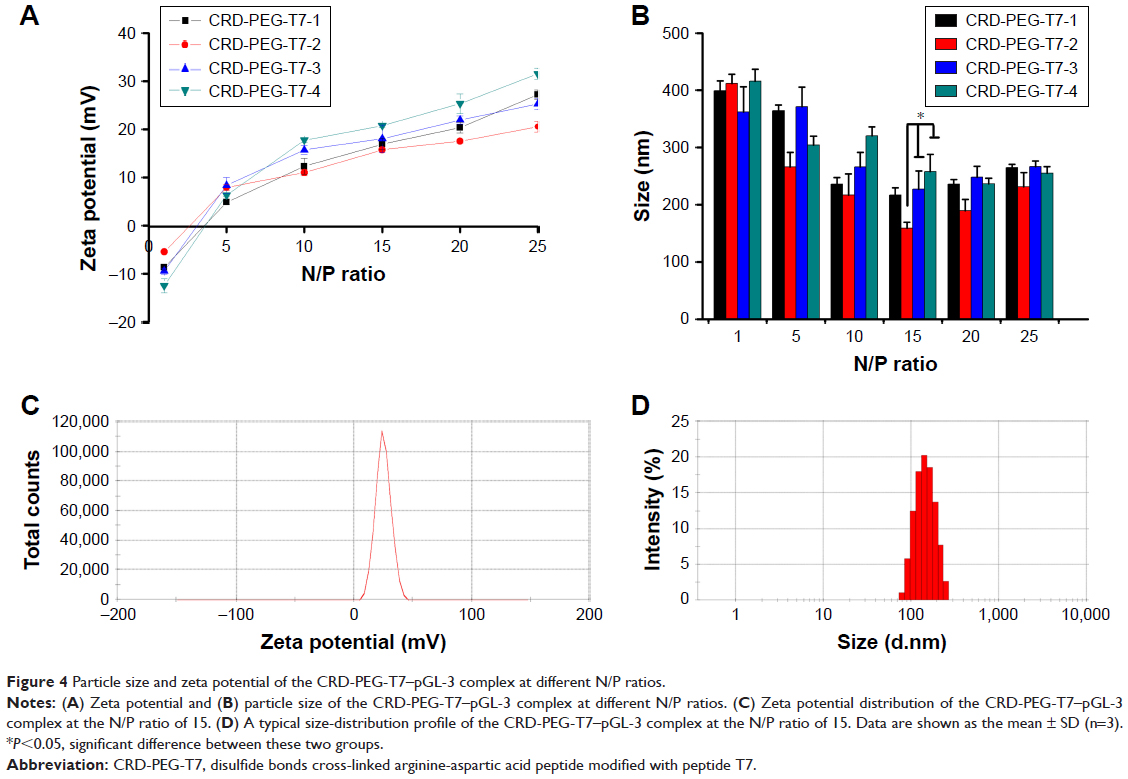 | Figure 4 Particle size and zeta potential of the CRD-PEG-T7–pGL-3 complex at different N/P ratios. |
Furthermore, an agarose gel electrophoresis assay was conducted to examine the DNA condensation ability of CRD-PEG and CRD-PEG-T7. As shown in Figure 5, DPM was completely retarded when the N/P ratio reached 2 for both CRD and T7 modified CRD, thus indicating that these polymers enjoyed favorable binding affinity to DNA and the binding affinity was narrowly influenced by T7 modification. As previous studies demonstrated that the cytoplasm could reduce disulfide bonds and weaken the binding affinity between DNA and the disulfide cross-linked peptides,25 we hypothesized that the complexes were stable in the extracellular environment relative to intracellular homeostasis, which means that the cytoplasm could trigger DNA release both in vitro and in vivo.
