Back to Journals » Clinical Ophthalmology » Volume 15
Pentosan Polysulfate Sodium-Associated Pigmentary Retinopathy: Risk Factors and Fundus Findings
Authors Leung EH, Sharma S, Levie-Sprick A, Lee GD, Cho H, Mukkamala SK
Received 23 September 2021
Accepted for publication 16 November 2021
Published 24 December 2021 Volume 2021:15 Pages 4809—4816
DOI https://doi.org/10.2147/OPTH.S340041
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Ella H Leung,1 Sahana Sharma,2 Ana Levie-Sprick,1 Gregory D Lee,1 Hyung Cho,1 Krishna Mukkamala1
1Georgia Retina, Atlanta, GA, USA; 2Vanderbilt University, Nashville, TN, USA
Correspondence: Ella H Leung
Georgia Retina, 833 Campbell Hill St NW, Suite 300, Marietta, GA, 30060, USA
Tel +1-770-218-1888
Email [email protected]
Purpose: To evaluate the risk factors and fundus findings of patients with potential PPS-associated retinopathy.
Patients and Methods: A retrospective chart review was performed of patients exposed to PPS who had a dilated fundus examination at a large retina-only practice from 2018– 21. Multimodal images were evaluated by masked reviewers.
Results: A total of 148 patients were included, of whom 33 (22%) had PPS-associated retinopathy, and 115 (78%) did not. The mean age was 60.3 years old, and the mean follow-up was 11.8 months. The PPS-associated retinopathy group had higher mean cumulative doses of PPS (1600g± 849 vs 864g± 852, P < 0.0001, Mann–Whitney test) and longer duration of PPS use (13.6 years vs 7.48, P < 0.0001). There was no statistically significant difference based on a history of kidney or liver disease or the dosage per day for the weight, body mass index, body surface area, or lean body weight. Of the patients with PPS-associated retinopathy whose genetic results were available, 15 of 16 (93%) were heterozygous for variants of uncertain significance.
Conclusion: A longer duration of PPS use and higher cumulative dosage of PPS were associated with an increased risk of developing PPS-associated pigmentary retinopathy. The role of genetic mutations in patients exposed to PPS is still to be determined.
Keywords: pentosan polysulfate sodium, pigmentary retinopathy, pharmacotoxicity, inherited retinal dystrophies, genetics
Introduction
Interstitial cystitis (IC), also known as bladder pain syndrome, is characterized by bladder wall irritation, urinary urgency, and bladder or pelvic pain.1 According to the American Urology Association, the first-line treatments include stress and pain management with behavioral modification. Second-line treatments include oral pentosan polysulfate sodium (PPS, Elmiron, Janssen Pharmaceuticals, Titusville, NJ) and pelvic floor physical therapy.2
Pentosan polysulfate sodium is a heparin-like compound that adheres to the bladder wall mucosa. In initial clinical trials, it improved the pain score in 29–34% of patients.3 PPS is partially desulfated by the liver and spleen, partially depolymerized in the kidney, and partly excreted through the kidneys and gastrointestinal tract.3,4 It is possible that the sulfation pathway can be saturated, which may lead to accumulation of the drug in the body. PPS has recently been associated with pigmentary maculopathy, but the association has been debated.5–11 The purpose of the study was to evaluate the risk factors and fundus findings for patients with pentosan polysulfate (PPS)-associated retinopathy.
Methods
A retrospective chart review was performed of patients with a history of PPS exposure and a dilated fundus examination between January 2018- September 2021 at a large retina-only private practice group. The exclusion criteria included pre-existing diagnoses of inherited retinal dystrophies, pathogenic variants in genes associated with retinopathies, age less than 18 years old, incomplete dosage information, and exposure to other medications associated with maculopathies. The Sterling Institutional Review Board (IRB) waived the IRB approval for the retrospective study. The study and data handling complied with all local and federal laws, the Health Insurance Portability and Accountability Act (HIPAA) regulations, and the tenants of the Declaration of Helsinki.
Patient demographic information was obtained, including ocular and systemic comorbidities, best corrected visual acuities, and duration and dose of PPS. The evaluation and management of patients were individualized based on the recommendations of the treating physician, but efforts were made to obtain multimodal imaging with fundus autofluorescence, optical coherence tomographies (OCTs), and fundus photos on all patients and to specifically ask patients about their dosage information and visual symptoms at their clinical visits. The data was analyzed with Prism (V9.0.0, GraphPad Software, San Diego, California USA), with a P value < 0.05 being considered statistically significant. The data was analyzed for normal distributions with the Shapiro–Wilks test, and the corresponding parametric or non-parametric tests were applied.
