Back to Journals » International Journal of Nanomedicine » Volume 14
PEGylated and CD47-conjugated nanoellipsoidal artificial antigen-presenting cells minimize phagocytosis and augment anti-tumor T-cell responses
Authors Song S, Jin X, Zhang L, Zhao C, Ding Y, Ang Q, Khaidav O, Shen C ![]()
Received 24 November 2018
Accepted for publication 27 February 2019
Published 8 April 2019 Volume 2019:14 Pages 2465—2483
DOI https://doi.org/10.2147/IJN.S195828
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Shilong Song, Xiaoxiao Jin, Lei Zhang, Chen Zhao, Yan Ding, Qianqian Ang, Odontuya Khaidav, Chuanlai Shen
Department of Microbiology and Immunology, Medical School, Southeast University, Nanjing, Jiangsu Province 210009, People’s Republic of China
Purpose: Antigen-presenting cells (APCs) are powerful tools to expand antigen-specific T cells ex vivo and in vivo for tumor immunotherapy, but suffer from time-consuming generation and biosafety concerns raised by live cells. Alternatively, the cell-free artificial antigen-presenting cells (aAPCs) have been rapidly developed. Nanoscale aAPCs are recently proposed owing to their superior biodistribution and reduced embolism than conventional cell-sized aAPCs, but pose the challenges: easier cellular uptake and smaller contact surface area with T cells than the cell-sized counterparts. This study aimed to fabricate a new “stealth” nano-aAPCs with microscale contact surface area to minimize cellular uptake and activate antigen-specific T cells by combination uses of ellipsoidal stretch, PEGylation, and self-marker CD47-Fc conjugation.
Methods: The spherical polylactic-co-glycolic acid nanoparticles were fabricated using a double-emulsion method, and then stretched twofold using film-stretching procedure followed by PEGylation and co-coupling with CD47-Fc, H-2Kb/TRP2180-188-Ig dimers, and anti-CD28. The resulting PEGylated and CD47-conjugated nanoellipsoidal aAPCs (EaAPCPEG/CD47) were co-cultured with macrophages or spleen lymphocytes and also infused into melanoma-bearing mice. The in vitro and in vivo effects were evaluated and compared with the nanospherical aAPCs (SaAPC), nanoellipsoidal aAPCs (EaAPC), or PEGylated nanoellipsoidal aAPC (EaAPCPEG).
Results: EaAPCPEG/CD47 markedly reduced cellular uptake in vitro and in vivo, as compared with EaAPCPEG, EaAPC, SaAPC, and Blank-NPs and expanded naïve TRP2180-188-specific CD8+ T cells in the co-cultures with spleen lymphocytes. After three infusions, the EaAPCPEG/CD47 showed much stronger effects on facilitating TRP2180-188-specific CD8+ T-cell proliferation, local infiltration, and tumor necrosis in the melanoma-bearing mice and on inhibiting tumor growth than the control aAPCs.
Conclusion: The superimposed or synergistic effects of ellipsoidal stretch, PEGylation, and CD47-Fc conjugation minimized cellular uptake of nano-aAPCs and enhanced their functionality to expand antigen-specific T cells and inhibit tumor growth, thus suggesting a more valuable strategy to design “stealth” nanoscale aAPCs suitable for tumor active immunotherapy.
Keywords: PLGA nanoparticles, artificial antigen-presenting cells, phagocytosis, cancer active immunotherapy
Introduction
Antigen-presenting cells (APCs), most notably dendritic cells (DCs), are powerful tools to expand antigen-specific T cells both ex vivo and in vivo, but limited by the time-consuming and cost-intensive generation when scaled up, nonspecific stimulation, and biosafety concerns raised by live cells.1,2 As an alternative strategy, the cell-free artificial antigen-presenting cells (aAPCs) were proposed by co-coupling antigenic peptide-loaded major histocompatibility complexes (pMHCs, antigen signal) and anti-CD28 (costimulatory signal) onto scaffolds of biomaterials to imitate natural APCs.3 They are more amenable to rapid manufacturing in a large-scale manner with highly uniform quality and little concern of biosafety. Therefore, numerous biomimetic aAPCs have been developed and are rapidly optimized on their physical and biochemical properties, such as size, shape, charge, surface modifications, signal strength, and the composition of new signal combinations for the establishment of therapeutic cellular immunity.4–8 Classically, most research studies focus on cell-sized and spherical aAPCs using a variety of biomaterials from liposomes9 to paramagnetic beads,10 non-degradable11,12 and biodegradable polymeric microparticles,13–15 and achieved intriguing prospects. But nanoscale aAPCs have also been reported recently owing to their advantages over cell-sized aAPCs: superior tissue distribution and drainage properties; and reduced risk of embolism and tissue infarction, so they are more suitable for in vivo use.10,16 However, the nano-aAPCs face two challenges: easier engulfment by phagocytes and smaller surface area for contact with T cells than the cell-sized counterparts, thus greatly reduce their direct interactions between aAPCs and antigen-specific T cells in vivo.
During past decades, a variety of biomimetic techniques have been developed to prevent phagocytosis in the drug and vaccine delivery systems of micro- and nanoparticles (MNPs). First, the “stealth particles” can be constructed by coating poly(ethylene glycol) (PEG),17 lipid bilayer,18 or CD47-Fc19 onto the surface of MNPs as well-known. The PEGylated nanoparticle carriers can decrease the adsorption of nonspecific serum proteins, reduce engulfment, and prolong their circulation time in vivo.20,21 CD47 can interact with signal regulatory protein-α on phagocytes to inhibit phagocytosis at low density22,23 and has been used as a “self-marker” in nanoparticle drug delivery systems in human.19 Second, the particle shape also markedly influences their phagocytosis and circulation time as well as particle–cell contact area in vivo.24 Compared to spherical and flattened disc-shaped particles, ellipsoidal particles showed the most efficient particle attachment and lowest in vitro internalization rates.25,26 As reported, both ellipsoidal polylactic-co-glycolic acid nanoparticles (PLGA-NPs) and PEGylated spherical PLGA-NPs presented much less cellular uptake by macrophages than the conventional ones. Moreover, the combination of PEGylation and ellipsoidal stretch enable the PLGA-NPs much stronger inhibition to phagocytosis.27 Based on these established nanotechnologies, an ellipsoidal nano-aAPC system has recently been developed, which significantly reduced the uptake by macrophages and endothelial cells in vitro and the clearance by liver and spleen in vivo.28 Consequently, the nanoellipsoidal aAPCs (EaAPC) activate antigen-specific cytotoxic T lymphocytes (CTLs) both ex vivo and in vivo more efficiently than the spherical counterparts, partially because that the nonspherical nano-aAPCs possess a micron length scale radius of curvature on a long axis to approximate the interfacial geometry of micro-aAPCs.28,29 Meanwhile, the CD47-conjugated spherical nano-aAPCs have also been developed with the enhanced effects of anti-phagocytosis and tumor inhibition.30
In this study, PEGylation, CD47 conjugation, and ellipsoidal stretch were integrated into the generation of new “stealth” nano-aAPCs with microscale contact surface to minimize internalization and augment anti-tumor CTL responses in melanoma-bearing mice. PLGA, a biocompatible, biodegradable, and nontoxic polymer widely used in drug and vaccine delivery systems in human,31–33 was employed to fabricate the spherical NPs and then was further stretched to the ellipsoidal NPs, followed by surface modification with PEGylation and surface co-coupling with H-2Kb/TRP2180-188-Ig dimers, anti-CD28, and CD47-Fc. The resulting PEGylated and CD47-conjugated nanoellipsoidal aAPCs, termed EaAPCPEG/CD47, possessed much more powerful function to minimize engulfment of phagocytes both in vitro and in vivo and elicited more robust expansion of TRP2-specific CTLs and anti-tumor effects in melanoma-bearing mice, as compared with the nanospherical aAPCs (SaAPC), EaAPC, and PEGylated nanoellipsoidal aAPC (EaAPCPEG). This study provided a new nanoscale aAPC system by modulating the particle shape and improving the surface modification and confirmed its first evidence of an enhanced in vivo function to minimize phagocytosis and augment anti-tumor T-cell responses.
Materials and methods
Mice, cell lines, and peptides
Female C57BL/6J mice at 6–7 weeks of age were purchased from the Comparative Medicine Center of Yangzhou University (Yangzhou, China) and maintained at the specific-pathogen-free Laboratory Animal Centre of Southeast University (Nanjing, China). All mice were used in experiments at 8–12 weeks of age. Animal welfare and experimental procedures were approved by the Animal Ethics Committee of Southeast University and performed in accordance with the Guidelines for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No 8023, revised 1978). The mouse melanoma B16F10 and macrophage RAW264.7 cell lines were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences. The TRP2180-188 (SVYDFFVWL) peptide was synthesized by Chinapeptides Biotech (Suzhou, Jiangsu, China) with a purity of >95%.
Preparation and characterization of spherical, ellipsoidal, and PEGylated ellipsoidal PLGA-NPs
PLGA 50/50 with an average molecular weight of 10 kD was obtained from Daigang Company (Jinan, China). The spherical PLGA-NPs were fabricated using a double-emulsion water-in-oil-in-water (W1/O/W2) method as previously described.34 Briefly, 200 mg of PLGA was dissolved in 6 mL of dichloromethane (Sigma-Aldrich Co., St Louis, MO, USA) and emulsified with a microtip probe sonicator (Microson XL 2000; Misonix Incorporated, Farmingdale, NY, USA) for 30 seconds at 40% amplitude to create the primary emulsion, followed by mixing with 50 mL of 1% polyvinylalcohol (PVA; Sigma-Aldrich Co.) solution and sonicated for 2 minutes to form the secondary emulsion. The resulting emulsification was poured into 100 mL of 0.5% PVA solution and stirred for 4 hours at room temperature (RT) with a magnetic stirrer until the organic solvent was completely evaporated. The solution was then centrifuged thrice at 4,000 rpm for 5 minutes to precipitate non-nano-dimensional materials. The nanoparticles were collected by centrifugation at 12,000 rpm for 15 minutes and washed twice with deionized water to remove the PVA. Then the particles were lyophilized and stored at −20°C until use.
The ellipsoidal nanoparticles were further prepared using a film stretching method as described.35 The lyophilized spherical PLGA-NPs were dissolved in the mixture of 10% PVA and 2% glycerol solution, and dried on a flat surface for 48 hours at RT. The resulting film was cut into sections of 7×7 cm and mounted on a customized aluminum installation. Each film section was stretched slowly to 14 cm in one dimension at 75°C, then cooled down to RT, and dissolved in ultrapure water. After centrifuging at 12,000 rpm for 10 minutes at 4°C, the particles were collected and washed five times to remove free PVA from the surface of stretched particles. The resulting ellipsoidal particles were lyophilized and stored at −20°C until use.
To generate the “stealth” ellipsoidal PLGA-NPs, the hetero-functional PEG (NH2-PEG2000-COOH; Ponsure Biotechnology, Shanghai, China) was further conjugated onto the ellipsoidal NPs as described.36 About 5 mg of lyophilized ellipsoidal NPs was resuspended in 10 mL of isotonic 0.1 M MES buffer saline (pH 5.5) and reacted with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (0.19 g) and N-hydroxysuccinimide (NHS; Sigma-Aldrich Co.) (0.057 g) for 1 hour. NPs were then centrifuged at 120,000 rpm for 10 minutes to remove excess EDC/NHS and the water-soluble byproduct. The resulting NHS-activated particles were resuspended in 5 mL of deionized water and reacted with NH2-PEG2000-COOH for 12 hours at RT. After centrifugation, the resulting PEGylated ellipsoidal PLGA-NPs were washed with deionized water to remove unbound PEG, then lyophilized and stored at −20°C until use.