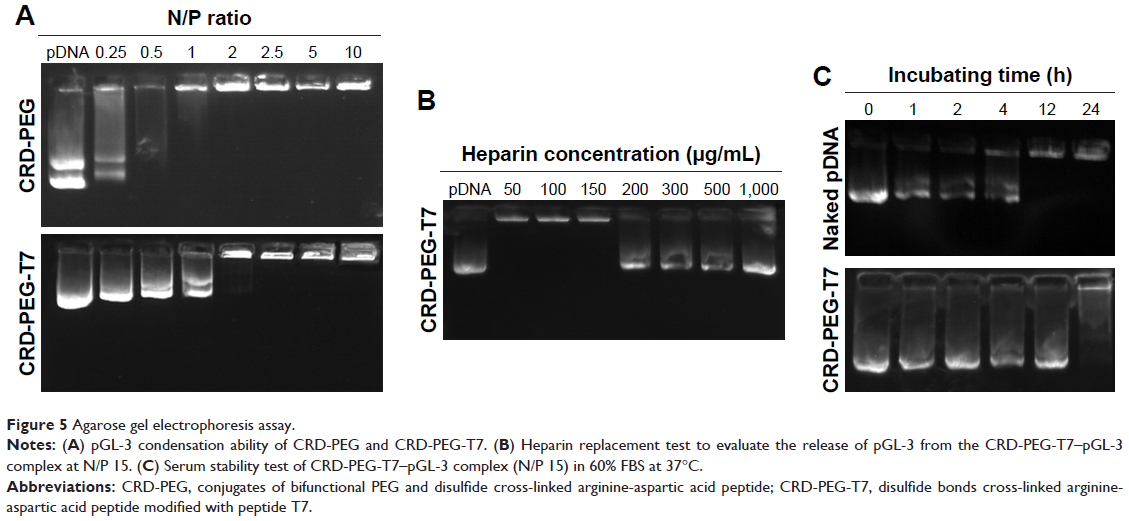 | Figure 5 Agarose gel electrophoresis assay. |
To determine the DNA-releasing capability from the CRD-PEG-T7–pGL-3 complex, a heparin replacement test was conducted. Heparin, a highly negatively charged sulfated glycosaminoglycan, was utilized to replace the pGL-3 in this study. Figure 5B shows that pGL-3 began being replaced when the heparin concentration reached 200 μg/mL, and was completely replaced when heparin concentration was up to 1,000 μg/mL.
The serum stability test is conducted to evaluate the protective ability of CRD-PEG-T7 in the presence of FBS. As shown in Figure 5C, naked pDNA was completely degraded in 60% FBS between 4 and 12 hours, whereas it was prevented from degrading for up to 24 hours when complexed with CRD-PEG-T7. As cellular uptake of the complexes generally occurred several hours after transfection, it would be sufficient for the protective effect provided by CRD-PEG-T7.
Cellular uptake and intracellular distribution
Cellular uptake is essential for successful gene delivery and superior transfection efficiency. In this study, the YOYO-1-labeled complex was used to present cellular uptake of the complexes in DU145 cells, which were prepared through complexing YOYO-1-labeled pGL-3 (YOYO-1-pGL-3) and CRD-PEG-T7 at an N/P ratio of 15. As is shown in