Two independent retina specialists graded the multimodal images, masked to the medical histories; in the event of discordance, a third, independent, masked retina specialist evaluated the images, and a consensus was reached after reviewing the medical records. Patients were grouped as “PPS-retinopathy” if they met the 6 point grading criteria described by Barnes et al:10 bilateral densely packed parafoveal hyperpigmentation and subretinal yellow deposits, patchy retinal pigment epithelial (RPE) atrophy, peripapillary hypoautofluorescent halo, hyperautofluorescent spots less than 2 venule widths, lack of typical drusen, parafoveal hypo- and hyperautofluorescent spots centered around the fovea on fundus autofluorescence (FAF), and focal RPE thickening or elevation with hyper-reflectance on near infrared images on OCT.6,10,12 Patients with presumed PPS-retinopathy were then categorized into mild, moderate, and severe. Mild disease was defined as RPE mottling and speckled hyper and hypoautofluorescence on fundus autofluorescence but no atrophy on FAF or OCT; moderate disease included well demarcated areas of atrophy on FAF that correlated with chorioretinal atrophy in the RPE and outer retina at least 250μm in diameter with a continuous zone of RPE attenuation and photoreceptor degeneration; severe PPS-associated retinopathy was associated with chorioretinal atrophy involving the central fovea.14
Fundus autofluorescence images were processed in Adobe Photoshop CS6 (Adobe Systems Inc.) and stacked using image landmarks; Image J software was used to measure the areas of macular atrophy. In order to minimize potential distortion, magnification, and rotation between fundus autofluorescence images obtained on different machines, the average of 3 measurements, measured in vertical disc diameters, was used.16
