Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Pazopanib in the management of advanced soft tissue sarcomas
Authors Cranmer L ![]() , Loggers E, Pollack S
, Loggers E, Pollack S
Received 24 February 2016
Accepted for publication 17 April 2016
Published 9 June 2016 Volume 2016:12 Pages 941—955
DOI https://doi.org/10.2147/TCRM.S84792
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Lee D Cranmer,1 Elizabeth T Loggers,2 Seth M Pollack2
1Division of Medical Oncology, University of Washington, Seattle, WA, USA; 2Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA
Abstract: Therapy of soft tissue sarcomas represents an area of significant unmet need in oncology. Angiogenesis has been explored as a potential target both preclinically and clinically, with suggestions of activity. Pazopanib is a multitargeted tyrosine kinase inhibitor with prominent antiangiogenic effects. In a Phase II study, pazopanib demonstrated activity in strata enrolling patients with leiomyosarcomas, synovial sarcomas, or other sarcomas but not those enrolling adipocytic sarcomas. PALETTE, the pivotal Phase III trial, demonstrated improved progression-free survival versus placebo in pazopanib-treated patients previously treated for advanced soft tissue sarcomas. No survival benefit was observed, and adipocytic sarcomas were excluded. Health-related quality-of-life assessments indicated significant decrements in several areas affected by pazopanib toxicities, but no global deterioration. Cost-effectiveness analyses indicate that pazopanib therapy may or may not be cost-effective in different geographic settings. Pazopanib provides important proof-of-concept for antiangiogenic therapy in soft tissue sarcomas. Its use can be improved by further biological studies of its activity profile in sarcomas, studies of biological rational combinations, and clinicopathologic/biological correlative studies of activity to allow better drug targeting.
Keywords: pazopanib, soft tissue sarcoma, cost-effectiveness, PALETTE, angiogenesis, tyrosine kinase inhibitor
Pazopanib development and properties
Angiogenesis has been identified as a key factor in neoplasia and one that is potentially amenable to therapeutic intervention.1 This led to a detailed study of the pathways involved in this process and the development of therapeutics intended to intervene with it. Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) are central players in the process of angiogenesis, and their study has led to new approaches to cancer treatment. In addition, stem cell factor (SCF), closely related to PDGF, has been implicated in a variety of other processes critical to neoplasia, and its exploitation has led to a number of important breakthroughs in cancer treatment.
Five members make up the VEGF ligand family: placenta growth factor and VEGF-A, -B, -C, and -D. These ligands bind to three distinct VEGF receptors (VEGFRs): VEGFR-1 (fms-like tyrosine kinase 1), -2 (human kinase insert domain receptor), and -3 (fms-like tyrosine kinase 4).2,3 Activation of VEGFR-2 is a key angiogenic signal.3
The PDGF pathway is activated through binding of one of five dimeric ligands to a target receptor.2,4 These ligands are either one of four homodimers, designated AA, BB, CC, and DD, or the heterodimer AB. The PDGF receptors (PDGFRs) consist of three dimeric receptors made up of two subunits, designated α and β.4 Binding of PDGF leads to receptor dimerization, yielding the functional receptors αα, αβ, and ββ. Dimerization leads to autophosphorylation of intracellular tyrosines, initiating signal transduction. These receptors convey signals involved in a wide variety of normal and pathologic processes, promoting cell proliferation, migration, and differentiation.
SCF is closely related to the PDGF family.2,4 It exists in both soluble and membrane-bound forms. The receptor for SCF (designated c-Kit or CD117) forms dimers and is activated by a mechanism very similar to that for PDGFR. In normal physiology, SCF regulates hematopoiesis, cell survival, cell proliferation, and cell migration, depending on the cellular context. Inappropriate activation of this pathway is pathogenetically linked to a number of conditions, including acute myeloid leukemia, systemic mastocytosis, and gastrointestinal stromal tumors (GIST).4 Studies of the role of SCF in GIST, and the identification of imatinib as a means to alter this process, have represented key breakthroughs in the development of targeted therapeutic strategies for cancer.5