Figure 6, the percentage of YOYO-1–positive cells was 47.06%±8.85% of the CRD-PEG–YOYO-1-pGL-3 complex and 70.69%±4.75% of the CRD-PEG-T7–YOYO-1-pGL-3 complex, showing a significant difference between these two groups. The significantly increased cellular uptake indicates that the peptide T7-modified vector had a potentially higher gene transfection efficiency.
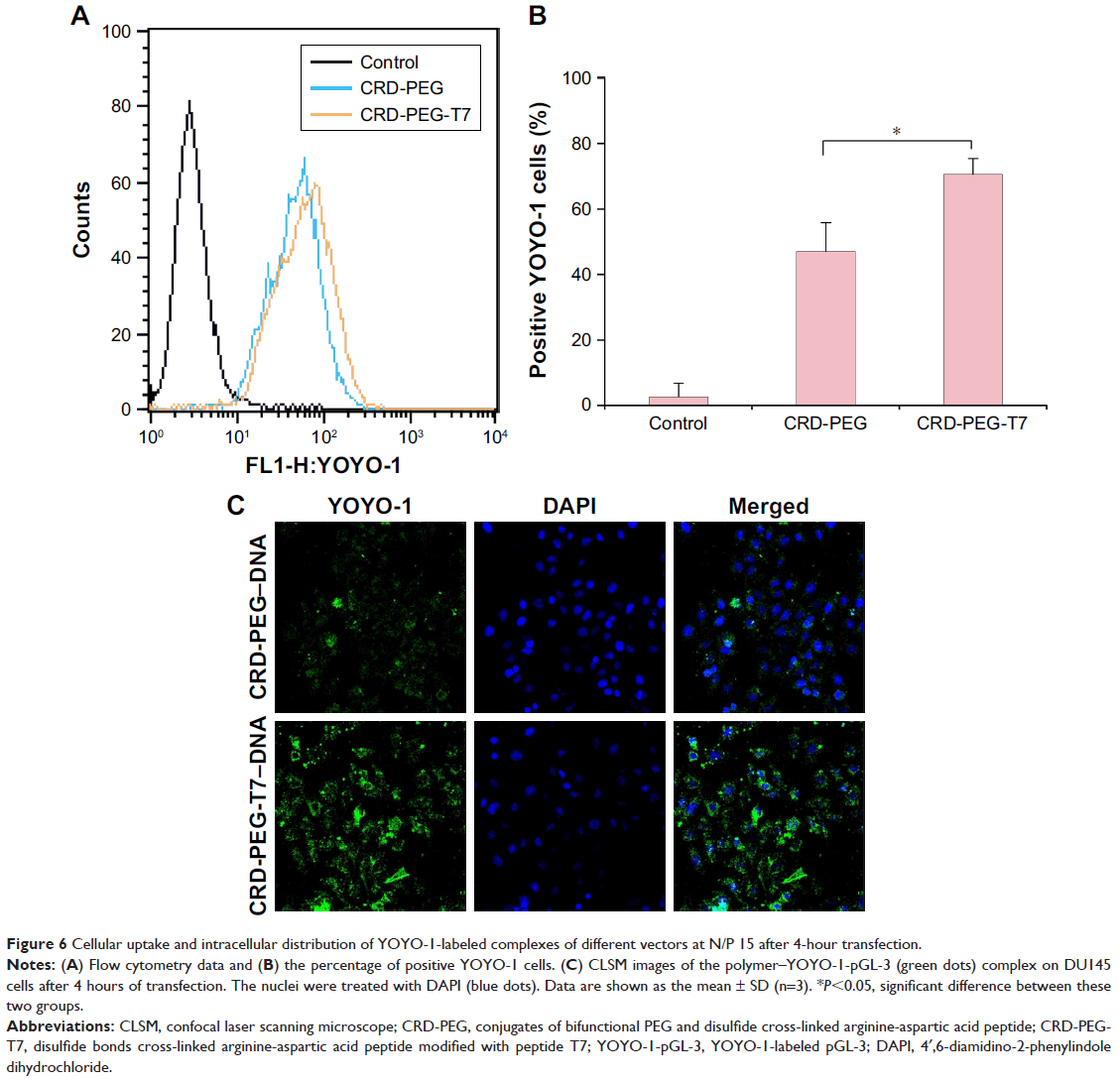 | Figure 6 Cellular uptake and intracellular distribution of YOYO-1-labeled complexes of different vectors at N/P 15 after 4-hour transfection. |
Figure 6C shows the regions where the complexes were distributed in DU145 cells 4 hours post transfection. The green dots represent the YOYO-1-labeled complex, and the blue dots were cell nuclei dyed by DAPI. After 4-hour incubation, the complexes were distributed mainly in the cytoplasm, and the fluorescence intensity of the CRD-PEG-T7–YOYO-1-pGL-3 complex was significantly higher than that of the CRD-PEG–YOYO-1-pGL-3 complex.
Cytotoxicity
The cytotoxicity of the CRD-PEG-T7–pGL-3 complex of various concentrations in DU145 cells and PC3 cells was evaluated by a CCK-8 assay. Figure 7 shows the cell viability remained as high as 60% at 24 hours after incubation even when the concentration rose to 200 μg/mL, indicating favorable biocompatibility with DU145 cells and PC3 cells.