The morphology of spherical and ellipsoidal PLGA-NPs was characterized by using scanning electron microscopy (SEM, ZEISS EVO 18; Carl Zeiss Meditec AG, Jena, Germany). The particle size distribution and zeta potential were measured by dynamic light scattering and laser Doppler anemometry with Nano ZS90 Zeta sizer (Malvern Instruments, Malvern, UK). The immobilization of PEG on the surface of PEGylated ellipsoidal NPs was verified by 1H-NMR (AVANCE DRX-500; Bruker, Switzerland).
Generation and phenotypic analyses of SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47
The lyophilized spherical, ellipsoidal, or PEGylated ellipsoidal PLGA-NPs were sterilized using UV for 30 minutes, and then dispersed in 0.1 M MES buffer at 2 mg mL−1 followed by ultrasonication in water bath for 5 minutes. Each type of NPs was activated by EDC/NHS as described above. The NHS-activated NPs were added dropwise into the polyethyleneimine (PEI) solution with magnetic stirring for 2 hours at RT, and then washed twice by centrifugation at 12,000 rpm to remove free PEI. For the generation of SaAPC, EaAPC, and EaAPCPEG, the resulting PEI-conjugated spherical NPs, ellipsoidal NPs, and PEGylated ellipsoidal NPs (10 mg) were co-incubated, respectively, with H-2Kb/TRP2180-188-Ig dimers (10 μg; BD Biosciences, San Jose, CA, USA) and anti-CD28 (10 μg; eBiosciences, San Diego, CA, USA) overnight at 4°C. To prepare the EaAPCPEG/CD47, 10 mg of PEGylated ellipsoidal NPs were co-incubated with H-2Kb/TRP2180-188-Ig dimers (10 μg), anti-CD28 (10 μg), and CD47-Fc (160 ng; R&D Systems, Inc., Minneapolis, MN, USA) overnight at 4°C. The resulting each type of aAPCs was blocked with BSA overnight at RT, and then washed thrice by centrifugation at 12,000 rpm for further use.
In order to analyze the phenotypes, each type of aAPCs was stained with PE-anti-H-2Kb (eBiosciences), FITC-anti-hamster IgG (binding to anti-CD28; eBiosciences), and APC-anti-human IgG1 (binding to CD47-Fc; Miltenyi Biotec, Bergisch Gladbach, Germany) for 30 minutes at 4°C; after washing twice, the aAPCs were acquired on a FACSCalibur flow cytometer (BD Biosciences).
Phagocytosis of macrophage in vitro
The SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47 were prepared as described, but additionally co-coupled with PE-streptavidin (BD Biosciences) (10 mg) onto the surface to generate PE-labeled aAPCs. RAW264.7 macrophages were seeded into a 24-well flat-bottom plate (1×105 cells per well) (Falcon Plate; Corning Incorporated, Corning, NY, USA), maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Thermo Fisher Scientific), and incubated in a humidified incubator with 5% CO2 and at 37°C for 24 hours to allow cell attachment. Then, the PE-labeled SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47 as well as PE-unlabeled SaAPC were added into the cell wells (200 μg per well), respectively, for another 6 hours of incubation in the dark. After that, the cell wells were washed five times gently with PBS (10 mM, pH 7.4) to remove the free aAPCs which did not enter into cells or did not contact with cells, followed by the incubation with 4% paraformaldehyde solution for 20 minutes 4°C in dark. After staining with DAPI (Sigma-Aldrich Co.) for 5 minutes at 4°C in dark, the cell wells were washed with PBS and imaged by confocal laser scanning microscope (CLSM, FV1000; Olympus Corporation, Tokyo, Japan).
In addition, at the end of 6-hour incubation of cells with each type of PE-labeled aAPCs, the medium was immediately discarded and the wells were washed twice with PBS to remove free PE-labeled aAPCs. Then the cells were harvested from wells by 0.25% trypsin-EDTA digestion first and centrifugation later. After washing twice with PBS, the cells were acquired on FACSCalibur flow cytometer (BD Biosciences). The percentage of PE-positive cells and their fluorescence intensity were calculated using FlowJo software (Tree Star, Ashland, OR, USA).
Expansion of TRP2180-188-specific CD8+ T cells by aAPCs ex vivo
Spleens were obtained from naïve C57BL/6J mice and lymphocytes were enriched by density gradient centrifugation using Mouse Lymphocyte Separation Solution (Shanghai Lianke Biotech, Shanghai, China). Then, the spleen lymphocytes (105 cells per well) were co-cultured with Blank-NPs (blocked with BSA), SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47 (200 μg per well) in a 96-well round-bottom plate. Co-cultures were maintained in RPMI-1640 medium supplemented with 10% FBS in the presence of IL-2 (50 U mL−1) and incubated in a humidified incubator with 5% CO2 and at 37°C for 8 days. Then, the co-cultures were harvested to detect the frequency of TRP2180-188-specific CD8+ T cells by pMHC multimer staining and flow cytometry. Briefly, the cells were incubated with anti-mouse CD16/CD32 (2.4G2; eBiosciences) for 30 minutes at 4°C to block the Fc receptors, then incubated with a mixture of H-2Kb/TRP2-Ig dimers with APC-anti-mouse IgG1 (A85-1; BD Biosciences) for 1 hour at 4°C in the dark. After washing, FITC-anti-mouse CD8a (53-6.7; eBiosciences) and PE-anti-mouse CD3e (145-2C11; eBiosciences) were added and the mixture was incubated for an additional 30 minutes at 4°C. In parallel, the isotype controls were also stained by using PE-Armenian hamster IgG, FITC-rat IgG2bκ, or FITC-rat IgG2aκ (eBiosciences). Finally, the frequencies of TRP2180-188-specific CD8+ T cells were quantified by flow cytometry.
Co-localizations of aAPCs with immune cells in vivo
On day 0, C57BL/6J mice were injected subcutaneously (s.c.) in the right flank with B16 cells at a dose of 5×105 cells per mouse. Mice were then randomized into five groups and infused via tail vein with PE-labeled SaAPC, EaAPC, EaAPCPEG, EaAPCPEG/CD47, and Blank-NPs (sphere) on day 11 (1 mg NPs per mouse). Six hours later, spleens were collected in dark and embedded in freezing medium (O.C.T, Sakura Finetek USA Inc, Torrance, CA, USA). Sections with a thickness of 5–7 μm were prepared, fixed with isopropanol and acetone (1:1 ratio), and then blocked with 10% mouse serum in PBS for 2 hours at 4°C. After washing twice with sterile PBS, the sections were incubated with FITC-anti-mouse CD4 (RM4-5), FITC-anti-mouse CD8 (H35-17.2), FITC-anti-mouse CD19 (MB19-1), FITC-anti-mouse CD11c (N418), or FITC-anti-mouse F4/80 (m2F8) (all from eBiosciences) at 4°C for overnight. After washing with PBS, the sections were further stained with DAPI (Sigma-Aldrich Co.) for 5 minutes and finally visualized under CLSM (FV1000; Olympus Corporation). In parallel, the isotype controls were also stained using FITC-rat IgG2bκ or FITC-rat IgG2aκ (eBiosciences). In addition, spleens from the treated melanoma mice were processed into single-cell suspensions, freshly stained with the FITC-labeled mAbs, and analyzed by flow cytometry.
Expansion of TRP2180-188-specific CD8+ T cells and tumor inhibition by aAPCs infusion in vivo
On day 0, B6 mice were injected s.c. in the right flank with B16 cells at a dose of 5×105 cells per mouse. Mice were then randomized into five groups and infused via tail vein with SaAPC, EaAPC, EaAPCPEG, EaAPCPEG/CD47, or Blank-NPs (sphere) on days 7, 9, and 11 (1 mg NPs per mouse per time point). Tumor growth was monitored at 3-day interval using a digital caliper, and the tumor volume was calculated by the modified ellipsoidal formula: V = (length × width2)/2.18 Mice were monitored daily for survival and were sacrificed when the tumor length exceeded 20 mm.
On day 28, peripheral blood, spleens, and tumors were collected from each mouse. Peripheral blood mononuclear cells and splenocytes (SPCs) were then prepared routinely. The single-cell suspensions derived from tumor tissues were prepared as described,2 with the modification as below: tumor tissues minced into small pieces were digested with collagenase type VIII (1.5 mg mL−1; Sigma-Aldrich Co.) for 2 hours at 37°C with gentle agitation. The cell suspensions were incubated with anti-mouse CD16/CD32 for 30 minutes at 4°C to block the Fc receptors, then incubated with a mixture of H-2Kb/TRP2180-188-Ig dimers and APC-labeled anti-mouse IgG1 for 1 hour at 4°C in the dark. After washing, FITC-anti-mouse CD8a and PE-anti-mouse CD3e were added and the mixture was incubated for an additional 30 minutes at 4°C. In parallel, the isotype controls were also stained. Finally, the frequencies of TRP2180-188-specific CD8+ T cells in the CD8+ T-cell populations were quantified by flow cytometry.
Functional analyses of melanoma antigen-specific CD8+ T cells after aAPC infusion
On day 14, spleen lymphocytes were prepared from each melanoma-bearing mouse after three infusions of aAPCs or Blank-NPs, and co-cultured in 24-well plate (1×106 cells well−1) with Phorbol 12-Myristate 13-Acetate (PMA)/ionomycin and Brefeldin A (BFA)/monensin mixture (Multi Sciences, Shanghai, China) for 4 hours or stimulated by TRP2180-188 peptide (20 μg mL−1) for 16 hours followed by the addition of protein transport inhibitor BFA for another 5 hours at 37°C under 5% CO2 and humidified conditions. Then, the cells were harvested, blocked with anti-mouse CD16/CD32 for 20 minutes at 4°C, incubated with FITC-anti-mouse CD8 and APC-anti-mouse CD3e (145-2C11) for 30 minutes at 4°C. After fixation/permeabilization, the cells were further incubated with PE-anti-mouse IFN-γ (XMG1.2) (eBiosciences) for another 30 minutes at 4°C and analyzed by flow cytometry.
In parallel, spleen lymphocytes from each mouse were used as effector cells. The mouse melanoma cell line B16 (H-2Kb+/TRP2+) and myeloma cell line SP2/0 (H-2Kb-/TRP2−) were used as target cells (1×104 cells/well). The cytotoxicity assay was then performed as previously described.12
Histochemical staining of melanoma tissues
On day 28, melanoma tissues were dissected from each melanoma-bearing mouse after three infusions of aAPCs or Blank-NPs, fixed in 4% formaldehyde, embedded in paraffin, and sectioned at a thickness of 5–7 μm. For the assessment of inflammation and cellular necrosis, the sections were stained with H&E routinely and visualized under light microscope (Nikon Corporation, Tokyo, Japan). To further detect in situ apoptotic tumor cells, the sections were stained with Tunel Kit (R&D Systems, Inc.) according to the manufacturer’s protocol. Briefly, the sections were deparaffinized and rehydrated following a standard procedure, treated with Proteinase K for 5 minutes at RT, and then stained by the mixture containing ddH2O, FITC-12-dUTP labeling mix, equilibration buffer, and recombinant TdT enzyme for 1 hour at 37°C in dark. After washing with PBS, the sections were further stained with DAPI (Sigma-Aldrich Co.) for 5 minutes and finally visualized under fluorescence microscope (Olympus Corporation).