Pazopanib was identified through screening of compounds for inhibition of VEGFR-2.6 After initial identification from a screening library of a molecule that inhibited VEGFR-2, molecular optimization was undertaken. Screening during this process was undertaken by assessing the ability of the synthetic molecular moieties to inhibit VEGFR-1, -2, and -3 by direct enzymatic assays. Further confirmation of the biological effect of the selected molecule was confirmed through bioassays assessing human umbilical vein endothelial cell proliferation, tumor xenografts, and the Matrigel plug assay. The binding to VEGFR-2 was not absolutely specific: the compound could also bind to VEGFR-1 and -3, PDGFR-α and -β, fibroblast growth factor receptors-1 and -3, c-kit, Interleukin-2 receptor-inducible T-cell kinase, leukocyte-specific protein tyrosine kinase, and transmembrane glycoprotein receptor tyrosine kinase (c-fms).7
Clinical development of pazopanib in forms of cancer other than sarcomas
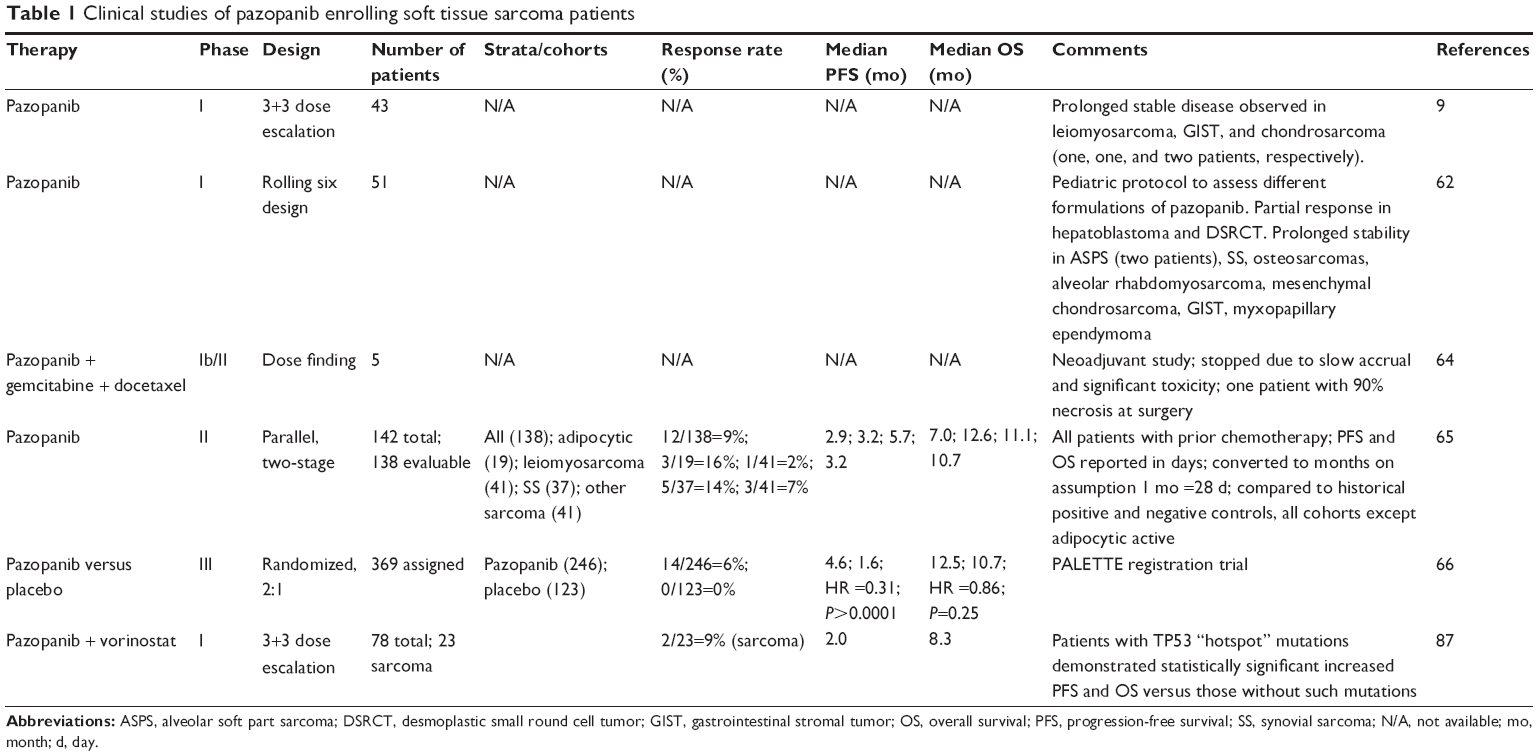
Preclinical studies indicated that pharmacodynamic inhibition of VEGFR-2 by pazopanib correlated with the steady-state plasma concentration, above a certain threshold.8 In mouse xenograft models, inhibition of VEGFR-2 and antitumor effects strongly correlated. A Phase I dose-escalation study enrolled 43 patients in the initial dose-escalation component of the study, with an additional 20 patients enrolled in the dose-expansion phase (Table 1).9 Pharmacokinetic results indicated a plateau in steady-state exposure at doses ≥800 mg once daily. Similar to with other angiogenic agents, hypertension was the most frequent grade 3 adverse event. The most frequent drug-related adverse events were hypertension, diarrhea, hair depigmentation, and nausea. These were generally mild to moderate. Proteinuria, another finding commonly seen in the use of angiogenic therapy, occurred in 52% of the patients; this was not, however, associated with azotemia.
 | Table 1 Clinical studies of pazopanib enrolling soft tissue sarcoma patients |
No maximally tolerated dose was identified.9 An oral dose of 800 mg once daily was selected for further study. This was based on pharmacodynamic and pharmacokinetic criteria. Doses >800 mg daily did not yield further increases in drug exposure. Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) revealed significant alterations in tumoral blood flow. Clinical activity was evident in several patients and seemingly correlated with hypertension as a potential pharmacodynamic marker.
Assessment of preliminary clinical activity was an important secondary objective of this study. Partial responses according to Response Evaluation Criteria In Solid Tumors (RECIST) criteria were observed in two patients with renal cell carcinoma (RCC) and in one patient with pancreatic islet cell tumor. Pertinent to this discussion, prolonged stable disease was reported in one patient with leiomyosarcoma, two patients with chondrosarcoma, and one patient with GIST.
In the further development of pazopanib, advanced unresectable or metastatic RCC has been the subject of a great deal of investigation. This was driven, in part, by the well-described role of angiogenesis in RCC and the approval of several other agents targeting angiogenesis for treatment of RCC.10 A randomized, double-blind, Phase III trial of pazopanib in this population led to approval of the drug for RCC.7,11,12 Progression-free survival (PFS) was the primary end point of the study and was improved by the test agent (median 9.2 versus 4.2 months; hazard ratio [HR] =0.46; P<0.0001). The objective response rate was also markedly greater in the pazopanib-treated group at 30%, versus 3% for those receiving placebo. Extensive crossover from placebo to pazopanib group occurred during the study, interfering with overall survival (OS) analysis. Post hoc analyses, adjusted for crossover, suggested a survival benefit.12
Pazopanib was later compared in a randomized, noninferiority trial to sunitinib in metastatic RCC.13 Pazopanib was not inferior to sunitinib in terms of PFS, the primary end point of study. OS was similar in both arms. Pazopanib performed better in terms of an assessment of health-related quality-of-life (HRQoL).
Pazopanib has also been explored in a randomized trial as maintenance therapy for patients with advanced ovarian carcinoma who did not progress during first-line chemotherapy.14 Maintenance pazopanib therapy yielded improved PFS versus placebo (median 17.9 versus 12.3 months; HR =0.77; P=0.0021). No significant survival difference was noted. There was a higher incidence of treatment discontinuation in the group receiving pazopanib (33.3%) versus those receiving placebo (5.6%).