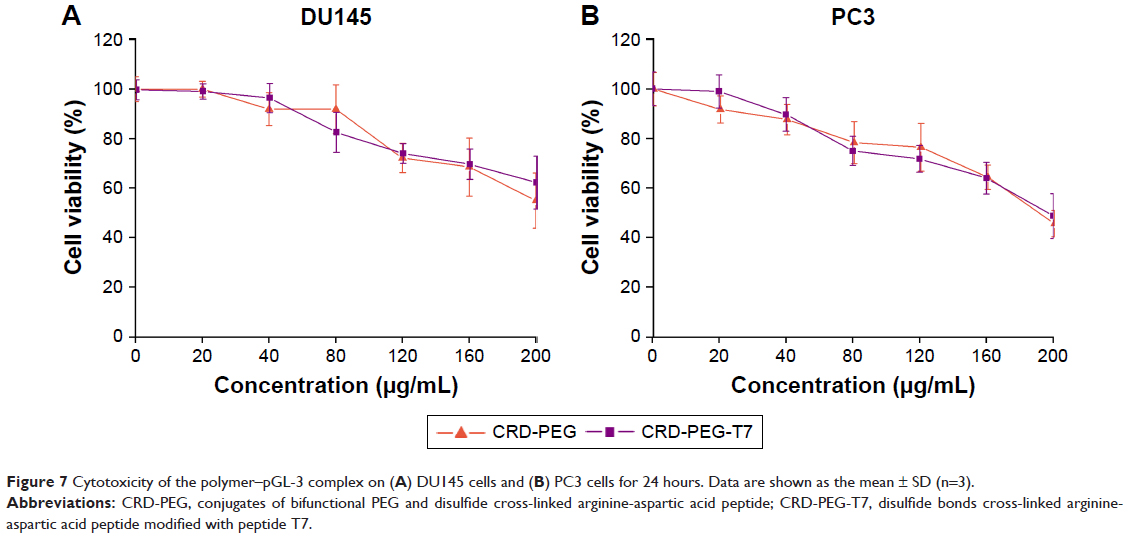 | Figure 7 Cytotoxicity of the polymer–pGL-3 complex on (A) DU145 cells and (B) PC3 cells for 24 hours. Data are shown as the mean ± SD (n=3). |
Gene transfection efficiency
Gene transfection efficacy of the complex in DU145 cells was measured using pEGFP and luciferase as reporter genes. As shown in Figure 8, the fluorescent intensity was influenced by the N/P ratio. The peptide T7-modified complex induced higher EGFP intensity at the N/P ratio of 15 (Figure 8A) when the luciferase activity of the CRD-PEG-T7–pDNA complex (2.91541×105) was four-fold higher than that of the CRD-PEG–pDNA complex (7.0286×104) in DU145 cells, exhibiting a significant enhancement in gene expression level.
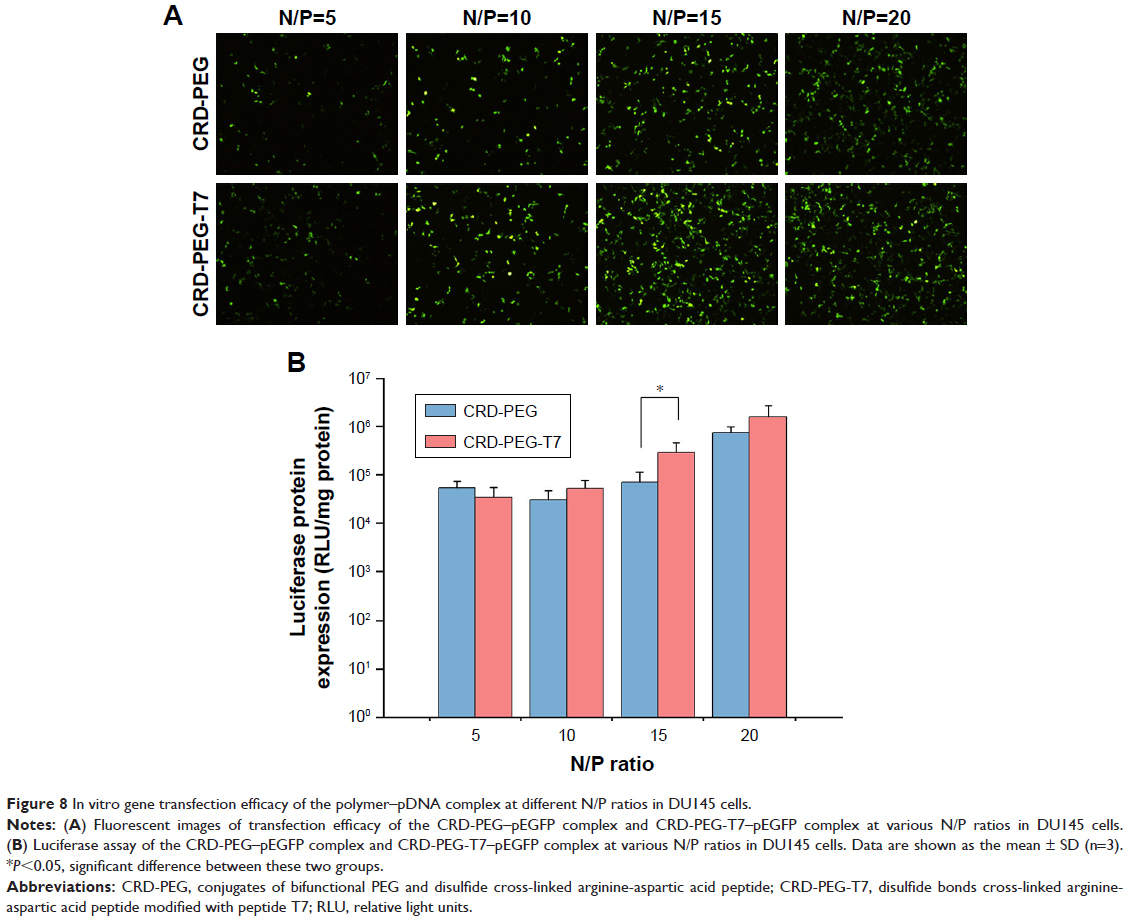 | Figure 8 In vitro gene transfection efficacy of the polymer–pDNA complex at different N/P ratios in DU145 cells. |
Cell migration assay
The Transwell migration assay was conducted in vitro to evaluate the effect of DPM expression on the migratory tendency of DU145 cells. The violet regions in Figure 9 were migrated cells stained by crystal violet solution, showing that the migratory ability of cells treated with the complex containing DPM was reduced, as represented by the decreased number of migrated cells when compared with the untreated cells. Additionally, the number of migrated cells in the CRD-PEG-T7–DPM group (245±30) was significantly decreased as compared with that in the CRD-PEG–DPM group (291±27).