Results
A total of 168 patients met the inclusion criteria. Twenty patients were excluded; one patient had a pre-existing history of cone-rod dystrophy, 7 were heterozygous for pathogenic genetic variants associated with retinopathy, 7 had incomplete dosage information, 4 were on hydroxychloroquine, and one was a child. There were therefore 148 patients included in the study.
The demographic information is summarized in Table 1. The mean age was 60.3 years old (range: 24–93), and the mean follow-up was 11.8±24 months from the initial ophthalmic exam. A total of 33 patients met the diagnostic criteria for PPS-associated retinopathy, of whom 12 (36%) had mild retinopathy, 21 (64%) had moderate, and none had severe disease. While the past medical histories were similar between the two groups, the retinopathy group had a greater proportion of females (100% vs 86%, P=0.023), longer follow-up (32.7±34 vs 5.74±17 months, P<0.0001), and greater family history of macular degeneration (28% vs 12%, P=0.028).
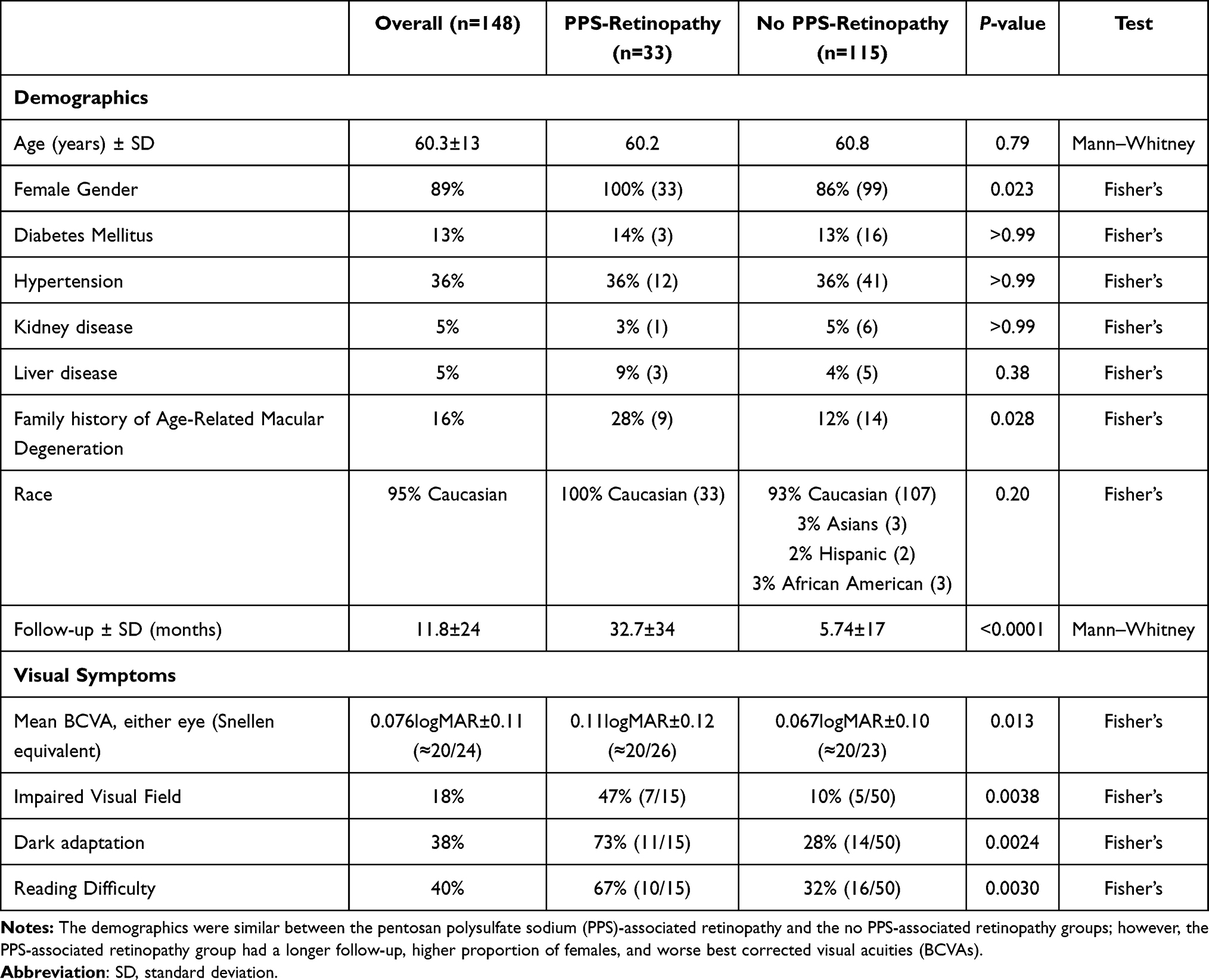 |
Table 1 Demographics Information |
A total of 115 (78%) patients did not fulfill all the criteria for PPS-associated retinopathy and were classified as “no-PPS-retinopathy.” In analyzing the multimodal images, 96% (110/115) met at least one grading criteria for PPS-associated retinopathy, most commonly absence of typical drusen, with 16% (18/115) having hyperpigmentation in the macula. While a peripapillary halo helped to differentiate between an inherited retinal dystrophy from PPS-associated retinopathy, 15 of the 21 patients (71%) in the PPS-associated retinopathy group with peripapillary involvement also had peripapillary atrophy, making it difficult to assess for a halo.