This strategy is not uniformly successful. A similar study was undertaken in patients with non-small cell lung cancer who had nonprogressive disease after 4–6 cycles of systemic chemotherapy.15 The primary end point was OS. A total of 102 out of 600 planned patients were randomized to either pazopanib or placebo maintenance. The study was discontinued at an interim analysis due to not achieving prespecified PFS benchmarks.
Even if not achieving their efficacy end points, studies conducted with pazopanib compared to placebo provide useful information regarding pazopanib’s toxicity profile.11,12,14,15 Grade 3–4 hypertension is seen in 30%–40% of patients when the drug is administered according to the 800 mg daily dosing schedule. Neutropenia is seen in approximately 8%–10% of patients. Liver abnormalities and enzymatic elevations are seen in approximately 5%–10% of patients. Treatment discontinuation due to adverse events is seen in 20%–35% of patients. Thus, while toxicity with this agent may be manageable, it is not a benign drug, and decisions regarding its use must carefully weigh its benefits and potential risks/toxicities.
Correlative studies of angiogenesis in soft tissue sarcomas
Like other areas of investigation focusing on sarcomas, studies of angiogenesis are confounded by the wide heterogeneity of diseases classified as sarcomas. Angiogenesis may be more or less important in the pathogenesis of specific subtypes of sarcomas, yet most of the correlative data available are based on aggregation. This increases the variability of the resulting data set for a given test and decreases the power of a given study to detect an effect.
That being said, several lines of evidence support the role of angiogenesis as an important factor in sarcoma pathogenesis and a promising target for therapeutic intervention.16–18 Studies of tumoral angiogenic factors, such as VEGF and PDGF expression, have correlated their expression with higher tumor grade19–21 and, in some cases, worsened survival outcomes.22 Similarly, high levels of circulating VEGF have also been associated with higher tumor grade; changes in association with treatment have been associated with response.23,24 Compared to normal subjects, sarcoma patients demonstrate high levels of circulating angiogenic factors, including VEGF.25
Hierarchical clustering analysis of sarcoma specimens using microarrays specific for angiogenesis-related genes demonstrated distinct profiles for the sarcoma specimens versus normal controls.25 Interestingly, similar histologic specimens shared angiogenic profiles more similar to one another than to other sarcoma subtypes. These data indicate that, while alterations in angiogenesis may be common in soft tissue sarcomas, different patterns of angiogenesis alteration may increase or decrease the susceptibility of different sarcoma subtypes to interventions targeting angiogenesis.
Studies of antiangiogenesis agents other than pazopanib in soft tissue sarcomas
Human studies of a number of angiogenesis inhibitors have been undertaken in sarcomas (Table 2). Thalidomide26 has been investigated in a small study of 17 patients with gynecologic sarcomas and carcinosarcomas without significant evidence of activity.27–29 Sorafenib,30 a multitargeted tyrosine kinase inhibitor with a spectrum of activity similar to pazopanib, has undergone several Phase II studies in soft tissue sarcomas.31–33 The study of Maki et al32 suggested possible activity of the drug in vascular sarcomas, including angiosarcomas. More recently, clinical trials in other sarcoma subtypes have explored the activity of sorafenib in specific sarcoma subtypes. These include chordoma,34 solitary fibrous tumor,35 angiosarcoma,36 epithelioid hemangioendothelioma,37 and, although not a soft tissue tumor, osteosarcoma.38 None of the studies indicated high response rates, although some patients experienced more prolonged disease control than might otherwise be expected.
 | Table 2 Phase II trials of antiangiogenic agents other than pazopanib in soft tissue sarcomas |
Sunitinib is another multitargeted tyrosine kinase inhibitor with antiangiogenic properties.39,40 Its spectrum of target inhibition overlaps that of other tyrosine kinase inhibitors, such as sorafenib and pazopanib. Several Phase II studies in soft tissue sarcomas have indicated the agent may have activity, especially as measured by prolonged periods of disease stability.41,42 A Phase II study of sunitinib in uterine leiomyosarcoma did not reveal significant activity.43 In contrast, studies of sunitinib’s activity in alveolar soft part sarcoma indicated potentially substantial activity.44,45
Two lesser known antiangiogenic agents have also been explored in soft tissue sarcomas. Semaxanib (SU5416) is a multitargeted tyrosine kinase inhibitor related to sunitinib.46,47 In a Phase II, single-arm study enrolling 26 evaluable soft tissue sarcoma patients, one patient experienced a minor response to treatment and five had stable disease lasting for at least 3 months. Median PFS was 2 months.48 In another study, 13 patients with advanced soft tissue sarcoma were enrolled. No responses were observed. Median PFS was 1.8 months and median OS was 22.8 months.49 Development of this drug was subsequently abandoned.
Cediranib is a multitargeted, tyrosine kinase inhibitor with prominent activity against the VEGFR family.50 In addition, the drug has activity against PDGFR-β and c-kit. In a pediatric Phase I dose-escalation study, objective responses were observed in patients with Ewing sarcoma, synovial sarcoma, and osteosarcoma.51 Because of preliminary activity identified in prior studies, a single-arm, Phase II study of cediranib in alveolar soft part sarcoma was undertaken.52,53 Of the 43 evaluable patients enrolled, 15 demonstrated a partial response, equating to a response rate of 35%. This included responses in patients previously treated with sunitinib. The drug is being further investigated for alveolar soft part sarcoma. Its activity against soft tissue sarcomas more generally is not yet defined.