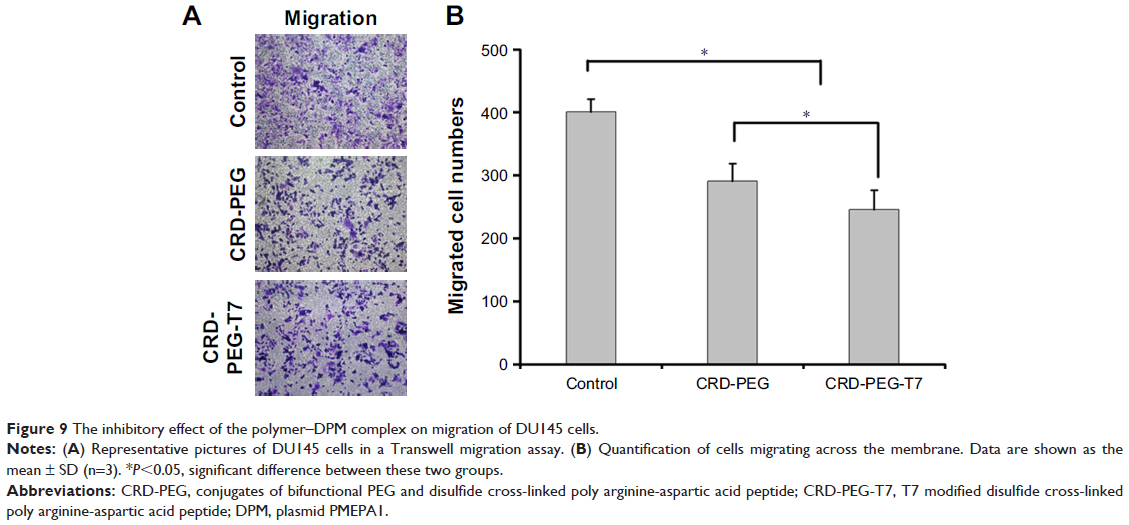 | Figure 9 The inhibitory effect of the polymer–DPM complex on migration of DU145 cells. |
In vivo distribution
In vivo distribution is an essential indicator for the tumor-targeting effect. In this study, we used hydrosoluble cyanine dye Cy-7, which has weak background signals and strong fluorescent intensity, as an indicator for biodistribution. All the nude mice were imaged at 1, 4, 12, and 24 hours post caudal vein injection of naked Cy-7, the Cy-7-CRD-PEG-T7–pGL-3 complex, and the Cy-7-CRD-PEG–pGL-3 complex. As shown in Figure 10, no fluorescent signal was detected at any set time point in the mice treated with saline. In contrast, a significantly stronger fluorescent signal was observed at the tumor site in the mice treated with the T7-modified complex 4 hours after injection, and the signal could be detected even 24 hours after injection, compared with the Cy-7-CRD-PEG–pGL-3 group. However, the fluorescent signals of naked Cy-7 gradually weakened 24 hours after injection, and they were mainly detected in the liver and lungs, with little accumulation at the tumor site.