There was a statistically significant difference in the cumulative PPS dose and duration of PPS between those with PPS-associated retinopathy and those without (1600g±849 vs 864g±852, P<0.0001, Mann–Whitney; and 13.6 years vs 7.48, P<0.0001, respectively, Table 2, Figure 1). The risk of developing retinopathy increased over time and with increased dosage of PPS (Table 3). In multiple linear regression analyses, there was collinearity of the variables.
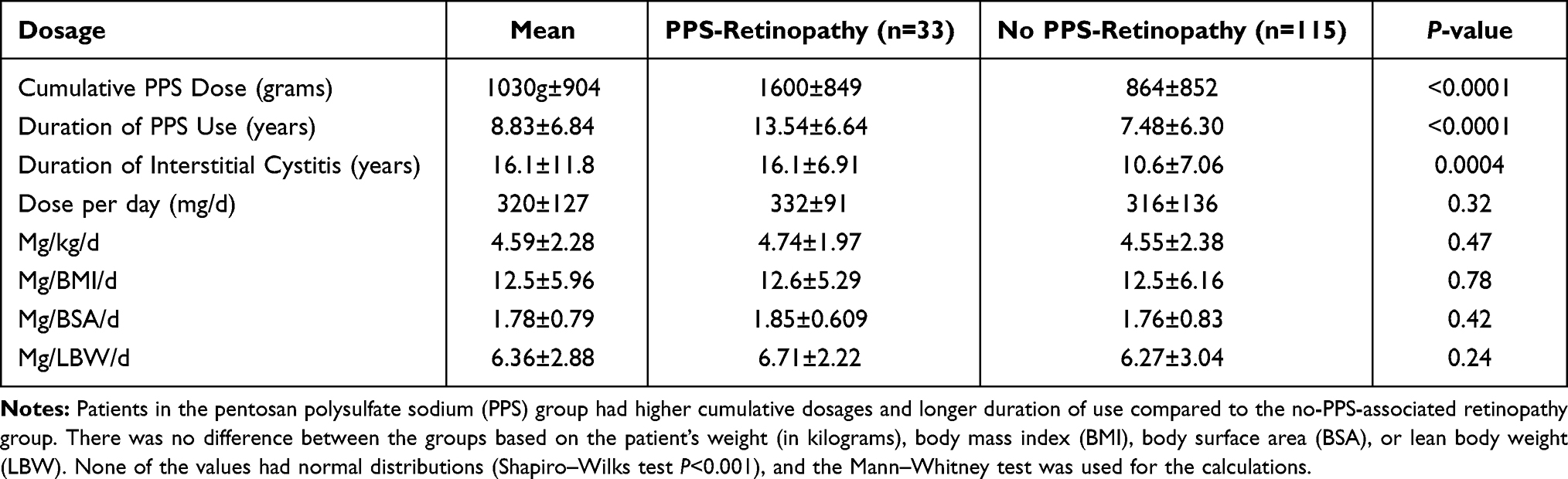 |
Table 2 Dosage of Pentosan Polysulfate |
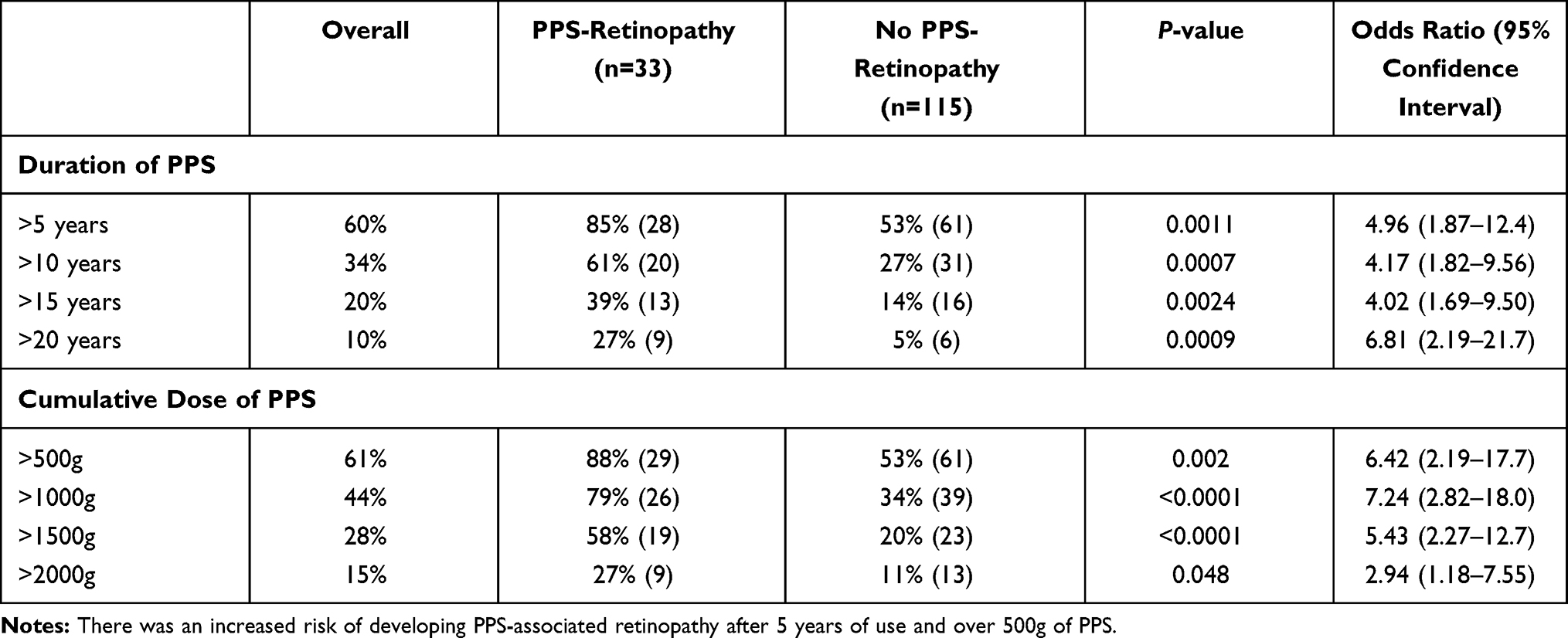 |
Table 3 Risk of Retinopathy and the Duration and Dose of Pentosan Polysulfate Sodium |
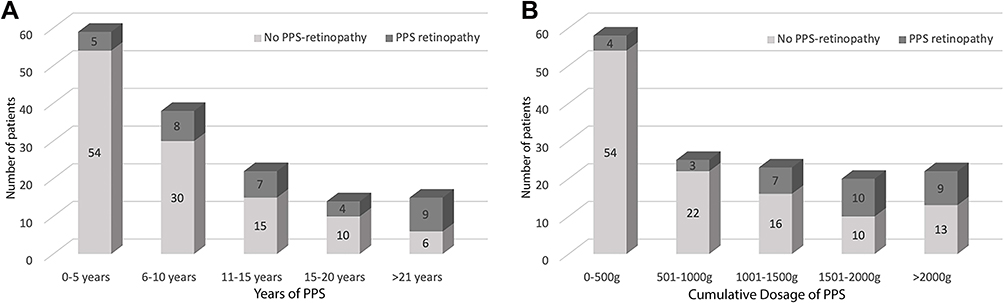 |
Figure 1 PPS dosage and usage. Notes: (A) groups the number of patients grouped based on the duration of PPS use, and (B) groups the patients based on the different cumulative dosages of PPS use. There was a relatively higher proportion of patients with higher dosages and duration in the PPS-associated retinopathy group. |
Of the 65 (44%) patients with documented visual symptoms (15 in the PPS-retinopathy group and 50 in the no-PPS-retinopathy group), patients with PPS- retinopathy were more likely to report visual field defects (47% vs 10%, P=0.0038), difficulty with dark adaptation (73% vs 28%, P=0.0024), and reading difficulty (67% vs 32%, P=0.0030). Patients with PPS-associated retinopathy had slightly worse visual acuities (mean of 0.11logMAR±0.12, ≈20/26 Snellen equivalent, vs 0.067logMAR±0.10 ≈20/23, P=0.013).