In order to evaluate the local infiltration of TRP2180-188-specific CD8+ T cells, in situ TRP2-dimer staining was performed on the melanoma sections. After dewaxing hydration, the sections were immersed in 0.01 M boiled citric acid buffer for 10 minutes later for antigen retrieval, then blocked with blocking buffer (10% normal mouse serum and 5% BSA in PBS) for 2 hours at 4°C. The H-2Kb/TRP2180-188-Ig dimers were incubated with PE-anti-mouse IgG1 (1 μg each) for 1 hour at 4°C, and diluted 50 times with blocking buffer. Then, the tumor sections were incubated overnight at 4°C in dark with FITC-anti-mouse CD8 (H35-17.2) and the mixture of TRP2-dimer and PE-anti-mouse IgG1. After washing, the sections were further stained with DAPI (Sigma-Aldrich Co.) for 5 minutes and finally visualized under CLSM (FV1000; Olympus Corporation).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, CA, USA). Tumor growth was statistically evaluated by Wilcoxon signed-rank tests. To obtain the mouse survival curve, a Kaplan–Meier graph was constructed, and a log-rank comparison of the groups was used to calculate the P-values. For other experiments, a two-tailed unpaired Student’s t-test was used to determine differences across groups. All data were presented as mean ± SD. P<0.05 was considered significant.
Results and discussion
Generation and characterization of spherical, ellipsoidal, and PEGylated ellipsoidal PLGA-NPs
The spherical PLGA-NPs and ellipsoidal PLGA-NPs were successfully prepared in-house. The spherical NPs showed a spherical shape with a smooth surface while ellipsoidal NPs showed a twofold stretched ellipsoidal and smooth morphology as revealed by SEM images (Figure 1A). Size analyses showed that spherical NPs have a mean diameter of 214.3±26.6 nm with polydispersity of 0.126 as measured by dynamic light scattering, while the PEI-modified spherical NPs showed a little bigger mean diameter of 248.2±34.1 (Figure 1B). Ellipsoidal NPs cannot be measured by dynamic light scattering since the Stokes–Einstein equation for this measurement assumes a spherical shape in media.37
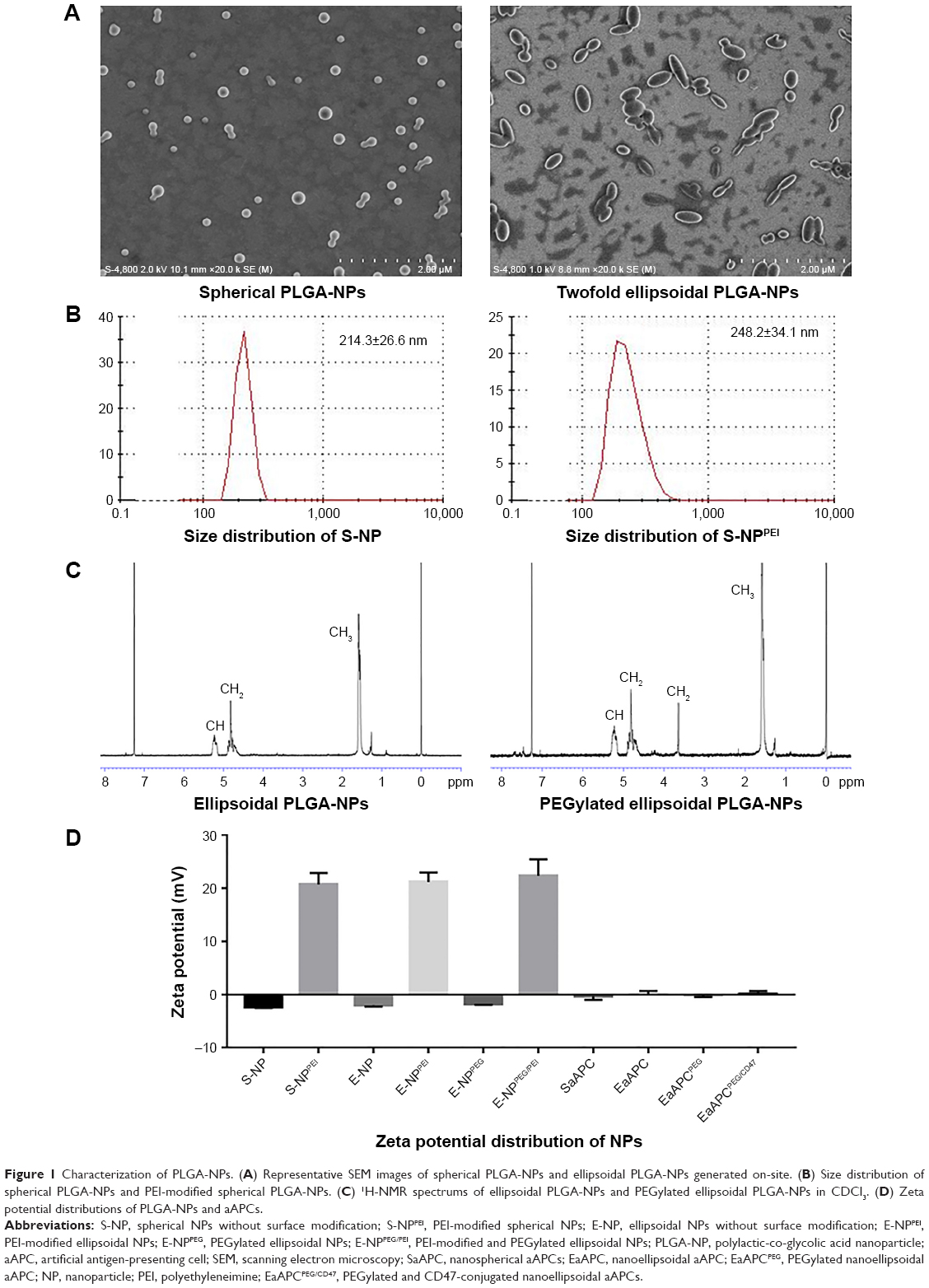 | Figure 1 Characterization of PLGA-NPs. (A) Representative SEM images of spherical PLGA-NPs and ellipsoidal PLGA-NPs generated on-site. (B) Size distribution of spherical PLGA-NPs and PEI-modified spherical PLGA-NPs. (C) 1H-NMR spectrums of ellipsoidal PLGA-NPs and PEGylated ellipsoidal PLGA-NPs in CDCl3. (D) Zeta potential distributions of PLGA-NPs and aAPCs. |
PEGylated ellipsoidal PLGA-NPs were further fabricated by activating ellipsoidal NPs via EDC/NHS technique first and PEGylation later. The chemical composition of the PEGylated NPs was confirmed by 1H-NMR (Figure 1C). The peak of spectrum at 1.55 ppm belonged to the methyl protons (−CH3) of the D- and L-lactic acid repeat units; the peaks at 4.8 and 5.2 ppm were due to the methylene groups (−CH2) of glycolic acid and methine groups (-CH) of lactic acid, respectively. The presence of methyl group (−CH3) and methylene group (−CH2) indicated the content of PLGA. The peak at 3.6 ppm is the striking feature corresponding to the methylene groups (−CH2) of the ethylene glycol monomer, thus confirming the successful PEG conjugation to PLGA for the PEGylated ellipsoidal PLGA-NPs, while the PEG-free ellipsoidal PLGA-NPs lost the peak at 3.6 ppm.
Zeta potential is an important parameter for the surface charge and capability of NPs to covalently couple proteins. For the spherical NPs, ellipsoidal NPs, and PEGylated ellipsoidal NPs, the mean zeta potential was −2.3±0.2, −1.7±0.3, and −2.1±0.3 mV, respectively. These comparable zeta potentials less than zero are due to the anionic carboxyl groups on the terminal of PLGA and NH2-PEG2000-COOH. After further modification with PEI, the spherical NPs, ellipsoidal NPs, and PEGylated ellipsoidal NPs had the mean zeta potential of 20.7±2.4, 21.7±3.1, and 22.3±2.2 mV, respectively (Figure 1D), implying the abundant cationic NH2 groups on the surface of PEI-conjugated NPs and a high capacity to covalently coupled proteins. These results also indicated that the stretching and PEGylation did not influence the surface charge of PEI onto PLGA-NPs.
Fabrication and phenotypic analyses of SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47
SaAPC, EaAPC, or EaAPCPEG was generated by co-coupling H-2Kb/TRP2-Ig and anti-CD28 onto the spherical, ellipsoidal, or PEGylated ellipsoidal NPs, while EaAPCPEG/CD47 was fabricated by co-coupling H-2Kb/TRP2-Ig, anti-CD28, and CD47-Fc onto PEGylated ellipsoidal NPs (Figure 2). To confirm the immobilization of multiple immune molecules on the surface of aAPCs, fluorescence-labeled antibody staining and flow cytometry were carried out. The histograms (Figure 3A) showed that each type of surface molecules was effectively coupled onto each type of aAPCs with the strong fluorescence signals. Furthermore, the flow cytometric dot plots (Figure 3B) revealed that around 52.3%–57.3% of SaAPC, EaAPC, or EaAPCPEG co-displayed both H-2Kb/TRP2-Ig dimers and anti-CD28, and 53.7%–56% of EaAPCPEG/CD47 co-displayed the three types of immune molecules, as determined by three-color staining and showed in each two-color dot plots. Each batch of aAPCs was routinely evaluated in this manner prior to use. Of note is that EaAPC and EaAPCPEG (ellipsoid) showed the fluorescence intensity similar to SaAPC (sphere) for each surface protein, although the surface area increased by 16% after twofold stretch for a spherical particle in a diameter of 200 nm.28 The possible cause is that the dosage of each surface protein was the same in the generation of both SaAPC and EaAPC and did not reach saturation for the surface area of spherical or ellipsoidal NPs.