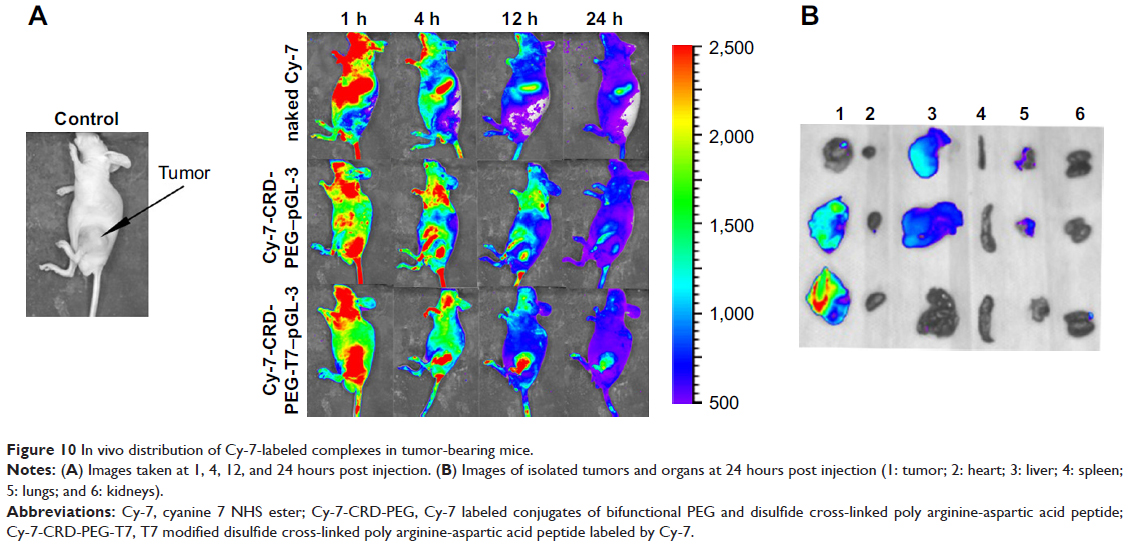 | Figure 10 In vivo distribution of Cy-7-labeled complexes in tumor-bearing mice. |
The tumors and organs were isolated 24 hours after injection to investigate the biodistribution of the complexes. As shown in Figure 10B, stronger fluorescent signals were observed at the tumor area of the mice treated with the peptide T7-modified complex, as compared with the mice treated with unmodified particles. No fluorescent signal was observed in the tumors of mice treated with naked Cy-7. These results indicate that the present gene delivery system achieved tumor targeting after injection.
Discussion
In this study, R7D6 monomers were cross-linked with cysteine under the oxidizing environment of low-concentration H2O2 solutions. Disulfide bonds were formed between sulfhydryl groups of R7D6. Cysteine was used to halt the chain reaction of polymerization. The synthesis of CRD-PEG-T7 is shown in Figure 3A. Such cationic CPPs cross-linked by disulfide bonds featured rapid gene release in the cytoplasm with low cytotoxicity.10,12 Therefore, adjusting the molar ratio of R7D6 to Cys could regulate the cross-linkage degree and Mw of the polymers. The oxidizing agent H2O2 was easier to remove, when compared to other oxidizing agents including dimethyl sulfoxide and iodine. The 1H-NMR spectra of CRD-PEG (Figure 3C) showed the distinctive peak of the MAL group at 8.5 ppm, which disappeared in the spectra of CRD-PEG-T7 (Figure 3D), whereas the PEG skeleton still had a sharp peak at 3.3 ppm. Peaks at 7.3 ppm were attributed to protons of peptide T7, indicating that peptide T7 was successfully conjugated to CRD-PEG. According to the results of 1H-NMR in Figure 3, we successfully synthesized CRD-PEG-T7 and the gene delivery vector was able to form nano-scale particles with a positively charged surface of pDNA. The Z-average size of the CRD-PEG-T7-2–pDNA complex (N/P ratio=15) was 158.8 nm, making it possible for it to accumulate in the tumor area according to enhanced permeability and retention (EPR) effect.26
Figure 2 illustrates the formation of the CRD-PEG-T7–DPM complex and its cellular uptake process by BMPCa cells. The results of cellular uptake showed that the CRD-PEG-T7 gene delivery vector had a relatively high cellular uptake efficacy. It is probably because the high expression of TfR on tumor cells facilitated DU145 cells to actively identify and take up the complex, thereby increasing the cellular uptake efficacy. Han et al27 reported that, compared with unmodified carriers, the T7-modified gene carrier was more efficiently internalized via targeting the TfR on tumor cells. In terms of the percentage of positive YOYO-1 cells, the present study demonstrated that the cellular uptake efficiency of the CRD-PEG-T7 gene delivery system was 1.5-fold higher than that of the CRD-PEG system.