Bevacizumab is a monoclonal antibody binding VEGF, preventing the ligand from binding to its receptors.54 This leads to a potent antiangiogenic effect. Its antineoplastic effects may be heightened when administered in combination with cytotoxic chemotherapy. Studies in soft tissue sarcoma patients have been in combination. An important Phase II study combined the drug with doxorubicin, considered the standard, first-line therapy for soft tissue sarcomas.55 Seventeen patients were enrolled. Two achieved a partial response, equating to a response rate of 12%. This did not meet prespecified criteria of 40%. Eleven patients had stable disease of ≥12 weeks. Critically, in six of the 17 enrolled patients, a significant decrement in the left ventricular ejection fraction occurred. One patient died from pneumothorax, which has been reported in association with bevacizumab therapy.56,57 The authors concluded that the significant proportion of patients with prolonged stable disease warranted further study, but the cardiac toxicity of the combination was unacceptable at the dosage used in this particular trial.
Bevacizumab has also been explored by several groups in combination with gemcitabine and docetaxel.58–60 The combination of gemcitabine and docetaxel demonstrated superiority in a randomized trial to gemcitabine monotherapy and subsequently entered wide use in the management of soft tissue sarcomas.61 One Phase II study of the triplet was initially intended as a randomized trial to assess the effect of bevacizumab.58 Due to slow accrual, the design was changed to a single-arm, open-label trial. Seventeen partial responses were observed by RECIST 1.1 criteria, equating to a 49% response rate. The primary end point, PFS at 6 months, was 65%, not achieving the prespecified level to consider the trial positive. The authors argued, however, that the 76% PFS at 3 months and the 49% response rate were higher than would be expected for the backbone regimen of gemcitabine and docetaxel.
Another study was initially planned as a dose finding study of the triplet.60 A total of 38 chemotherapy-naïve patients with soft tissue sarcoma were enrolled. Nine patients received increasing doses of gemcitabine in three cohorts, followed by a dose expansion cohort of 27 patients. The regimen was found to be tolerable. Five complete and six partial responses were reported, and 18 patients exhibited stable disease lasting for a median of 6 months.
Initial human studies of pazopanib in sarcomas
The initial in-human, Phase I study of pazopanib identified an appropriate dose for further development and provided some tantalizing evidence of activity in patients with soft tissue sarcomas (Table 1).9 Since children may display different pharmacokinetic behavior, a Phase I study to assess the pharmacokinetics and pharmacodynamics of pazopanib in patients aged 2–22 years was undertaken.62 A study of 51 patients was undertaken to compare a powder suspension of the product versus the intact tablet.
Dose-limiting toxicities included elevation of lipase, amylase, ALT, proteinuria, and hypertension. One patient in the study with an occult brain metastasis developed intracranial hemorrhage. Among the patients enrolled, one with hepatoblastoma and one with desmoplastic small round cell tumor experienced partial responses to therapy. In addition, eight patients displayed stable disease for at least 6 months, including two patients with alveolar soft part sarcoma and one patient each with synovial sarcoma, osteosarcoma, alveolar rhabdomyosarcoma, mesenchymal chondrosarcoma, GIST, and myxopapillary ependymoma.
Ten pediatric patients receiving pazopanib as part of this Phase I trial underwent DCE-MRI. All eight evaluable patients demonstrated decreases in fractional tumor blood volume in response to pazopanib. Plasma blood biomarker levels were also assessed prior to treatment and after treatment initiation. Plasma soluble VEGFR-2 decreased significantly after pazopanib initiation, and the change from the pretreatment baseline correlated strongly with the trough concentration of pazopanib at day 15. Collectively, these data indicate that pazopanib can be safely administered to pediatric patients with the anticipated toxicities and that pharmacodynamic effects of the drug are as would be predicted. Only a small proportion of patients derived clear benefit by the relatively crude measure of clinical response by RECIST, but prolonged disease control was evident in more patients.
A variety of Phase I studies have examined combination regimens combining pazopanib and cytotoxic chemotherapy. Several of these are pertinent when considering soft tissue sarcoma management. Pazopanib has been successfully combined with gemcitabine.63 The adverse events appeared to be consistent with that associated with the two components of this regimen. There was no pharmacokinetic interaction between the two drugs. No sarcoma patients participated.
In another study, pazopanib was combined with gemcitabine and docetaxel in the neoadjuvant setting prior to surgical resection.64 Only five sarcoma patients were enrolled. Two of these patients had a leiomyosarcoma, and the other three had an undifferentiated pleomorphic sarcoma. Three patients discontinued treatment because of toxicity. No clinical responses were observed, although one patient demonstrated >90% pathologic response to treatment at the time of surgery. The authors concluded that the regimen displayed significant toxicity.
A key Phase II study was undertaken of pazopanib in patients with intermediate or high-grade, advanced soft tissue sarcomas who were either ineligible for cytotoxic chemotherapy or had received fewer than three prior cytotoxic regimens for advanced disease and had documented progression.65 The primary end point of the study was the rate of freedom from progression at 12 weeks. Pazopanib was administered orally at the standard dosing of 800 mg daily. Secondary end points included response rate, safety, and OS. The study was designed as four parallel two-stage cohorts. The cohorts consisted of adipocytic soft tissue sarcomas, leiomyosarcoma, synovial sarcomas, and other soft tissue sarcoma subtypes. For each cohort, a total of 17 evaluable patients were to be enrolled, and if more than three patients were progression-free at 12 weeks, the cohort would be expanded to a total of 37 evaluable patients. If more than ten patients were progression-free and alive at 12 weeks, this was considered presumptive evidence for a 40% rate of PFS at 12 weeks. The study was designed with a type I error rate of 10% and a power of 90%.
A total of 142 patients were enrolled in the study. Among patients with adipocytic sarcomas, only three of the first 17 patients were alive and progression-free at 12 weeks, and this stratum was discontinued. Two patients who had been assigned to other strata were subsequently identified as having adipocytic sarcomas; this yielded a progression-free rate at 12 weeks of 26% (five of 19 patients).