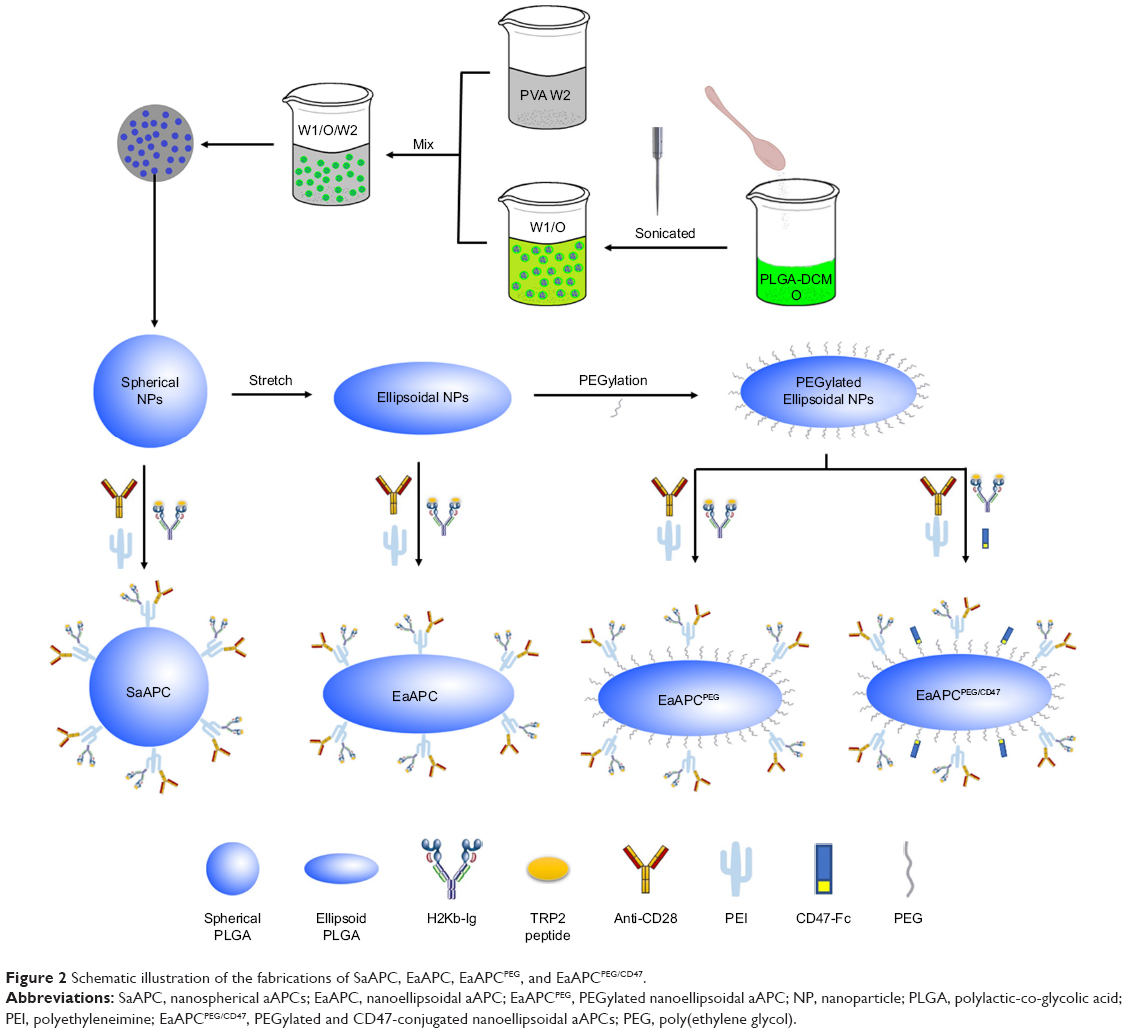 | Figure 2 Schematic illustration of the fabrications of SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47. |
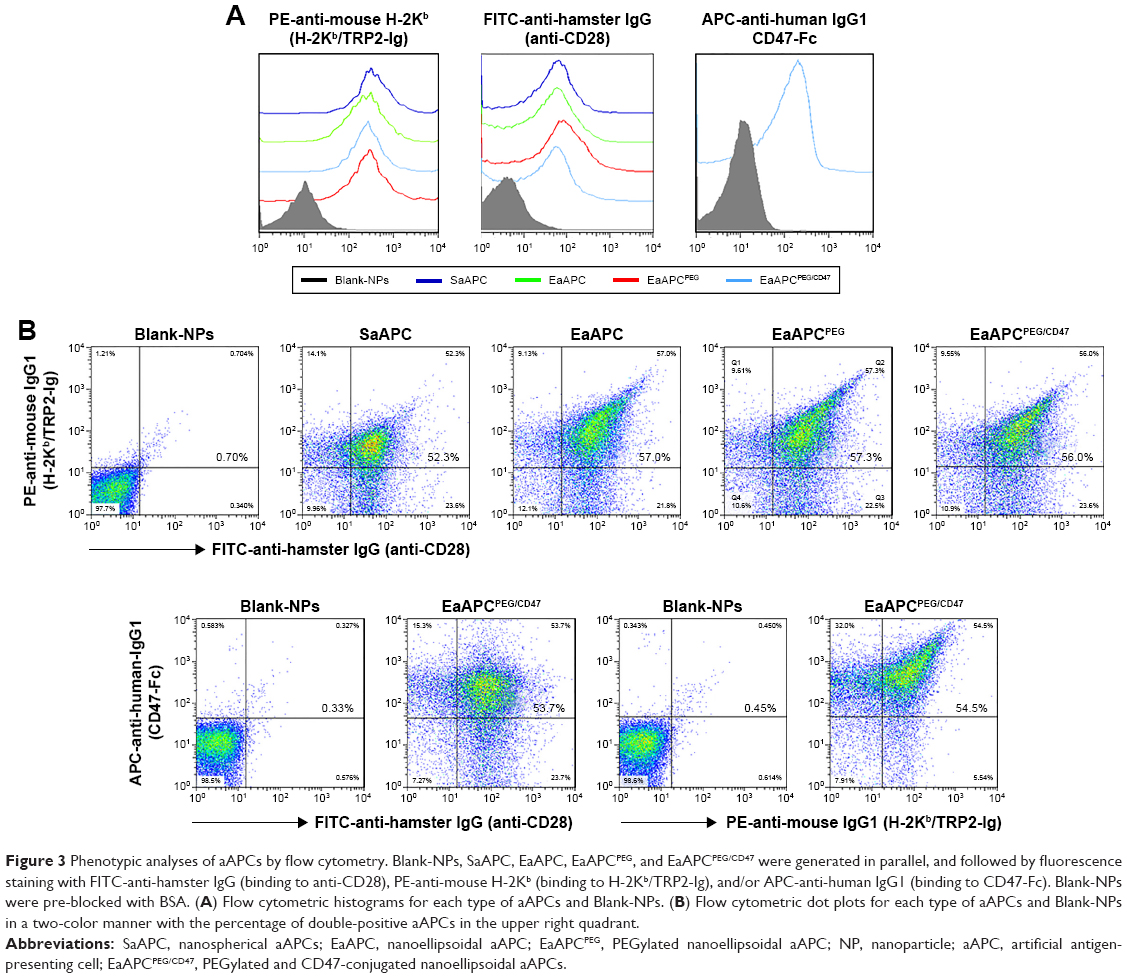 | Figure 3 Phenotypic analyses of aAPCs by flow cytometry. Blank-NPs, SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47 were generated in parallel, and followed by fluorescence staining with FITC-anti-hamster IgG (binding to anti-CD28), PE-anti-mouse H-2Kb (binding to H-2Kb/TRP2-Ig), and/or APC-anti-human IgG1 (binding to CD47-Fc). Blank-NPs were pre-blocked with BSA. (A) Flow cytometric histograms for each type of aAPCs and Blank-NPs. (B) Flow cytometric dot plots for each type of aAPCs and Blank-NPs in a two-color manner with the percentage of double-positive aAPCs in the upper right quadrant. |
EaAPCPEG/CD47 markedly reduced cellular uptake by macrophage in vitro
To visualize the engulfment of EaAPCPEG/CD47 by macrophage, PE-labeled SaAPC, EaAPC, EaAPCPEG, EaAPCPEG/CD47, and PE-free SaAPC were prepared, respectively, and co-incubated with RAW 264.7 cells in 24-well plate for 6 hours followed by confocal laser scanning analyses and flow cytometry. As shown in Figure 4A, the cells incubated with conventional SaAPC showed much stronger red fluorescence than the others, while the cells co-cultured with EaAPCPEG/CD47 display the weakest red fluorescence. According to the cytoskeleton of RAW 264.7 cells in bright images, the red fluorescence mostly accumulated inside the macrophages, implying the uptake of aAPCs by macrophages. As quantified by flow cytometry, the fluorescence intensity of cells incubated with SaAPC was the highest, but the value of EaAPC, EaAPCPEG, and EaAPCPEG/CD47 groups decreased by 33.1% (P<0.01), 50.7% (P<0.001), and 67.1% (P<0.0001), respectively, as compared with the SaAPC group (Figure 4B and C). The significant differences were also found among the EaAPC, EaAPCPEG, and EaAPCPEG/CD47 groups while EaAPCPEG/CD47 showed the least cellular uptake inside and/or onto the macrophages (Figure 4C). These data indicate that the nanoscale EaAPCPEG/CD47 possessed much more powerful capability to inhibit engulfment of phagocytes than the nanoscale SaAPC, EaAPC, and EaAPCPEG, owing to the superimposed effects of ellipsoidal stretch, PEGylation, and self-marker CD47.
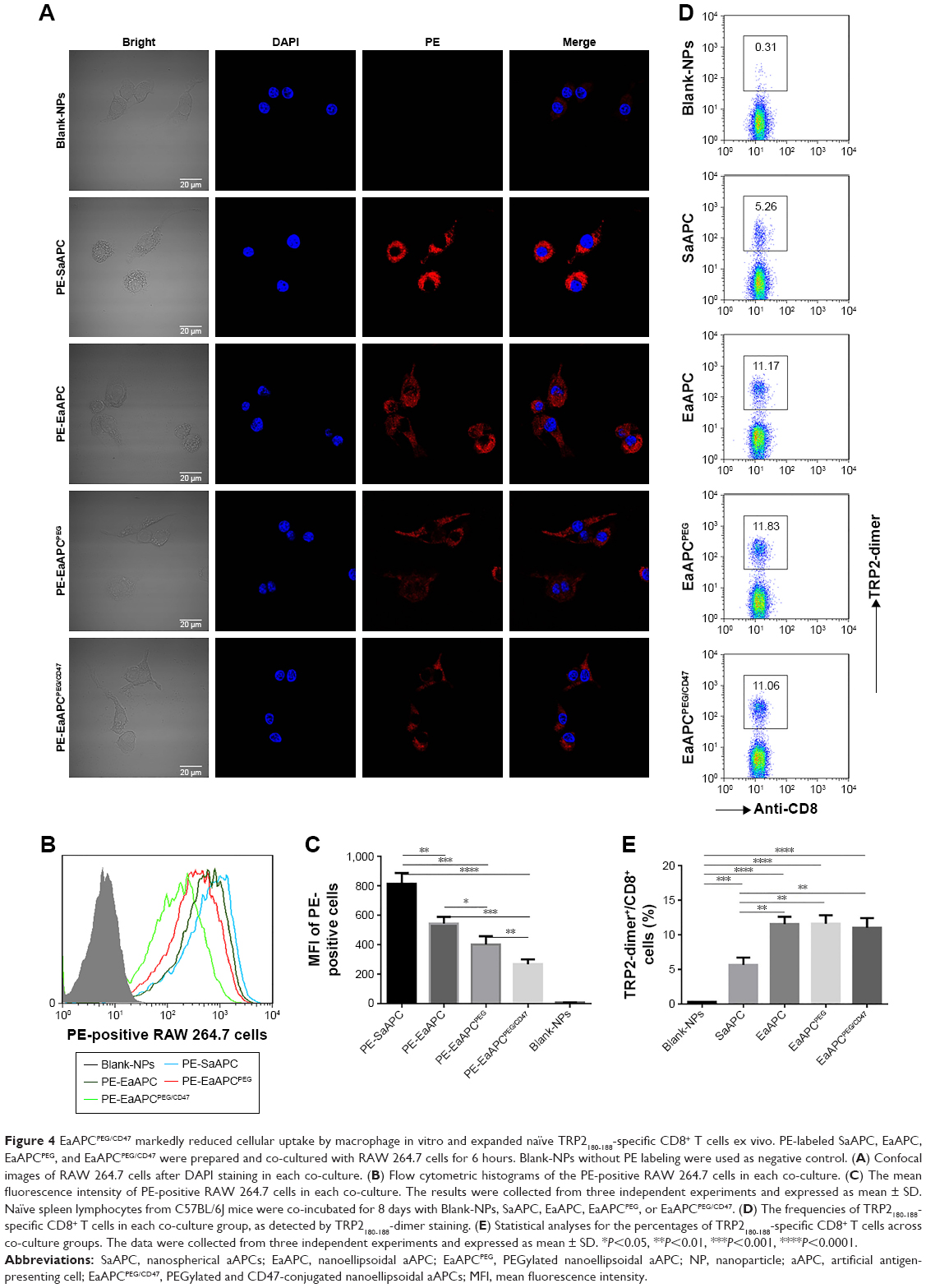 | Figure 4 EaAPCPEG/CD47 markedly reduced cellular uptake by macrophage in vitro and expanded naïve TRP2180-188-specific CD8+ T cells ex vivo. PE-labeled SaAPC, EaAPC, EaAPCPEG, and EaAPCPEG/CD47 were prepared and co-cultured with RAW 264.7 cells for 6 hours. Blank-NPs without PE labeling were used as negative control. (A) Confocal images of RAW 264.7 cells after DAPI staining in each co-culture. (B) Flow cytometric histograms of the PE-positive RAW 264.7 cells in each co-culture. (C) The mean fluorescence intensity of PE-positive RAW 264.7 cells in each co-culture. The results were collected from three independent experiments and expressed as mean ± SD. Naïve spleen lymphocytes from C57BL/6J mice were co-incubated for 8 days with Blank-NPs, SaAPC, EaAPC, EaAPCPEG, or EaAPCPEG/CD47. (D) The frequencies of TRP2180-188-specific CD8+ T cells in each co-culture group, as detected by TRP2180-188-dimer staining. (E) Statistical analyses for the percentages of TRP2180-188-specific CD8+ T cells across co-culture groups. The data were collected from three independent experiments and expressed as mean ± SD. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
As demonstrated, shape influences cellular uptake of the particles. Shape stretching minimizes the regions of high length-normalized curvature to the ends of the particles, and the adhesion anywhere along the length of the particles would inhibit phagocytosis attributed to the low curvature. Thus the non-spherical shapes with very high aspect ratios could reduce phagocytosis compared to the spherical particles with equal volume,38 and show a negative correlation between aspect ratio and uptake rate.26 In addition, PEGylation provides a hydrophilic steric shell onto particles, which shield NPs from aggregation, opsonization, and rapid recognition by the reticuloendothelial system39,40 and also improve the stability of colloidal suspension by inter-particle steric repulsion.39 CD47-mediated anti-phagocytosis might be due to the inhibition of myosin II accumulation and inactivation of actomyosin contraction.41 In this study, twofold ellipsoidal stretch, PEGylation, and self-marker CD47 conjugation were integrated into the new “stealth” aAPC system. As expected, each factor showed significant contribution to reducing cellular uptake and exerted the superimposed or synergistic effects.