While all patients in the PPS-associated retinopathy group had fundus findings associated with PPS retinopathy, 29 had additional fundus findings, like RPE mottling extending to the nasal peripheral retina, RPE mottling in the peripheral retina, and asymmetric disease, which may reflect some of the fundus autofluorescence images being wide-field and the spectrum of disease (Figure 2). Of the 9 patients with more than one fundus autofluorescence, five had areas of atrophy that expanded at a rate of approximately 0.084 disc areas per year. Of the 33 patients with PPS-associated retinopathy, four (12.1%) had received anti-vascular endothelial growth factor injections in the past, of whom 2 had achieved quiescence without recurrent subretinal fluid for over 2 years and one was maintained on an injection every 4 months.
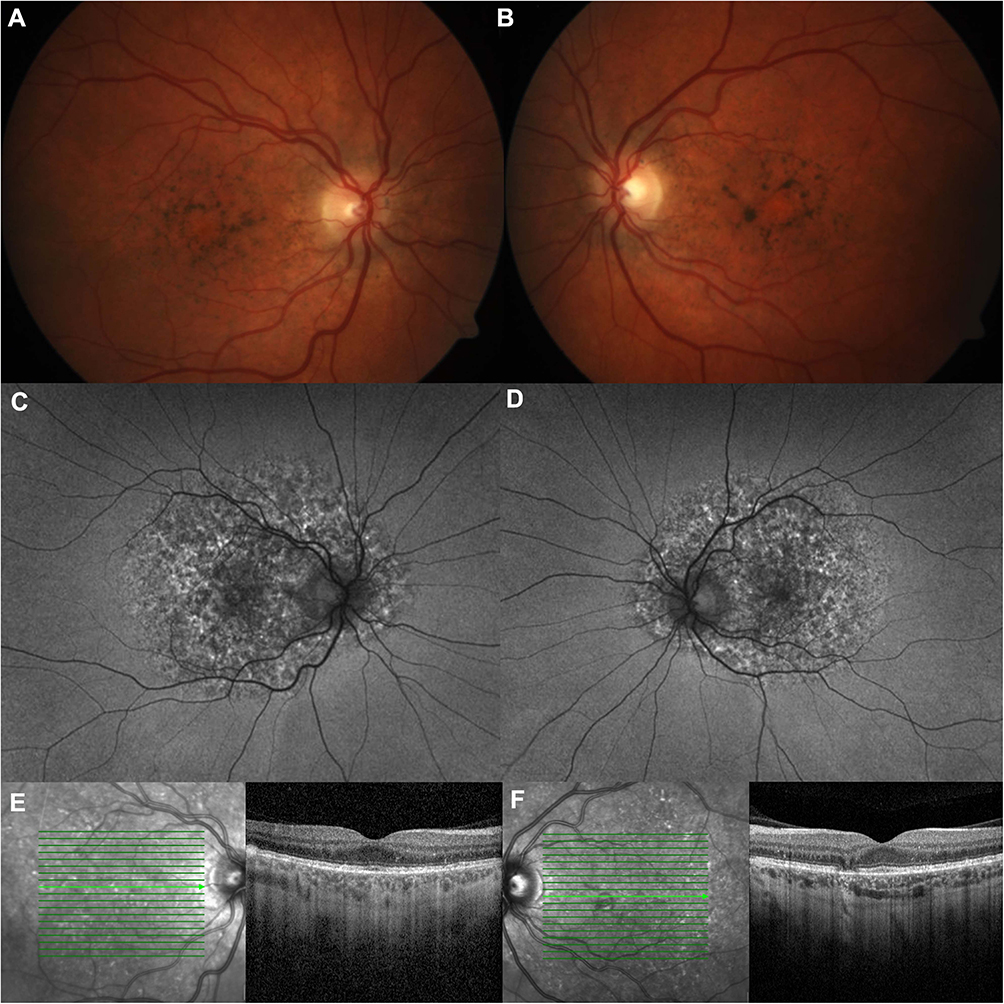 |
Figure 2 PPS-associated retinopathy. Notes: The patient is a 49 year old female with no family history of macular degeneration whose RPE mottling extends beyond the macula. She was on PPS for 8 years with a total cumulative dose of 1130g. She was heterozygous for variants of uncertain significance in CACNA2D4, PRPF8, and WHRN on genetic testing. On fundus photos of the left (A) and right eyes (B), she has hyperpigmentation and subretinal yellow deposits. On wide-field fundus autofluorescence (C and D), the hyper and hypoautofluorescent spots extend outside the macula. On optical coherence tomography (E and F), there is focal RPE thickening with hyperreflectance on near infrared images and atrophy. |
A total of 36 patients had undergone genetics testing (22 with PPS-retinopathy, 7 without PPS retinopathy, and 7 who were excluded due to having pathogenic variants). The majority underwent full-gene sequencing and deletion/duplication analyses with next-generation sequencing technology, evaluating a panel of 330 genes potentially involved in inherited retinal dystrophies. Fourteen of the 15 with genetic results available in the PPS-retinopathy were heterozygous for variants of uncertain significance (VUS); one had no mutations, and 6 had pending results. The patient with PPS-retinopathy with less than 3 years of PPS exposure was heterozygous for a RIMS1 mutation of uncertain significance. Two of the 7 patients in the no-PPS-retinopathy group who had undergone genetic testing had abnormal fundus findings not consistent with PPS-associated retinopathy; both were heterozygous for VUS. The other 5 results were pending.
Discussion
There have been some conflicting data regarding the association between pigmentary retinopathy and PPS, but many large studies have found an association between PPS and the presence of maculopathy.5–11,13 Ludwig et al did not find a statistically significant correlation between PPS usage and pigmentary retinopathy in an insurance database review,17 but the majority of the patients were on PPS for less than 1 year, only 29% had eye exams, and only 0.26% had more than 5 years of use.18 In a cohort study by Hanif et al, patients with IC exposed to PPS had a 25% prevalence of maculopathy, compared