Our study showed that the admirable gene transfection efficiency of the system was attributed to the significantly enhanced cellular uptake. Figure 8 shows that the gene transfection efficiency of the T7-modified complex was significantly enhanced when compared to the complex without T7 modification; this is partially due to the easily broken disulfide bonds in the reductive environment in cytoplasm of tumor cells. Liu et al11 reported the use of disulfide cross-linked dendrimers as the gene delivery vector and achieved a superior gene transfection efficacy with minimal cytotoxicity. It was reported15 that there is an enormous amount of glutathione (known as a reducing molecule) inside tumor cells, thus providing a favorable environment for disulfide bonds to break and inducing a rapid release of DNA, which was also validated by the result of the Transwell assay. Cell migration was significantly inhibited as a result of the expression of DPM and blockage of the TGF-β signaling pathway, which promoted tumor invasion and bone metastasis.7 Feng et al28 found that the inhibition of PMEPA1 expression contributed to tumor progression.
The in vivo distribution assay demonstrated that the peptide T7 complex mainly accumulated inside the tumor owing to the bone-targeting effect of the peptide sequence of aspartic acid, the tumor-targeting effect of peptide T7, and the EPR effect. Figure 1 illustrates the targeting mechanism of the CRD-PEG-T7–DNA complex to BMPCa cells in the bone tissue. A peptide sequence of aspartic acid, which could exclusively bind to hydroxyapatite (the main component of the bone tissue), has already been used as a bone-targeting agent.17,18 Therefore, with the assistance of the aspartic acid sequence, peptide T7, and the nanoscale size, the particles could accumulate exclusively at the bone metastatic tumor site. The accumulation of naked Cy-7 in the liver was probably due to the effect of the reticuloendothelial system.29
The result of a cytotoxicity assay showed that the CRD-PEG-T7 gene delivery system enjoyed favorable biocompatibility. Previous studies11 have reported that one of the advantages of such vectors is their low cytotoxicity. This means that T7 modification increased the targeting efficacy without inducing additional cytotoxicity to normal cells. These results demonstrate that CRD-PEG-T7, with a dual-targeting ability bestowed by the aspartic acid peptide and peptide T7, is appropriate for pDNA delivery.
Conclusion
In this study, we successfully developed a T7-modified cationic polypeptide CRD-PEG-T7 for pDNA delivery. This gene delivery system exhibited admirable gene transfection efficacy, favorable biocompatibility, and a dual-targeting effect owing to the conjugation of the aspartic acid peptide and T7 peptide, suggesting that it is a promising gene delivery vector for bone-metastatic PCa.