EaAPCPEG/CD47 markedly expanded naïve TRP2180-188-specific CD8+ T cells ex vivo
After 8-day co-cultures of naïve spleen lymphocytes from C57BL/6J mice with Blank-NPs, SaAPC, EaAPC, EaAPCPEG, or EaAPCPEG/CD47, the frequencies of TRP2180-188-specific CD8+ T cells were detected by H-2Kb/TRP2-Ig dimers staining and flow cytometry. As compared with the Blank-NP group (0.31%), TRP2180-188-specific CD8+ T cells were expanded around 17.0, 36.0, 38.2, and 35.7 times by SaAPC (5.26%), EaAPC (11.17%), EaAPCPEG (11.83%), and EaAPCPEG/CD47 (11.06%), respectively (Figure 4D). Under the current co-culture conditions in the presence of IL-2, SaAPC showed much weaker function to expand antigen-specific CTLs than the ellipsoidal ones, but no significant difference was found across the EaAPC, EaAPCPEG, and EaAPCPEG/CD47groups (Figure 4E).
In addition to anti-phagocytosis, the ellipsoidal MNPs have greater contact surface area and more frequent interactions with T cells than spherical ones,13 thus ellipsoidal nano- or micro-aAPCs caused improved anti-tumor T-cell responses in vivo compared to their spherical counterparts.13,28 In the co-cultures with naïve spleen lymphocytes, the nanoellipsoidal aAPCs (EaAPC, EaAPCPEG, and EaAPCPEG/CD47) expanded TRP2-specific CD8+ T cells more efficiently than the nanospherical aAPCs (SaAPC), but no significant differences were found across the EaAPC, EaAPCPEG, and EaAPCPEG/CD47 groups, although they showed significantly different capability inhibiting cellular uptake in their co-cultures with macrophages. This conflicting results may be due to the much fewer phagocytes in the co-cultures of lymphocytes with aAPCs; therefore, in vivo effects are worthy of further investigation.
EaAPCPEG/CD47 markedly reduced co-localizations with phagocytes and increased contacts with CD8+ T cells in vivo
In order to further confirm that EaAPCPEG/CD47 can reduce cellular uptake and directly interplay with target CD8+ T cells in vivo, spleens were collected from each melanoma-bearing mouse at 6 hours after intravenous (i.v.) injection of PE-labeled aAPCs or Blank-NPs. Frozen sections were prepared and followed by immune fluorescence staining. As shown by confocal micrographs (Figure 5), EaAPCPEG/CD47 was mainly distributed in the red pulp and marginal zone and showed many co-localizations with CD8+ T cells but much fewer co-localizations with macrophages and DCs, as compared with the Blank-NPs, SaAPC, EaAPC, or EaAPCPEG. Both EaAPCPEG and EaAPC showed fewer or similar co-localizations with CD8+ T cells but more contacts with macrophages and DCs relative to EaAPCPEG/CD47. In addition, CD4+ T cells and B cells showed negligible co-localization with the aAPCs or Blank-NPs.
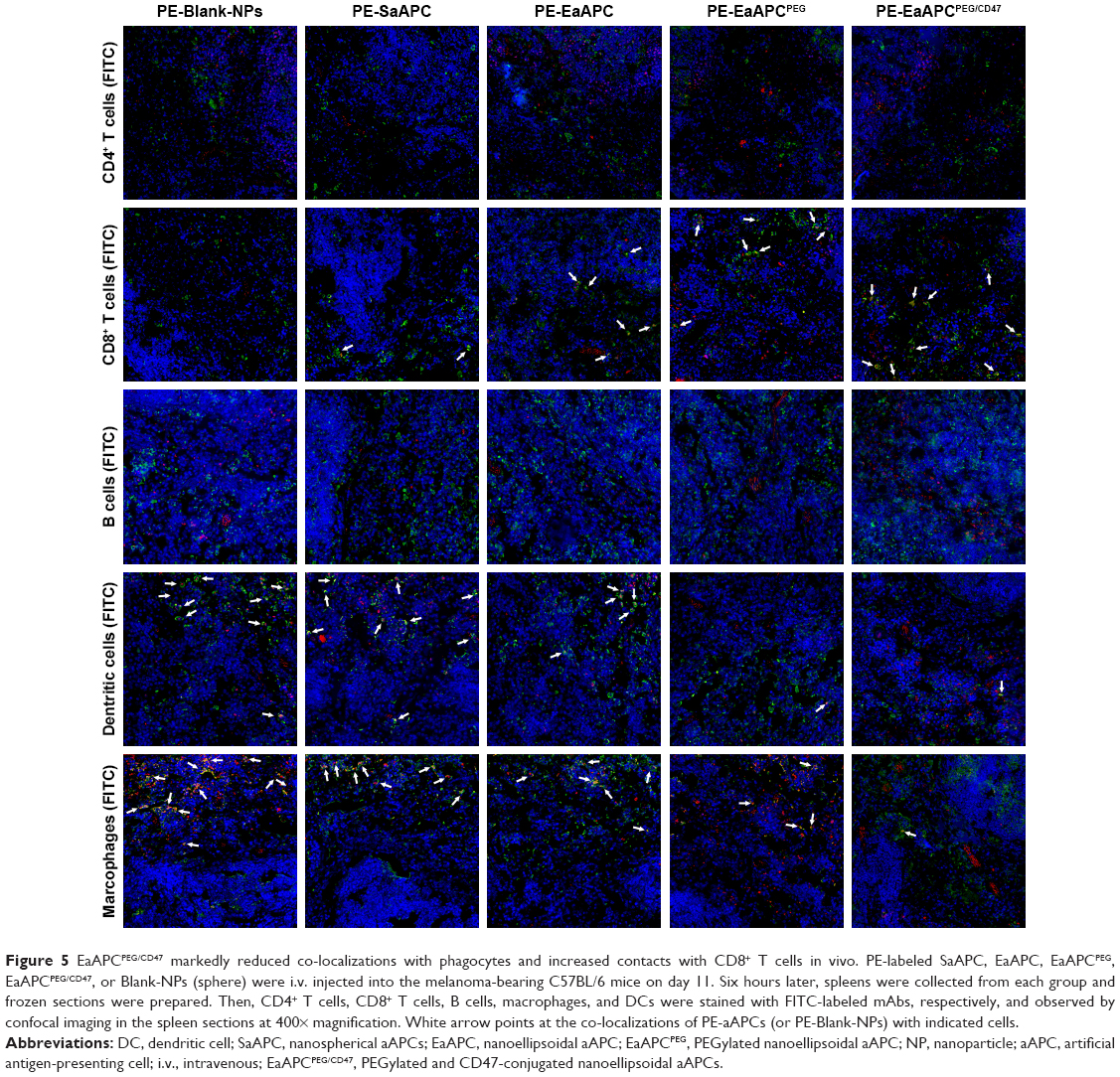 | Figure 5 EaAPCPEG/CD47 markedly reduced co-localizations with phagocytes and increased contacts with CD8+ T cells in vivo. PE-labeled SaAPC, EaAPC, EaAPCPEG, EaAPCPEG/CD47, or Blank-NPs (sphere) were i.v. injected into the melanoma-bearing C57BL/6 mice on day 11. Six hours later, spleens were collected from each group and frozen sections were prepared. Then, CD4+ T cells, CD8+ T cells, B cells, macrophages, and DCs were stained with FITC-labeled mAbs, respectively, and observed by confocal imaging in the spleen sections at 400× magnification. White arrow points at the co-localizations of PE-aAPCs (or PE-Blank-NPs) with indicated cells. |
In another similar experiment, the spleens were processed into single-cell suspensions and freshly stained with FITC-labeled mAbs specific for CD4+ T cells, CD8+ T cells, B cells, DCs, and macrophages, respectively, then analyzed by flow cytometry. As shown in Figure 6, the PE+/FITC+ signals (presumably aAPC-T-cell conjugates or phagocytes intaking aAPCs) were quantified with a visible percentage in the spleen cell suspensions. EaAPCPEG/CD47 showed significantly higher percentages of PE+/CD8-FITC+ signals but lower percentages of PE+/FITC-DC+ or PE+/FITC-Mφ+ signals than Blank-NPs, SaAPC, EaAPC, and EaAPCPEG. In parallel, each type of aAPCs showed comparable percentages co-localized with CD4+ T cells or B cells. To a certain extent, these quantitative data from spleen cell suspensions detected by flow cytometry confirmed the results from spleen sections obtained by confocal images in Figure 5.