In the remaining cohorts, the studies went on to complete the second stage of accrual. Among patients with leiomyosarcomas, the progression-free rate at 12 weeks was 44% (18 of 41 patients). One leiomyosarcoma patient displayed a partial response according to RECIST criteria. The progression-free rate at 12 weeks for synovial sarcoma patients was 49% (18 of 37 patients), and five of these patients exhibited a partial response. Among patients with other types of sarcomas not assignable to one of the other cohorts, the progression-free rate of 12 weeks was 39% (16 of 41 patients), and three patients in this cohort exhibited a partial response. PFS and OS among the three active cohorts collectively compared favorably to historical controls, referencing both presumably active and other regimens known to be inactive for sarcoma treatment. These promising results provided the rationale for undertaking a Phase III study of pazopanib in patients with soft tissue sarcoma.
Pazopanib expLorEd in sofT-Tissue sarcoma – a phasE III study
Pazopanib expLorEd in sofT-Tissue sarcoma – a phasE III study (PALETTE) was a multicenter, international, Phase III study to assess the activity of pazopanib in soft tissue sarcomas.66 The study was undertaken in patients at least 18 years of age with metastatic soft tissue sarcoma and progressive disease according to RECIST 1.0 criteria. Patients were required to have received at least one regimen of treatment containing an anthracycline and might have received up to four prior lines of systemic therapy for metastatic disease, or no more than two lines of combination treatment. Based on the results of the Phase II study,65 patients with adipocytic sarcomas were excluded. In addition, patients with the following sarcoma subtypes were excluded from participation: embryonal rhabdomyosarcoma, chondrosarcoma, osteosarcoma, Ewing sarcoma, primitive neuroectodermal tumor, GIST, dermatofibrosarcoma protuberans, inflammatory myofibroblastic sarcoma, malignant mesothelioma, and mixed mesodermal tumors of the uterus.
This was a randomized, placebo-controlled trial in which patients were randomized 2:1 to receive pazopanib or placebo, respectively. Patients were treated at the standard dose of pazopanib 800 mg once daily orally. No crossover was incorporated into the study design. The primary end point was PFS at 6 months. The study had a statistical power of 95% to detect a 15% difference in this parameter from an assumed rate of 15% in the control group, with a type I error rate of 5%. In addition, the study had a power of 90% at the type I error level of 5% to detect a 30% decrease in the HR of death, corresponding to an increase in median OS of 8 months anticipated in the control group to 12 months in those receiving pazopanib. Analysis of the primary end point was according to an intent-to-treat analysis.
This study randomized 369 patients, with 246 allocated to pazopanib and 123 receiving placebo. At the time of the primary analysis, the study achieved its primary objective: median PFS was 4.6 months among the group receiving pazopanib versus 1.6 months in those receiving placebo. This corresponded to an HR of 0.31 (95% confidence interval [CI], 0.24–0.40; P<0.0001). OS, while favoring those treated with pazopanib, was not significantly different between the groups. Pazopanib-treated patients displayed median OS of 12.5 months, versus 10.7 months in those receiving placebo (HR =0.86; 95% CI, 0.67–1.11; P=0.2514). Fourteen of the 246 patients (6%) receiving pazopanib experienced a partial response of their disease; none of the 123 patients receiving placebo did so.
Univariate Cox analyses demonstrated that better performance status (0 versus 1), fewer lines of systemic therapy (0–1 versus 2–4), and lower tumor grade (I–II versus III) were significant prognostic factors associated with prolonged PFS. Sex, age, histologic subtype of sarcoma, local regional versus disseminated disease, or the presence of liver metastases was not significant. Among all three histologic subtypes of sarcomas assessed (leiomyosarcoma, synovial sarcoma, or others), treatment with pazopanib was favored over placebo treatment in terms of PFS. In addition, normal hemoglobin at baseline was also associated with longer PFS and OS.67
Adverse events in the PALETTE study were graded according to the National Cancer Institute Common Toxicity Criteria for Adverse Events (CTC AE, version 3.0).66 Of 246 patients randomized to receive pazopanib, only seven did not receive this therapy. Of these 239 treated patients, 221 discontinued pazopanib, 167 of which were due to disease progression. Of the patients receiving pazopanib, 34 discontinued therapy due to adverse events, versus one patient assigned to placebo treatment. A total of 167 pazopanib-treated patients (70%) discontinued due to disease progression, versus 118 (96%) of the patients assigned to receive placebo.
Patients in the pazopanib-treated group experienced eight fatal serious adverse events. One of these, multiorgan failure, was possibly related to study drug treatment. Among placebo-treated patients, six fatal serious adverse events were reported. Grade 3–4 adverse events (serious or life-threatening) reported in >5% of the pazopanib-treated subjects included fatigue (14%), hypertension and parenthesis (7%), anorexia (6%), and diarrhea (5%). The most common (all-grade) adverse events for pazopanib-treated patients included fatigue, diarrhea, nausea, weight loss, and hypertension.
A number of adverse events associated specifically with pazopanib therapy or therapy with antiangiogenesis agents were also reported by the authors. A decreased left ventricular ejection fraction was reported in 16 pazopanib-treated patients (7%; versus 3/123 or 2% in the placebo-treated group). Notably, left ventricular ejection fraction improved in eight of these pazopanib-treated patients; while five continued pazopanib, the other three discontinued due to other reasons. Thromboembolic adverse events occurred in 13 (5%) pazopanib-treated patients versus three (2%) placebo-treated patients. Pneumothorax occurred in eight pazopanib-treated patients, versus one patient (1%) taking placebo.
HRQoL and cost-effectiveness of pazopanib therapy
There is no indication that pazopanib monotherapy is able to act as curative treatment in soft tissue sarcomas. The PALETTE trial administered pazopanib as palliative therapy after up to four lines of prior systemic therapy for advanced disease.66 In this setting, HRQoL and cost-effectiveness become prominent considerations in decisions to use, or not use, a specific therapy.