to 2.8% in the IC group without PPS.9 Jain et al found an association between maculopathy and over 7 years of PPS use in a review of insurance data.6 The estimated prevalence of PPS-associated retinopathy ranges from 2–25%.14,19 The current retrospective study of 148 patients exposed to PPS found that approximately 22% met the proposed grading criteria for PPS-associated retinopathy, with many having additional findings in the peripheral retina evident on wide-field fundus autofluorescence. Some of these findings such as asymmetry and nasal extension have been included in the extended spectrum of PPS-associated retinopathy.13
Some patients with PPS-associated retinopathy may unfortunately experience difficulty with dark adaptation, reading, contrast sensitivity, and changes in mesopic microperimetry.12,20 In the current study, the PPS-associated retinopathy group was more likely to have complaints of difficulty with their visual field, reading, dark adaptation, and visual acuity. Choroidal neovascular membranes and non-leaking cystoid macular edema may also occur, which may be managed with intravitreal bevacizumab or other anti-vascular endothelial growth factor injections.13,21
Similar to previously published studies, there was a statistically significant risk of developing PPS-associated retinopathy with higher cumulative dosages and longer duration of medication usage. A large multi-center retrospective study of patients with PPS-associated retinopathy found a median cumulative dose of 1.5kg and a median exposure of 14 years.13 Similarly, the present study found a mean cumulative dose of 1.6kg and a mean duration of 13.5 years in the PPS-associated retinopathy group. Vora et al performed a multivariable regression analysis of a cohort of 117 patients and found that the risk of developing PPS-associated maculopathy increased from 13% in those with 500–999g of exposure to 42% with over 1500g;7 the present study found similar incidences of 12% (3/25) and 50% (10/20), respectively. There was no statistically significant association between the presence or severity of pigmentary retinopathy and a history of liver or renal insufficiency, nor the female height, dosage per weight, BMI, BSA, or LBW, but the present study may not have been large enough to detect a difference.
While there appears to be an association between PPS use and pigmentary retinopathy, the causality and exact pathogenesis have yet to be established. Fundus autofluorescence and OCT demonstrate likely involvement of the photoreceptor layer and RPE.13 Greenlee et al suggested that PPS may antagonize the fibroblast growth factors 1, 2, and 4 signaling pathways that are important in the organization, development, and maintenance of the retina.5 PPS resembles glycosaminoglycans and may cause direct toxicity to the interphotoreceptor matrix, which is primarily composed of glycosaminoglycans, or it may inhibit retinal repair by damaging the supporting Muller glia.5,6 Others have suggested impairment of the heparanase activity.13 It is also possible that there may be a genetic component.