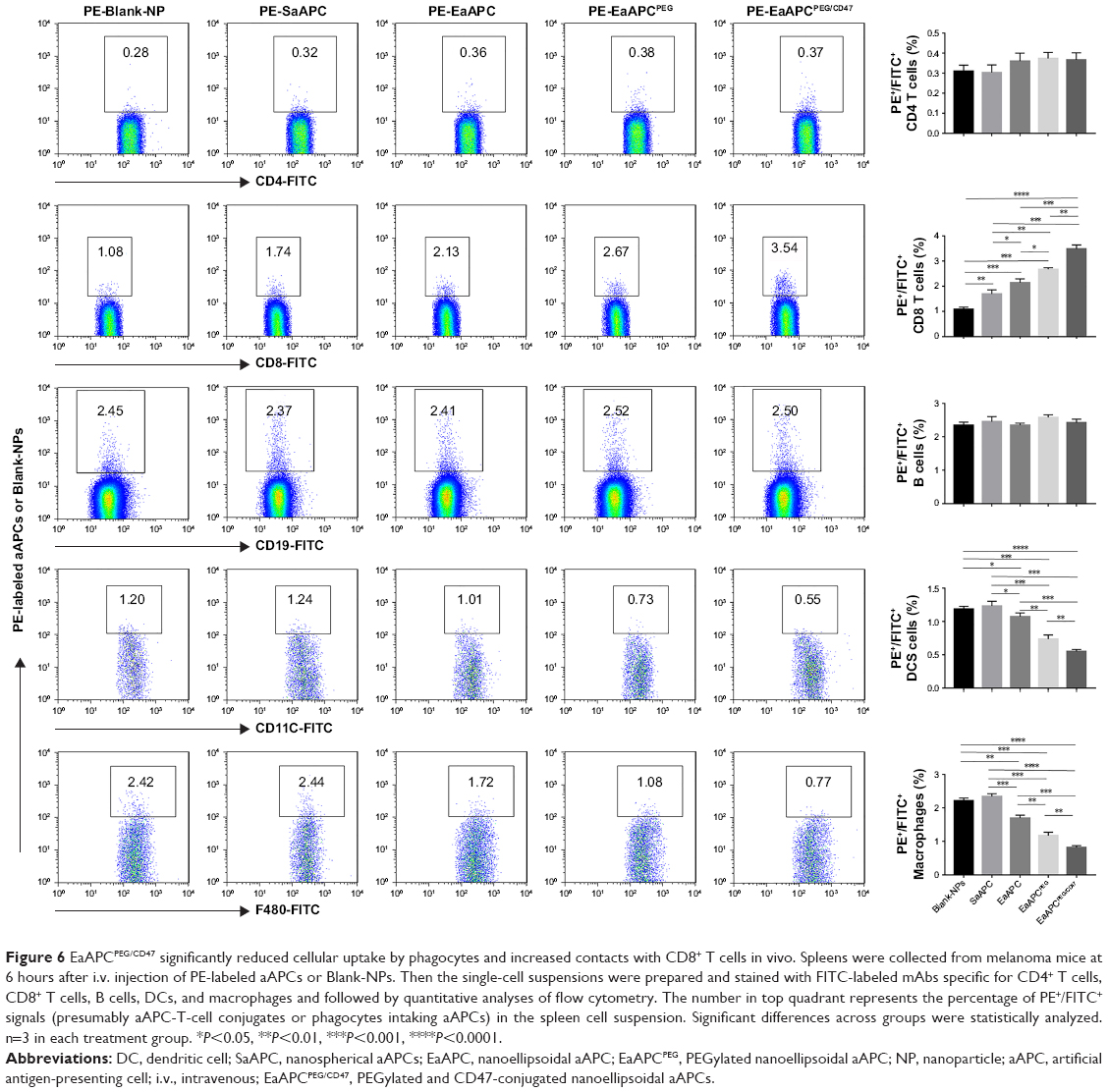 | Figure 6 EaAPCPEG/CD47 significantly reduced cellular uptake by phagocytes and increased contacts with CD8+ T cells in vivo. Spleens were collected from melanoma mice at 6 hours after i.v. injection of PE-labeled aAPCs or Blank-NPs. Then the single-cell suspensions were prepared and stained with FITC-labeled mAbs specific for CD4+ T cells, CD8+ T cells, B cells, DCs, and macrophages and followed by quantitative analyses of flow cytometry. The number in top quadrant represents the percentage of PE+/FITC+ signals (presumably aAPC-T-cell conjugates or phagocytes intaking aAPCs) in the spleen cell suspension. Significant differences across groups were statistically analyzed. n=3 in each treatment group. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
EaAPCPEG/CD47 markedly inhibited melanoma growth and augmented antigen-specific CTL responses after in vivo injections
To further evaluate the in vivo effects of EaAPCPEG/CD47 on antigen-specific CTLs and tumor, C57BL/6J mice were injected with B16 cells s.c. on day 0, then randomized into five groups and infused with SaAPC, EaAPC, EaAPCPEG, EaAPCPEG/CD47, or Blank-NPs (sphere) i.v. on days 7, 9, and 11 as an early therapeutic regimen (Figure 7A). On day 7, the average size of subcutaneous melanoma was 2.668±0.184 mm3 before treatment. Then the mice infused with EaAPCPEG/CD47 showed more remarkable reduction in melanoma growth relative to other aAPC groups and Blank-NPs group. The significant differences were found between groups, with a group order from tumor growth slowly to fast: EaAPCPEG/CD47 < EaAPCPEG < EaAPC < SaAPC < Blank-NPs, as determined by Wilcoxon signed-rank tests (Figure 7B and E). On day 28, mice treated with EaAPCPEG/CD47 achieved the smallest tumor burden with the mean size of 802±28 mm3, while the mice treated with EaAPCPEG, EaAPC, SaAPC, or Blank-MPs presented an average tumor size of 996±33, 1,134±37, 1,345±42, and 1,775±51 mm3, respectively, with significant differences across groups (Figure 7C and E). Moreover, in the EaAPCPEG/CD47, EaAPCPEG, EaAPC, SaAPC, and Blank-NP groups, the median survival times (MSTs) were 51, 47, 44, 38, and 28 days, respectively, and prolonged by 82.1% (P<0.001), 67.9% (P<0.001), 57.1% (P<0.001), and 35.7% (P<0.01), respectively, when compared with the Blank-NP group (Figure 7D). The significant differences were also found across groups, with a group order from MST long to short: EaAPCPEG/CD47 > EaAPCPEG > EaAPC > SaAPC > Blank-NPs (Figure 7E).
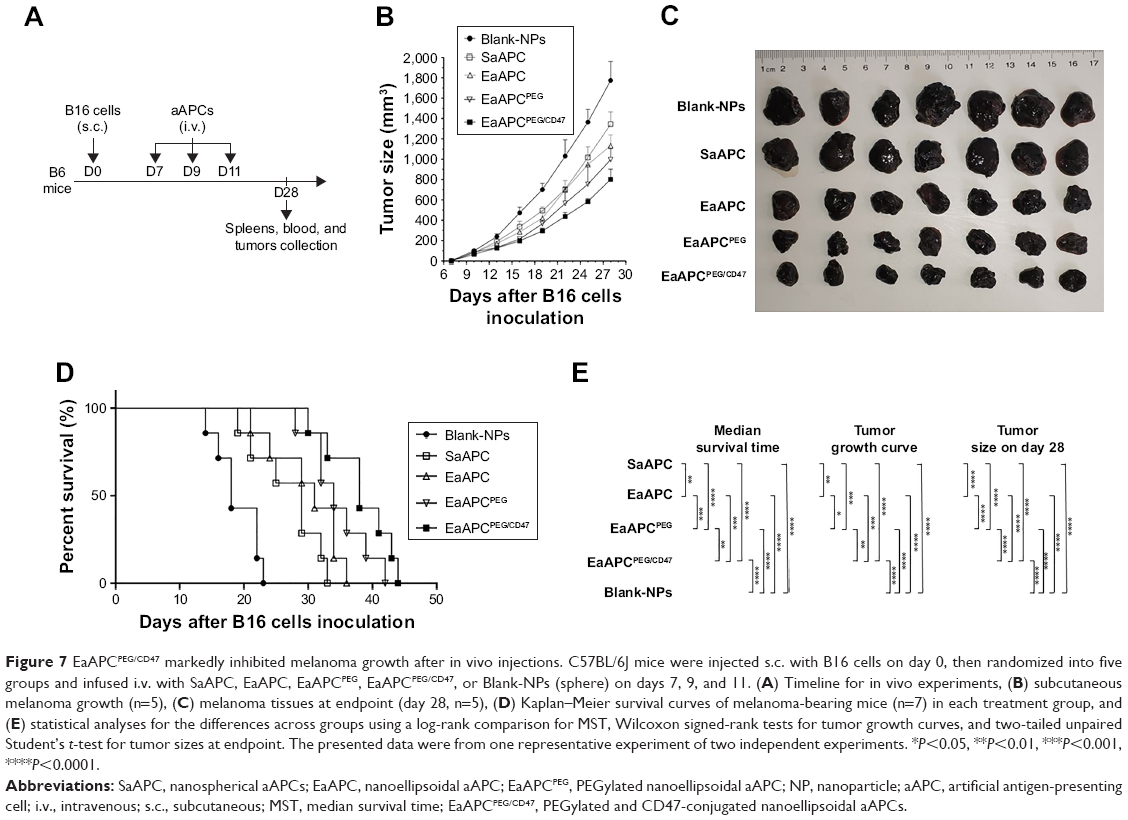 | Figure 7 EaAPCPEG/CD47 markedly inhibited melanoma growth after in vivo injections. C57BL/6J mice were injected s.c. with B16 cells on day 0, then randomized into five groups and infused i.v. with SaAPC, EaAPC, EaAPCPEG, EaAPCPEG/CD47, or Blank-NPs (sphere) on days 7, 9, and 11. (A) Timeline for in vivo experiments, (B) subcutaneous melanoma growth (n=5), (C) melanoma tissues at endpoint (day 28, n=5), (D) Kaplan–Meier survival curves of melanoma-bearing mice (n=7) in each treatment group, and (E) statistical analyses for the differences across groups using a log-rank comparison for MST, Wilcoxon signed-rank tests for tumor growth curves, and two-tailed unpaired Student’s t-test for tumor sizes at endpoint. The presented data were from one representative experiment of two independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
At the endpoint (day 28, 17 days after final infusion of aAPCs), the TRP2180-188-specific CTLs were detected in peripheral blood, spleens, and tumors in each group, by TRP2-dimer staining and flow cytometry. In peripheral blood, the percentages of TRP2180-188-specific CD8+ T cells in CD8+ T-cell population were increased by 17.5-folds (4.20%±0.32%), 13.3-folds (3.19%±0.21%), 9.8-folds (2.34%±0.27%), and 5.8-folds (1.38%±0.23%) in EaAPCPEG/CD47, EaAPCPEG, EaAPC, and SaAPC groups, respectively, as compared with the percentage in Blank-NP group (0.24%±0.06%) (Figure 8A). In spleen lymphocytes suspensions, the frequencies of TRP2180-188-specific CD8+ T cells in the indicated groups were enhanced 15.8-folds (4.17%±0.23%), 12.7-folds (3.34%±0.21%), 8.5-folds (2.24%±0.17%), and 6.3-folds (1.66%±0.13%), respectively, relative to the Blank-NP group (0.26%±0.07%) (Figure 8B). To further investigate the local infiltration of melanoma antigen-specific CTLs in tumor tissues, the single-cell suspensions derived from tumor tissues were prepared and detected. The percentages of TRP2180-188-specific CD8+ T cells were markedly increased by 25.2-folds (3.53%±0.27%), 20.9-folds (2.93%±0.25%), 17.45-folds (2.43%±0.21%), and 10.4-folds (1.46%±0.16%) in the EaAPCPEG/CD47, EaAPCPEG, EaAPC, and SaAPC treatment groups, respectively, when compared to the Blank-NP group (0.14%±0.03%) (Figure 8C). Furthermore, the immunohistochemical staining also confirmed that the EaAPCPEG/CD47 group showed the most extensive infiltration of TRP2180-188-specific CD8+ T cells in the tumor tissues among these groups, as determined by in situ staining of TRP2-dimer and confocal laser scanning (Figure 9). Meanwhile, the H&E staining and Tunel staining revealed much more necrotic tumor cells and apoptotic cells in the melanoma tissues from the EaAPCPEG/CD47 group (Figure 10).