HRQoL assessment was a preplanned component of PALETTE as an exploratory end point.66,68 HRQoL was assessed at 4, 8, and 12 weeks after initiating treatment using the European Organization for the Research and Treatment of Cancer Quality-of-Life Questionnaire (QLQ-C30). Completion of the questionnaires was 94% at 4 weeks, decreasing to 81% by the completion of this component of the study at week 12. The authors did not identify specific factors associated with decreased questionnaire completion, although they noted that the compliance rates were consistent with other studies. All scores for the general health status of patients receiving pazopanib or placebo were not significantly different over time; subscores related to diarrhea, loss of appetite, nausea/vomiting, fatigue, and role functioning favored the placebo treatment group. The authors argued that the global HRQoL was preserved, while being associated with improvement in PFS, despite decrements in specific subscales due to pazopanib toxicity.
Cost-effectiveness is another means of analyzing the utility of pazopanib; several such analyses have been undertaken.69–71 These analyses are hampered by their need to make a variety of assumptions, including those related to relative effectiveness, costs of medication and medical care in different geographical settings, and different societal prioritizations regarding acceptable costs for health care. An analysis of hypothetical use of pazopanib in Britain found that health system-associated costs of pazopanib as second-line therapy in soft tissue sarcoma were £62,000 (US$88,570 on February 16, 2016) per quality-adjusted life year (QALY).69 The authors assumed a desirable value of £30,000 per QALY, and thus placebo was favored as more cost-effective. However, pazopanib demonstrated a favorable cost-effectiveness ratio when compared to trabectedin, ifosfamide, or gemcitabine combined with docetaxel.
A study by the same group using very similar methodologies examined the use of pazopanib in Canada for the same indication.70 Here, the health care-associated costs of pazopanib therapy as second-line therapy in soft tissue sarcoma were Canadian dollars 163,336 (US$117,544 on February 16, 2016) per QALY. The difference versus Britain reflects high medication acquisition costs in Canada. In addition, different acceptable threshold values per QALY are important factors. In Britain, pazopanib may not meet an acceptable QALY threshold; in Canada it may do so. Of course, these analyses are conducted in reference to placebo therapy, which equates to supportive care only. Critical to this analysis are the other systemic therapy alternatives, which themselves leave much to be desired.
A study from Spain attempted to address the alternatives more realistically.71 Here, pazopanib therapy was contrasted with trabectedin therapy for second-line treatment of advanced soft tissue sarcoma. Pazopanib therapy was markedly less expensive at €21,861 (US$24,327 on February 1, 2016) per QALY versus €45,338 (US$50,452 on February 1, 2016) for trabectedin. This was due to the lower drug acquisition cost, simpler oral administration, and less expensive adverse event management for pazopanib as compared to trabectedin.
Reported activity of pazopanib in specific sarcoma subtypes
Studies of HRQoL and cost-effectiveness highlight important facts regarding pazopanib in soft tissue sarcoma: pazopanib has some significant toxicity; it is an expensive drug; and, when administered without targeting to patients requiring salvage therapy, it is only modestly active. The heterogeneity of soft tissue sarcomas interferes with systematic assessment of the activity of new agents. Often, preliminary evidence of activity comes not from clinical trials but rather from case reports and small case series. Pazopanib is no exception. Activity of the drug has been reported in Ewing sarcoma,72–74 epithelioid hemangioendothelioma,75,76 synovial sarcoma,77 solitary fibrous tumor,78 rhabdomyosarcoma,79 epithelioid sarcoma,80 retroperitoneal liposarcoma,81 and hemangiopericytoma.82 This list is not exhaustive and will undoubtedly expand, as numerous trials of pazopanib in sarcomas are in progress.
What would, however, be more desirable is a more systematic approach to identifying those most likely to benefit from this agent. The PALETTE trial did not indicate histologic differences in responsiveness to this therapy: patients with leiomyosarcoma, synovial sarcoma, and other sarcomas demonstrated equivalent benefit.66 Some histologies were specifically excluded, either because they had other specific therapies, they were highly nonresponsive to systemic treatments, or the antecedent Phase II trial did not indicate significant activity.65 The latter factor explains the exclusion of adipocytic sarcomas (liposarcomas) from the definitive, Phase III trial. That being said, at least two trials studying liposarcoma treatment with pazopanib have completed accrual, and more are in progress (see trials #NCT01506596 and NCT01692496 at ClinicalTrials.gov).
A retrospective analysis compared 38 patients with histologies eligible for the PALETTE study with nine patients with ineligible histologies. Patients with ineligible histologies demonstrated shorter PFS and OS, though this did not achieve statistical significance. The authors hypothesized that these data reflect the fundamental difference in responsiveness between PALETTE-eligible and -ineligible patients.
Development of hypertension occurred in approximately 39% of patients in Phase II and Phase III trials of pazopanib in soft tissue sarcoma.83 This had been proposed previously as a potential biomarker9 and was explored retrospectively as a potential biomarker of benefit. If true, this would provide a very useful and easily implemented means to predict benefit from pazopanib. However, hypertension developing during the first 5 weeks was not, in multivariate analysis, associated with PFS or OS. This was true when considering the association of clinical benefit with either initial development of hypertension or the maximal difference in systolic and diastolic blood pressures. Interestingly, hypertension also did not correlate with the development of adverse events that might be hypothesized as being hypertension-related.
The group that conducted the PALETTE study and its prior Phase II study identified a subset of participating patients who were long-term responders.67 This group of patients demonstrated PFS of at least 6 months and/or OS of at least 18 months. The authors identified good performance status, low or intermediate tumor grade, and a normal hemoglobin level at baseline as being associated with improved long-term outcomes. These data suggest that there is a subset of patients that have biologically favorable disease, from the perspective of pazopanib therapy. Investigation of the outcomes among placebo-treated patients in the Phase III trial could provide important supportive data to determine whether these parameters are predictive of improved pazopanib effect or are simply prognostic.