Genetics testing was performed in 67% of those with PPS-associated retinopathy, and additional genetic testing is being performed in both groups. Of the 15 genetics results currently available in the PPS-associated retinopathy group, 14 (93%) were heterozygous for a variety of variants of uncertain significance. In comparison, an estimated 20–36% of the general population may be healthy carriers of at least one mutation associated with autosomal recessive inherited retinal diseases,23 and a prior study found mutations in ABCA4 in 1 patient and 5 patients with VUS (29%).22 It is possible that the patients who met the grading criteria for PPS-associated retinopathy may be carriers of inherited retinal dystrophies with variable expressivities or phenotypes, their pigmentary retinopathy or their genetic variants might be incidental findings, or they may be predisposed to developing pigmentary retinopathy with the use of certain medications.
There is currently no treatment for PPS-associated maculopathy, and the retinopathy can progress even after discontinuing PPS.24 In a multicenter study of 11 patients who had discontinued PPS, 77% continued to experience progression, with the RPE atrophy growing at a rate of 0.32mm per year, and 9% developing worsening vision over 12 months.15 Of the small number of patients with more than one fundus autofluorescence in the present study, the growth of atrophy (0.084 disc area/year) was similar, after accounting for the average vertical disc diameter of 1.88mm;25 however, there was no control cohort for comparison.
The limitations of the present study include possible selection and recall bias. Patients were referred by their urologists for baseline evaluations as well as comprehensive ophthalmologists or optometrists for evaluation of possible retinopathy, which may have initially led to a selection bias and higher estimated prevalence rate that may decrease over time as more patients are referred for baseline screening examinations. The dosage information was based on patient reports and may be subject to recall bias since the medication was approved by the Federal Drug Administration in 1996, but the association with a pigmentary retinopathy was only discovered in 2018;8 longer follow-ups and large cohort studies are therefore needed. While the majority of patients had all of the multimodal imaging, 12% did not. Efforts were made to minimize the risk of improper classification, with masked reviewers evaluating the images; however, it is possible that some patients may have alternate etiologies for their fundus findings. Some patients declined genetics testing, some tests reported only pathogenic variants, the detection for genetic mutations rate varies from 50–75%, and not all retinal dystrophy genes may have been tested.23 More genetic testing may help elucidate the clinical significance of these VUS.
In conclusion, the retrospective study found an association between pigmentary retinopathy and longer duration and higher cumulative dose of pentosan polysulfate sodium. The role of genetic variations in predisposing patients to or accelerating the rate of progression of the pigmentary retinopathy is still to be determined. Warning labels have since been added to PPS medications, and a baseline evaluation is recommended within 6 months of commencing therapy.19 Given the potential risk of pigmentary retinopathy and vision loss in advanced cases, even after stopping the medication, patients who are prescribed pentosan polysulfate sodium should consider regular ophthalmic examinations, especially those with baseline retinopathy, possible genetic predispositions, and higher cumulative dosages.
Acknowledgments
The authors are extremely grateful to the contributions of David Chin Yee MD, Michael Jacobson MD, Morgan Jackson COA, Sean Koh MD, Scott Lampert MD, Mark Rivellese MD, Jay Stallman MD, Robert Stoltz MD PhD, Stephanie Vanderveldt MD, and Harpreet (Paul) Walia MD at Georgia Retina.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Interstitial Cystitis | CDC; 2020. Available from: https://www.cdc.gov/ic/index.html.
2. Hanno PM, Erickson D, Moldwin R, Faraday MM. American Urological Association. Diagnosis and treatment of interstitial cystitis/bladder pain syndrome: AUA guideline amendment. J Urol. 2015;193(5):1545–1553. doi:10.1016/j.juro.2015.01.086
3. Ortho-McNeil Pharmaceutical. Elmiron-100mg; 2006. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/020193s004lbl.pdf.
4. Humans IWG on the E of CR to. Mechanistic and Other Relevant Data. International Agency for Research on Cancer; 2016. https://www.ncbi.nlm.nih.gov/books/NBK350418/.