 | Figure 8 EaAPCPEG/CD47 markedly augmented melanoma antigen-specific CTL responses in melanoma-bearing mice. On day 28 (17 days after final infusion of aAPCs or Blank-NPs), TRP2180-188-specific CTLs were detected in peripheral blood, spleens, and tumors for each treatment group, by TRP2-dimer staining and flow cytometry. The percentages of TRP2180-188-specific CTLs in the CD8+ T-cell populations in PBMCs (A), spleen lymphocytes (B), and the cells suspensions derived from melanoma tissues (C) were presented, along with the statistical analyses of differences across groups using a two-tailed, unpaired Student’s t-test; n=5; *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. |
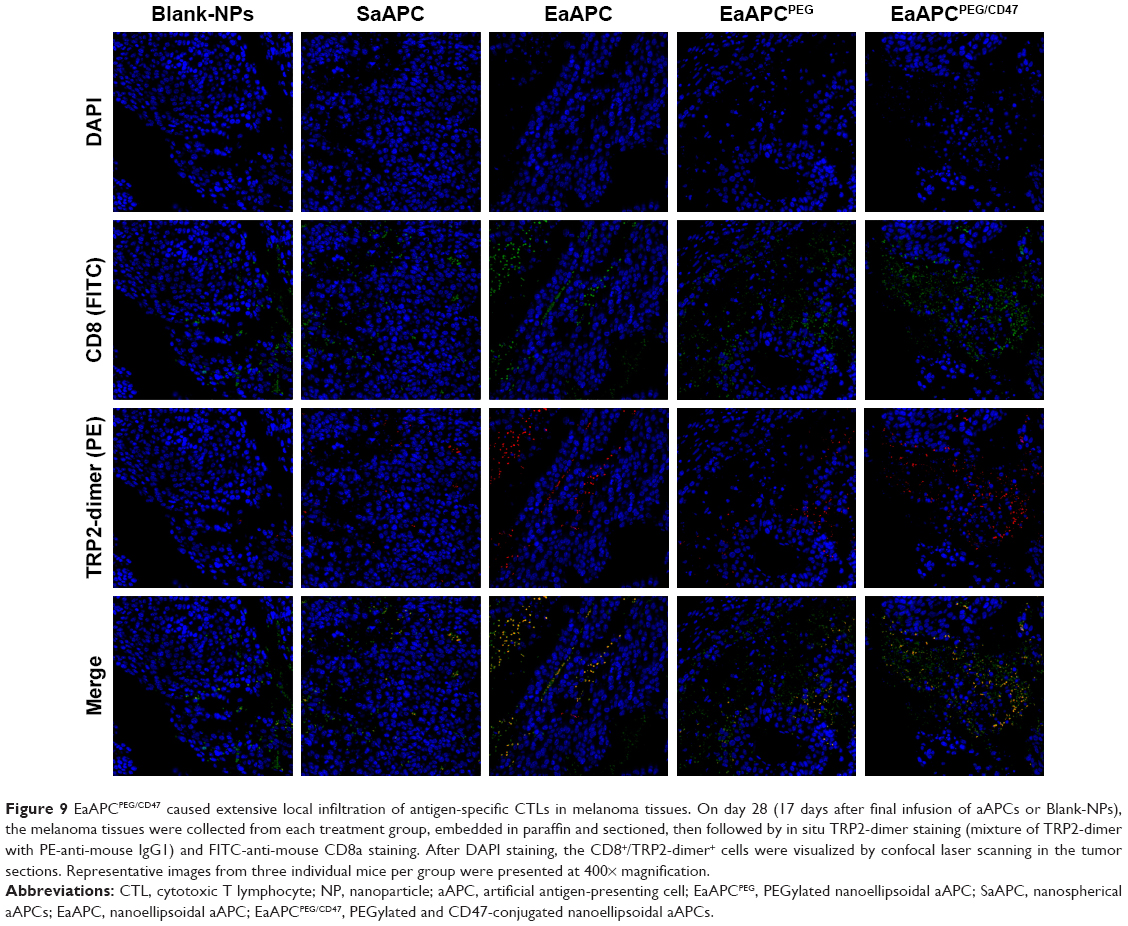 | Figure 9 EaAPCPEG/CD47 caused extensive local infiltration of antigen-specific CTLs in melanoma tissues. On day 28 (17 days after final infusion of aAPCs or Blank-NPs), the melanoma tissues were collected from each treatment group, embedded in paraffin and sectioned, then followed by in situ TRP2-dimer staining (mixture of TRP2-dimer with PE-anti-mouse IgG1) and FITC-anti-mouse CD8a staining. After DAPI staining, the CD8+/TRP2-dimer+ cells were visualized by confocal laser scanning in the tumor sections. Representative images from three individual mice per group were presented at 400× magnification. |
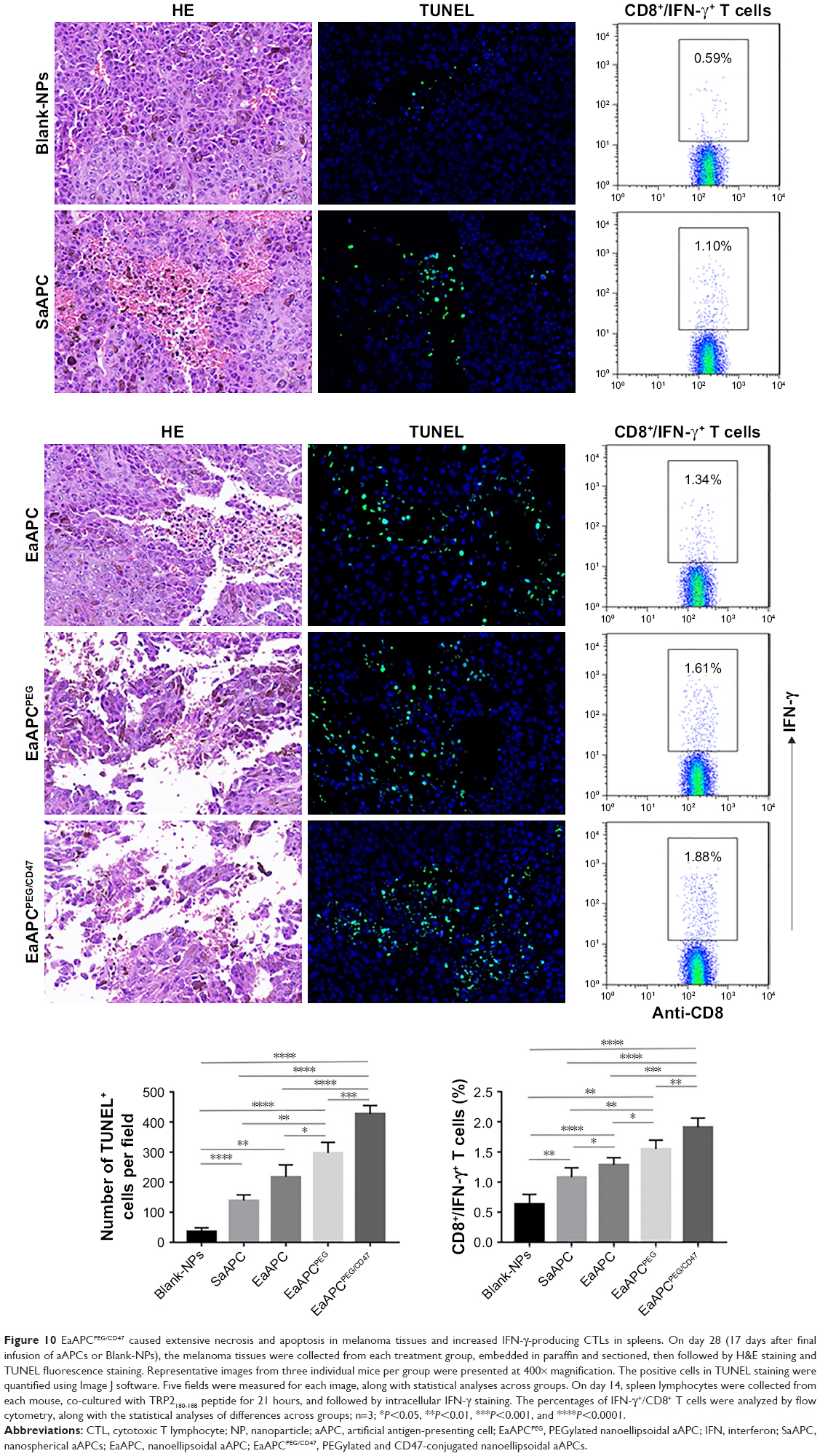 | Figure 10 EaAPCPEG/CD47 caused extensive necrosis and apoptosis in melanoma tissues and increased IFN-γ-producing CTLs in spleens. On day 28 (17 days after final infusion of aAPCs or Blank-NPs), the melanoma tissues were collected from each treatment group, embedded in paraffin and sectioned, then followed by H&E staining and TUNEL fluorescence staining. Representative images from three individual mice per group were presented at 400× magnification. The positive cells in TUNEL staining were quantified using Image J software. Five fields were measured for each image, along with statistical analyses across groups. On day 14, spleen lymphocytes were collected from each mouse, co-cultured with TRP2180-188 peptide for 21 hours, and followed by intracellular IFN-γ staining. The percentages of IFN-γ+/CD8+ T cells were analyzed by flow cytometry, along with the statistical analyses of differences across groups; n=3; *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. |
EaAPCPEG/CD47 significantly improved the functionality of melanoma antigen-specific CTLs after in vivo injections
On day 14, spleen lymphocytes were prepared from each mouse after three infusions of aAPCs, then co-cultured with TRP2180-188 peptide for 21 hours, and followed by intracellular INF-γ staining as well as flow cytometry. As shown in Figure 10 (right panel), after TRP2 peptide stimulation, the frequency of IFN-γ+/CD8+ T cells in the spleens of mice in the EaAPCPEG/CD47 (1.91%±0.15%) was significantly higher than EaAPCPEG (1.57%±0.12%), EaAPC (1.30%±0.10%), SaAPC (1.08%±0.155%), and Blank-NP (0.63%±0.16%) groups. The cytotoxicity assay of CTLs against melanoma cells showed that EaAPCPEG/CD47 elicited significantly greater cytotoxicity of SPCs against B16 cells at various E:T ratios than EaAPCPEG, EaAPC, SaAPC, or Blank-NPs. In the nonspecific cytolysis controls, SPCs from all groups showed <11% of baseline cytolysis against SP2/0 cells (Figure 11).