While in other types of cancer, histology has been a useful guide to treatment, this has not been the case in sarcomas. A recent paper describes the use of pazopanib in a Phase II window-of-opportunity study.84 The primary end point of the study is the metabolic response rate, as assessed through PET imaging. This is a very interesting approach. While it does not offer the possibility of assessing responsiveness prior to treatment initiation in an individual patient, it may indicate whether PET imaging can provide an early marker of benefit, allowing treatment discontinuation. A significant limitation is that the authors again limit the study according to histology: “[t]he study population should consist of patients with STS for which efficacy of pazopanib is assumed”.84 This limitation will prevent the acquisition of data regarding the activity of pazopanib in histologies for which its activity has not been adequately explored, but may in fact exist.85 Restrictions based on histology are only useful in so far as they correlate with biological responsiveness, or lack thereof, to a given treatment.
Studies of combination therapy incorporating pazopanib
Existing studies of pazopanib demonstrate that it has modest activity in patients with advanced soft tissue sarcoma. In addition to attempting to target the drug toward patients more likely to benefit, another approach to increasing its activity could be through combining it with complementary agents. This strategy has proven valuable in other types of cancer, for example, colorectal cancer.86 Enthusiasm for combination therapy with doxorubicin has likely been tempered by the adverse events seen with the bevacizumab/doxorubicin combination.55 Published results of a Phase I study of pazopanib given neoadjuvantly in combination with gemcitabine and docetaxel also reported significant toxicity.64 Many studies are in progress looking at combinations of pazopanib with other cytotoxic chemotherapy agents. It is likely that some of these are tolerable. Whether the underlying backbone chemotherapy regimens would make sense to apply in the setting of soft tissue sarcomas is not clear.
A hypothesis-driven combination approach was explored in a Phase I trial combining pazopanib with vorinostat, a histone deacetylase inhibitor.87 This was a Phase I dose-escalation study, using a conventional 3+3 design, but allowing cohort expansion to enroll additional patients for cancer types that evidenced clinical benefit. Analysis of TP53 in enrolled patients was undertaken to identify those with specific TP53 mutations to determine whether they were more or less susceptible to this combination.
No maximally tolerated dose is identified in the report. The recommended dose for Phase II studies is pazopanib 600 mg daily and vorinostat 300 mg daily. Both these drugs are administered orally. Four dose-limiting toxicities occurred, including thrombocytopenia, diarrhea, and skin rash. A significant number of patients required dose reductions, and patient withdrawals for toxicity occurred frequently. This led the authors to identify the recommended Phase II dose without achieving maximally tolerated dose. Hypertension, a toxicity associated with pazopanib therapy, was observed, but was neither frequent nor dose-limiting.
Four patients demonstrated partial responses, and eleven patients had stable disease for at least 6 months. The clinical benefit rate was 19%. Median PFS and OS among all 78 patients were 2.2 and 8.9 months, respectively. There was no difference based on prior exposure to VEGF-targeted therapy. In multivariate Cox analysis to predict patients qualifying as having clinical benefit (partial response or stable disease in at least 6 months), only hypoalbuminemia and lack of a mutation in one of the “hotspots” of TP53 were associated with a lower rate of clinical benefit.
Among 23 sarcoma patients enrolled, two had a partial response and four had a stable disease for at least 6 months, equating to a clinical benefit rate of 26%. Median PFS and OS were 2.0 and 8.3 months, respectively. In an analysis aggregating the 23 sarcoma patients and 14 colorectal cancer patients in the study, improved PFS and OS were reported for the group possessing a “hotspot” TP53 mutation versus those lacking such a mutation. While comparing those two groups poses some problems (eg, each group represents patients receiving a variety of different doses of the combination), the data serve to suggest testable hypotheses for further assessment.
Conclusions and advice to clinicians
At the time of its approval in 2012 for management of soft tissue sarcomas, pazopanib represented the first agent to be approved for this indication in decades. Since that time, at least two additional agents have been approved for soft tissue sarcoma management – eribulin and trabectedin. Eribulin demonstrated an improvement in OS in patients with liposarcomas versus dacarbazine chemotherapy.88 No benefit was seen in patients with leiomyosarcoma, and no benefit in PFS was observed. Trabectedin was approved based on improved PFS, but no OS benefit, in patients with liposarcomas and leiomyosarcomas compared to dacarbazine.89 Pazopanib likewise was approved based on a PFS, but no OS benefit, compared to a placebo.
At least in part, these approvals have been driven by the significant unmet need in the treatment of sarcomas. None of the approvals are in the first-line setting. The pertinence of the comparator arms in the studies leading to approval of all three agents can reasonably be questioned. It is doubtful that any of these agents would prove superior to doxorubicin treatment in the first-line setting.90
The present data set pertaining to pazopanib use demonstrates that the agent has clinically significant activity in soft tissue sarcomas, as reflected by improved PFS in patients with nonadipocytic soft tissue sarcomas.66 This activity was documented in a study population that had been exposed to up to four prior cytotoxic chemotherapy agents and was studied in reference to placebo. The study population might superficially seem like one with a relatively poor prognosis. This may not, in fact, be the case.
The results of the registration trial would suggest that the population enrolled was effectively selected for having more indolent disease than normal. Among the placebo patients, median OS was 10.7 months, after 56% of them had received two or more lines of treatment and 99% had at least received prior treatment incorporating an anthracycline.66 Estimated survival for patients receiving anthracycline-based therapy for soft tissue sarcomas was 51 weeks in a large meta-analysis, a finding confirmed in a randomized trial comparing doxorubicin with or without ifosfamide as first-line therapy.91,92 This would account for the reported OS time in the placebo arm of the pazopanib registration trial, even if one assumes that all patients only received 2–3-week cycles of conventional doxorubicin therapy prior to enrollment. In fact, the majority of patients likely had significantly more prior therapy than this. The requirements for prior therapy and the general requirements for participation in the clinical trial (especially with regard to performance status) serve to identify a population of patients with more indolent disease. It would be especially important for such a population to be enrolled if the drug is modestly active or has slow onset of action. Additionally, the choice of placebo as the comparator, without allowance for crossover, sets as low a bar as possible for pazopanib to exceed.