5. Greenlee T, Hom G, Conti T, Babiuch AS, Singh R. Re: pigmentary maculopathy associated with chronic exposure to pentosan polysulfate sodium. Ophthalmology. 2019;126(7):e51. doi:10.1016/j.ophtha.2018.12.037
6. Jain N, Li AL, Yu Y, VanderBeek BL. Association of macular disease with long-term use of pentosan polysulfate sodium: findings from a US cohort. Br J Ophthalmol. 2020;104(8):1093–1097. doi:10.1136/bjophthalmol-2019-314765
7. Vora RA, Patel AP, Melles R. Prevalence of Maculopathy Associated with Long-Term Pentosan Polysulfate Therapy. Ophthalmology. 2020;127(6):835–836. doi:10.1016/j.ophtha.2020.01.017
8. Pearce WA, Chen R, Jain N. Pigmentary Maculopathy Associated with Chronic Exposure to Pentosan Polysulfate Sodium. Ophthalmology. 2018;125(11):1793–1802. doi:10.1016/j.ophtha.2018.04.026
9. Hanif AM, Shah R, Yan J, et al. Strength of Association between Pentosan Polysulfate and a Novel Maculopathy. Ophthalmology. 2019;126(10):1464–1466. doi:10.1016/j.ophtha.2019.04.024
10. Barnes AC, Hanif AM, Jain N. Pentosan Polysulfate Maculopathy versus Inherited Macular Dystrophies: comparative Assessment with Multimodal Imaging. Ophthalmol Retina. 2020;4(12):1196–1201. doi:10.1016/j.oret.2020.05.008
11. Hadad A, Helmy O, Leeman S, Schaal S, Novel Multimethod A. Image Analysis to Quantify Pentosan Polysulfate Sodium Retinal Toxicity. Ophthalmology. 2020;127(3):429–431. doi:10.1016/j.ophtha.2019.10.013
12. Lyons RJ, Brower J, Jain N. Visual Function in Pentosan Polysulfate Sodium Maculopathy. Invest Ophthalmol Vis Sci. 2020;61(13):33. doi:10.1167/iovs.61.13.33
13. Jain N, Liao A, Garg SJ, et al. Expanded Clinical Spectrum of Pentosan Polysulfate Maculopathy: a Macula Society Collaborative Study. Ophthalmol Retina. 2021. doi:10.1016/j.oret.2021.07.004
14. Wang D, Velaga SB, Grondin C, et al. Pentosan Polysulfate Maculopathy: prevalence, Spectrum of Disease, and Choroidal Imaging Analysis Based on Prospective Screening. Am J Ophthalmol. 2021;227:125–138. doi:10.1016/j.ajo.2021.02.025
15. Dai Y, Jonas JB, Ling Z, Sun X. Ophthalmoscopic-Perspectively Distorted Optic Disc Diameters and Real Disc Diameters. Invest Ophthalmol Vis Sci. 2015;56(12):7076–7083. doi:10.1167/iovs.14-16033
16. Ludwig CA, Vail D, Callaway NF, Pasricha MV, Moshfeghi DM. Pentosan Polysulfate Sodium Exposure and Drug-Induced Maculopathy in Commercially Insured Patients in the United States. Ophthalmology. 2020;127(4):535–543. doi:10.1016/j.ophtha.2019.10.036
17. VanderBeek BL, Jain N. Pentosan polysulfate sodium exposure and drug-induced maculopathy in commercially insured patients in the United States (Ophthalmology. 2020;127:535-543). Ophthalmology. 2020;127(5):e35–e36. doi:10.1016/j.ophtha.2020.01.004
18. Paredes Mogica JA, De EJB. Pentosan Polysulfate Maculopathy: what Urologists Should Know in 2020. Urology. 2020;147:109–118. doi:10.1016/j.urology.2020.08.072
19. Uner OE, Shah MK, Jain N. PENTOSAN POLYSULFATE AND VISION: findings from an International Survey of Exposed Individuals. Retina Phila Pa. 2021;41(7):1562–1569. doi:10.1097/IAE.0000000000003078
20. De Larochellière E, Bourgault S. Pentosan polysulfate sodium-induced pigmentary maculopathy with non-leaking cystoid macular edema successfully treated with anti-VEGF therapy. Retin Cases Brief Rep. 2020;Publish Ahead of Print. doi:10.1097/ICB.0000000000001013
21. Hanany M, Rivolta C, Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci. 2020;117(5):2710–2716. doi:10.1073/pnas.1913179117
22. Hanif AM, Armenti ST, Taylor SC, et al. Phenotypic Spectrum of Pentosan Polysulfate Sodium–Associated Maculopathy: a Multicenter Study. JAMA Ophthalmol. 2019;137(11):1275–1282. doi:10.1001/jamaophthalmol.2019.3392
23. Barnett JM, Jain N. Potential new onset clinically detectable pentosan polysulfate maculopathy years after drug cessation. Retin Cases Brief Rep. 2020;Publish Ahead of Print. doi:10.1097/ICB.0000000000001090
24. Shah R, Simonett JM, Lyons RJ, Rao RC, Pennesi ME, Jain N. Disease Course in Patients With Pentosan Polysulfate Sodium-Associated Maculopathy After Drug Cessation. JAMA Ophthalmol. 2020;138(8):894–900. doi:10.1001/jamaophthalmol.2020.2349
25. Quigley HA, Brown AE, Morrison JD, Drance SM. The size and shape of the optic disc in normal human eyes. Arch Ophthalmol Chic. 1990;108(1):51–57. doi:10.1001/archopht.1990.01070030057028
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.