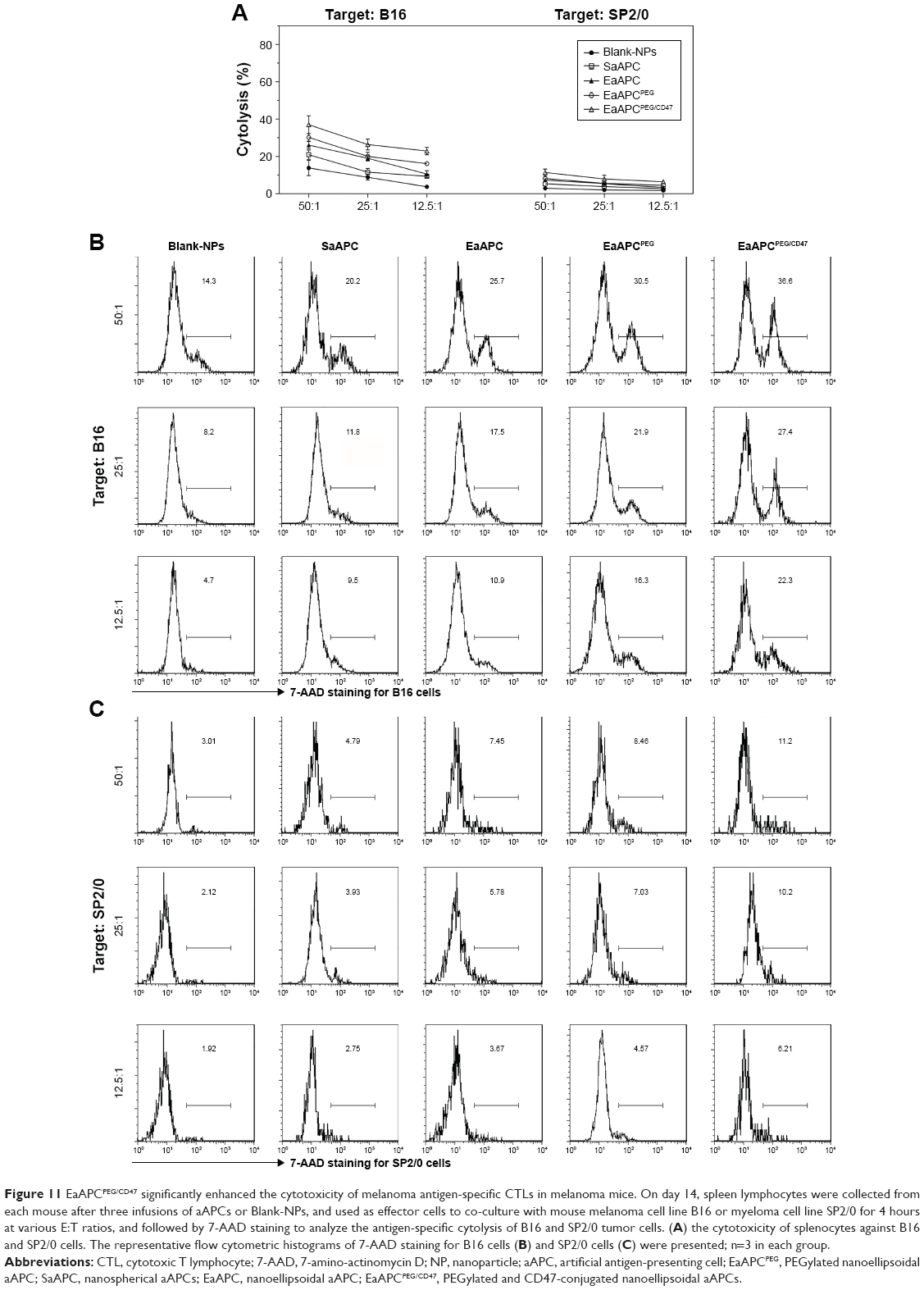 | Figure 11 EaAPCPEG/CD47 significantly enhanced the cytotoxicity of melanoma antigen-specific CTLs in melanoma mice. On day 14, spleen lymphocytes were collected from each mouse after three infusions of aAPCs or Blank-NPs, and used as effector cells to co-culture with mouse melanoma cell line B16 or myeloma cell line SP2/0 for 4 hours at various E:T ratios, and followed by 7-AAD staining to analyze the antigen-specific cytolysis of B16 and SP2/0 tumor cells. (A) the cytotoxicity of splenocytes against B16 and SP2/0 cells. The representative flow cytometric histograms of 7-AAD staining for B16 cells (B) and SP2/0 cells (C) were presented; n=3 in each group. |
Different from the previous studies of SaAPCs,10,16 CD47-modified SaAPCs,30 and SaAPCs,28 the present study initially illustrated the EaAPCPEG and EaAPCPEG/CD47. The results from in vivo experiments demonstrated that the superimposed or synergistic effects of ellipsoidal stretch, PEGylation, and self-marker CD47 conjugation onto nano-aAPC tailored much stronger in vivo functionality of the EaAPCPEG/CD47 to expand antigen-specific CTLs, facilitate their local infiltration, and eventually inhibit tumor growth, when compared to the control aAPCs with a function order from strength to weakness: EaAPCPEG/CD47 > EaAPCPEG > EaAPC > SaAPC. For the SaAPCs and EaAPCs, our results are in agreement with the previous investigations in which the twofold stretched ellipsoidal nano-aAPCs and micro-aAPCs expanded antigen-specific CTLs more efficiently than the spherical counterparts ex vivo and/or in vivo.13,28 The mechanism underlying the elevated functionality may be the increased aAPC–cell interactions along the long axis of ellipsoidal aAPCs as indicated by confocal imaging.13
Conclusion
This study initially integrated ellipsoidal stretch, PEGylation, and surface marker CD47 conjugation in the fabrication of nanoscale aAPCs. The superimposed or synergistic effects enacted the greater resistance to phagocytosis and stronger functionality to expand antigen-specific T cells and inhibit tumor growth, thus suggesting a more valuable strategy to design “stealth” nanoscale aAPCs suitable for tumor active immunotherapy.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81372448) and Science & Technology Support Program of Jiangsu Province (BE2017714). The sponsors had no role in study design, data collection and analysis, preparation of the manuscript, or decision to submit the article for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Bhargava A, Mishra D, Banerjee S, Mishra PK. Dendritic cell engineering for tumor immunotherapy: from biology to clinical translation. Immunotherapy. 2012;4(7):703–718. doi:10.2217/imt.12.40 | ||
Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22(8):1897–1906. doi:10.1158/1078-0432.CCR-15-1399 | ||
Kim JV, Latouche JB, Riviere I, Sadelain M. The ABCs of artificial antigen presentation. Nat Biotechnol. 2004;22(4):403–410. doi:10.1038/nbt955 | ||
Green JJ, Elisseeff JH. Mimicking biological functionality with polymers for biomedical applications. Nature. 2016;540(7633):386–394. doi:10.1038/nature21005 | ||
Meyer RA, Sunshine JC, Green JJ. Biomimetic particles as therapeutics. Trends Biotechnol. 2015;33(9):514–524. doi:10.1016/j.tibtech.2015.07.001 | ||
Perica K, Kosmides AK, Schneck JP. Linking form to function: biophysical aspects of artificial antigen presenting cell design. Biochim Biophys Acta. 2015;1853(4):781–790. doi:10.1016/j.bbamcr.2014.09.001 | ||
Wang C, Sun W, Ye Y, Bomba HN, Gu Z. Bioengineering of artificial antigen presenting cells and lymphoid organs. Theranostics. 2017;7(14):3504–3516. doi:10.7150/thno.19017 | ||
Rhodes KR, Green JJ. Nanoscale artificial antigen presenting cells for cancer immunotherapy. Mol Immunol. 2018;98:13–18. doi:10.1016/j.molimm.2018.02.016 | ||
Giannoni F, Barnett J, Bi K, et al. Clustering of T cell ligands on artificial APC membranes influences T cell activation and protein kinase C theta translocation to the T cell plasma membrane. J Immunol. 2005;174(6):3204–3211. | ||
Perica K, De Leon Medero A, Durai M, et al. Nanoscale artificial antigen presenting cells for T cell immunotherapy. Nanomedicine. 2014;10(1):119–129. doi:10.1016/j.nano.2013.06.015 | ||
Lu X, Jiang X, Liu R, Zhao H, Liang Z. Adoptive transfer of pTRP2-specific CTLs expanding by bead-based artificial antigen-presenting cells mediates anti-melanoma response. Cancer Lett. 2008;271(1):129–139. doi:10.1016/j.canlet.2008.05.049 | ||
Shen C, Cheng K, Miao S, et al. Latex bead-based artificial antigen-presenting cells induce tumor-specific CTL responses in the native T-cell repertoires and inhibit tumor growth. Immunol Lett. 2013;150(1–2):1–11. doi:10.1016/j.imlet.2013.01.003 | ||
Sunshine JC, Perica K, Schneck JP, Green JJ. Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials. 2014;35(1):269–277. doi:10.1016/j.biomaterials.2013.09.050 | ||
Zhang L, Wang L, Shahzad KA, et al. Paracrine release of IL-2 and anti-CTLA-4 enhances the ability of artificial polymer antigen-presenting cells to expand antigen-specific T cells and inhibit tumor growth in a mouse model. Cancer Immunol Immunother. 2017;66(9):1229–1241. doi:10.1007/s00262-017-2016-9 | ||
Kosmides AK, Meyer RA, Hickey JW, et al. Biomimetic biodegradable artificial antigen presenting cells synergize with PD-1 blockade to treat melanoma. Biomaterials. 2017;118:16–26. doi:10.1016/j.biomaterials.2016.11.038 | ||
Steenblock ER, Wrzesinski SH, Flavell RA, Fahmy TM. Antigen presentation on artificial acellular substrates: modular systems for flexible, adaptable immunotherapy. Expert Opin Biol Ther. 2009;9(4):451–464. doi:10.1517/14712590902849216 | ||
Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. doi:10.1016/j.addr.2015.09.012 | ||
Ashley CE, Carnes EC, Phillips GK, et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10(5):389–397. doi:10.1038/nmat2992 | ||
Massarelli E, Papadimitrakopoulou V, Welsh J, Tang C, Tsao AS. Immunotherapy in lung cancer. Transl Lung Cancer Res. 2014;3(1):53–63. doi:10.3978/j.issn.2218-6751.2014.01.01 | ||
Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134(4):2139–2147. doi:10.1021/ja2084338 | ||
Jiang W, Huang Y, An Y, Kim BY. Remodeling tumor vasculature to enhance delivery of intermediate-sized nanoparticles. ACS Nano. 2015;9(9):8689–8696. doi:10.1021/acsnano.5b02028 | ||
Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339(6122):971–975. doi:10.1126/science.1229568 | ||
Tsai RK, Rodriguez PL, Discher DE. Self inhibition of phagocytosis: the affinity of ‘marker of self’ CD47 for SIRPalpha dictates potency of inhibition but only at low expression levels. Blood Cells Mol Dis. 2010;45(1):67–74. doi:10.1016/j.bcmd.2010.02.016 | ||
Geng Y, Dalhaimer P, Cai S, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2(4):249–255. doi:10.1038/nnano.2007.70 | ||
Sharma G, Valenta DT, Altman Y, et al. Polymer particle shape independently influences binding and internalization by macrophages. J Control Release. 2010;147(3):408–412. doi:10.1016/j.jconrel.2010.07.116 | ||
Florez L, Herrmann C, Cramer JM, et al. How shape influences uptake: interactions of anisotropic polymer nanoparticles and human mesenchymal stem cells. Small. 2012;8(14):2222–2230. doi:10.1002/smll.201102002 | ||
Mathaes R, Winter G, Besheer A, Engert J. Influence of particle geometry and PEGylation on phagocytosis of particulate carriers. Int J Pharm. 2014;465(1–2):159–164. doi:10.1016/j.ijpharm.2014.02.037 | ||
Meyer RA, Sunshine JC, Perica K, et al. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small. 2015;11(13):1519–1525. doi:10.1002/smll.201402369 | ||
Sunshine JC, Green JJ. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine (Lond). 2013;8(7):1173–1189. doi:10.2217/nnm.13.98 | ||
Bruns H, Bessell C, Varela JC, et al. CD47 enhances in vivo functionality of artificial antigen-presenting cells. Clin Cancer Res. 2015;21(9):2075–2083. doi:10.1158/1078-0432.CCR-14-2696 | ||
Pagels RF, Prud’homme RK. Polymeric nanoparticles and microparticles for the delivery of peptides, biologics, and soluble therapeutics. J Control Release. 2015;219:519–535. doi:10.1016/j.jconrel.2015.09.001 | ||
Kapoor DN, Bhatia A, Kaur R, Sharma R, Kaur G, Dhawan S. PLGA: a unique polymer for drug delivery. Ther Deliv. 2015;6(1):41–58. doi:10.4155/tde.14.91 | ||
Han FY, Thurecht KJ, Whittaker AK, Smith MT. Bioerodable PLGA-based microparticles for producing sustained-release drug formulations and strategies for improving drug loading. Front Pharmacol. 2016;7:185. doi:10.3389/fphar.2016.00323 | ||
Iqbal M, Zafar N, Fessi H, Elaissari A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int J Pharm. 2015;496(2):173–190. doi:10.1016/j.ijpharm.2015.10.057 | ||
Champion JA, Katare YK, Mitragotri S. Making polymeric micro- and nanoparticles of complex shapes. Proc Natl Acad Sci U S A. 2007;104(29):11901–11904. doi:10.1073/pnas.0705326104 | ||
Betancourt T, Byrne JD, Sunaryo N, et al. PEGylation strategies for active targeting of PLA/PLGA nanoparticles. J Biomed Mater Res A. 2009;91(1):263–276. doi:10.1002/jbm.a.32247 | ||
Arnida, Janát-Amsbury MM, Ray A, Peterson CM, Ghandehari H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur J Pharm Biopharm. 2011;77(3):417–423. doi:10.1016/j.ejpb.2010.11.010 | ||
Champion JA, Mitragotri S. Shape induced inhibition of phagocytosis of polymer particles. Pharm Res. 2009;26(1):244–249. doi:10.1007/s11095-008-9626-z | ||
Avgoustakis K. Pegylated poly(lactide) and poly(lactide-co-glycolide) nanoparticles: preparation, properties and possible applications in drug delivery. Curr Drug Deliv. 2004;1(4):321–333. | ||
Gref R, Luck M, Quellec P, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18(3–4):301–313. | ||
Sosale NG, Spinler KR, Alvey C, Discher DE. Macrophage engulfment of a cell or nanoparticle is regulated by unavoidable opsonization, a species-specific ‘Marker of Self’ CD47, and target physical properties. Curr Opin Immunol. 2015;35:107–112. doi:10.1016/j.coi.2015.06.013 |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.