Acknowledgments
This study was sponsored by the National Natural Science Foundation of China (grant no 81772749) and Shanghai Rising-Star Program (grant no 18QB1400400).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Nakazawa M, Paller C, Kyprianou N. Mechanisms of therapeutic resistance in prostate cancer. Curr Oncol Rep. 2017;19(2):13–33. | ||
Clement JM, Sweeney CJ. Evolving treatment of oligometastatic Hormone-Sensitive prostate cancer. J Oncol Pract. 2017;13(1):9–18. | ||
Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 – an update. J Gene Med. 2013;15(2):65–77. | ||
Marangoni K, Neves AF, Rocha RM, et al. Prostate-specific RNA aptamer: promising nucleic acid antibody-like cancer detection. Sci Rep. 2015;5:121–134. | ||
Li H, Mohamed AA, Sharad S, et al. Silencing of PMEPA1 accelerates the growth of prostate cancer cells through AR, NEDD4 and PTEN. Oncotarget. 2015;6(17):15137–15149. | ||
Fournier PG, Juárez P, Jiang G, et al. The TGF-β signaling regulator PMEPA1 suppresses prostate cancer metastases to bone. Cancer Cell. 2015;27(6):809–821. | ||
Vazquez E, Roldán M, Diez-Gil C, et al. Protein nanodisk assembling and intracellular trafficking powered by an arginine-rich (R9) peptide. Nanomedicine. 2010;5(2):259–268. | ||
Futaki S, Hirose H, Nakase I. Arginine-rich peptides: methods of translocation through biological membranes. Curr Pharm Des. 2013;19(16):2863–2868. | ||
Yao C, Liu J, Wu X, et al. Reducible self-assembling cationic polypeptide-based micelles mediate co-delivery of doxorubicin and microRNA-34a for androgen-independent prostate cancer therapy. J Control Release. 2016;232:203–214. | ||
Liu H, Wang H, Yang W, Cheng Y. Disulfide cross-linked low generation dendrimers with high gene transfection efficacy, low cytotoxicity, and low cost. J Am Chem Soc. 2012;134(42):17680–17687. | ||
Tai Z, Wang X, Tian J, et al. Biodegradable stearylated peptide with internal disulfide bonds for efficient delivery of siRNA in vitro and in vivo. Biomacromolecules. 2015;16(4):1119–1130. | ||
Kim TI, Ou M, Lee M, Kim SW. Arginine-grafted bioreducible poly(disulfide amine) for gene delivery systems. Biomaterials. 2009;30(4):658–664. | ||
Won YW, Yoon SM, Lee KM, Kim YH. Poly(oligo-D-arginine) with internal disulfide linkages as a cytoplasm-sensitive carrier for siRNA delivery. Mol Ther. 2011;19(2):372–380. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
Son S, Namgung R, Kim J, Singha K, Kim WJ. Bioreducible polymers for gene silencing and delivery. Acc Chem Res. 2012;45(7):1100–1112. | ||
Low SA, Yang J, Kopeček J. Bone-targeted acid-sensitive doxorubicin conjugate micelles as potential osteosarcoma therapeutics. Bioconjug Chem. 2014;25(11):2012–2020. | ||
Jiang T, Yu X, Carbone EJ, Nelson C, Kan HM, Lo KW. Poly aspartic acid peptide-linked PLGA based nanoscale particles: potential for bone-targeting drug delivery applications. Int J Pharm. 2014;475(1–2):547–557. | ||
Oh S, Kim BJ, Singh NP, Lai H, Sasaki T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. 2009;274(1):33–39. | ||
Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121(2):159–176. | ||
Lee JH, Engler JA, Collawn JF. Receptor mediated uptake of peptides that bind the human transferrin receptor. Febs J. 2010;268(7):2004–2012. | ||
Han L, Li J, Huang S, et al. Peptide-conjugated polyamidoamine dendrimer as a nanoscale tumor-targeted T1 magnetic resonance imaging contrast agent. Biomaterials. 2011;32(11):2989–2998. | ||
Kuang Y, An S, Guo Y, et al. T7 peptide-functionalized nanoparticles utilizing RNA interference for glioma dual targeting. Int J Pharm. 2013;454(1):11–20. | ||
Havens AM, Pedersen EA, Shiozawa Y, et al. An in vivo mouse model for human prostate cancer metastasis. Neoplasia. 2008;10(4):371–IN4. | ||
Kim TI, Ou M, Lee M, Kim SW. Arginine-grafted bioreducible poly(disulfide amine) for gene delivery systems. Biomaterials. 2009;30(4):658–664. | ||
Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69(1):1–9. | ||
Han L, Huang R, Liu S, Huang S, Jiang C. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol Pharm. 2010;7(6):2156–2165. | ||
Feng S, Zhu X, Fan B, Xie D, Li T, Zhang X. miR-19a-3p targets PMEPA1 and induces prostate cancer cell proliferation, migration and invasion. Mol Med Rep. 2016;13(5):4030–4038. | ||
Cho HJ, Yoon IS, Yoon HY, et al. Polyethylene glycol-conjugated hyaluronic acid-ceramide self-assembled nanoparticles for targeted delivery of doxorubicin. Biomaterials. 2012;33(4):1190–1200. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.