Perhaps the most important accomplishment of the pazopanib registration trial is in its providing proof-of-concept for the hypothesis that targeted, antiangiogenic therapy can produce clinically meaningful benefit in soft tissue sarcomas. In most patients, the benefits are modest, but a proportion of patients experience apparent long-term benefit.67 This is at the cost of nontrivial toxicity that clearly has an adverse impact on some facets of HRQoL.68 Further, pazopanib therapy is expensive in all analyses conducted to date. Whether the potential benefits outweigh the financial costs to either health systems or societies is a question that can only be addressed through considering ethics, resources and competing societal needs.
Use of pazopanib is not likely to be static in the future. Further research may be able to clarify our data set regarding its use. This may improve the therapeutic index of the drug, changing the calculus regarding its use.
First, although pazopanib is considered to be an antiangiogenic agent, this may not be the key mechanism of action in soft tissue sarcomas. Different facets of pazopanib activity may be more or less important in different sarcoma subtypes or even in individual patients with the same histologic condition. As noted above, a panel of other antiangiogenic agents has been explored in soft tissue sarcoma treatment. The known spectrum of inhibition of these agents frequently overlaps that of pazopanib. Why then was pazopanib successful in demonstrating a clinically meaningful benefit whereas other, closely related agents were unable to do so?
The answer could be biological in nature, with pazopanib having some inhibitory property that other agents lack. Alternatively, pazopanib may have pharmacodynamic or pharmacokinetic properties that favor its activity. Finally, it is possible that the clinical trials undertaken with pazopanib were conducted in a manner that revealed its beneficial activity. Had other agents been studied in the same way, their activity might have been similarly identified. Studies focused on explaining why pazopanib was found to be active may then be useful in the further development of the drug in soft tissue sarcomas. Second, if studies of antiangiogenic therapy in other settings are to be used as a guide, pazopanib is likely to be most active in combination therapy.86,93,94 Identification of complementary combinations for sarcoma patients should take a high priority. Preferably, such combinations should first be identified through preclinical testing to identify biologically rational combinations. Combinations with nonoverlapping toxicity profiles would be especially desirable.
As anthracyclines are effectively standard, first-line therapy for patients with soft tissue sarcomas, one might consider a combination of pazopanib with doxorubicin. However, the enthusiasm for such combinations of antiangiogenic therapy with doxorubicin has been tempered by the study of bevacizumab with doxorubicin,55 in which exacerbation of doxorubicin’s cardiotoxicity by bevacizumab was reported.
The recent approval of trabectedin for salvage therapy of patients with leiomyosarcomas and liposarcomas provides an opportunity.89 Decreased cardiac ejection fraction is not a prominent side effect of trabectedin, having been reported in only 1% of the patients enrolled in the registration trial. Since virtually all patients enrolled in the trial had received prior anthracycline therapy, a residual effect of that prior anthracycline therapy cannot be excluded as a cause for this.
Trabectedin’s demonstrated activity in patients with leiomyosarcomas would complement that of pazopanib. Evaluation of this combination in those with advanced leiomyosarcomas would be appropriate. Evaluation in other histologies, including liposarcomas, could be undertaken using a Phase II trial design with parallel strata. Evaluation of this combination should take a relatively high priority, given the activity of both agents in leiomyosarcomas. The impetus for evaluation of pazopanib in combination with eribulin is less compelling, given pazopanib’s as-yet undemonstrated activity in liposarcomas, the target population for eribulin.
Third, further studies to focus the use of pazopanib on the population most likely to benefit may yield actionable information soonest. At present, histology has been the focus of these efforts. Histology however imposes severe limitations moving forward. On one hand, histologic classifications may be too general (such as “liposarcomas”, which may encompass numerous distinct histologic entities). Alternatively, histologic classifications may be very specific. Trials in the sarcoma world that are highly focused on exceedingly small subsets of patients are difficult to complete. Certainly, where an activity signal is obvious, such a trial should be undertaken. However, such signals are not clearly evident for pazopanib at this point.
What is needed instead are means to either use clinicopathologic or personalized biological data to guide decisions regarding pazopanib use. For clinicopathologic data, even modest changes in the probability of benefit in a given patient would potentially be useful, particularly data that would indicate a patient who is especially unlikely to benefit. Presenting this information in an accessible format, such as a web-based nomogram calculator, would be particularly useful to clinicians treating sarcoma patients. Preliminary data that could be used for this purpose already exist.67
Correlative biological studies may provide an alternative approach. The data of Fu et al indicated that “hotspot” mutations in TP53 of patients with either sarcoma or colorectal cancer enrolled in their Phase I clinical trial correlated with a higher probability of benefit.87 The study was especially notable in that these data were generated irrespective of histology. If the study could be replicated in sarcoma patients prospectively, it might provide an important tool for selecting patients nonhistologically for pazopanib therapy. This is merely an example. Any approach that allows antecedent assessment to indicate a high probability of benefit, or lack of benefit, might significantly improve the use of pazopanib.
In conclusion, pazopanib is now an approved multitargeted, tyrosine kinase inhibitor available for the management of patients with advanced soft tissue sarcomas. It has demonstrated activity in the salvage setting after prior cytotoxic chemotherapy, yielding an improvement in PFS but not OS. Some patients seem to derive long-term benefit. The drug has a relatively broad spectrum of action, with the exception of liposarcomas, in which its activity has not been clearly established. Pazopanib is associated with significant toxicities, including fatigue, nausea, vomiting, hypertension, thromboembolic events, and reduced cardiac ejection fraction. These toxicities can have an adverse impact on some aspects of HRQoL. Global HRQoL measures are unaffected. The utility of the drug in soft tissue sarcoma management may be further improved by detailed correlative studies to identify the key facets leading to its activity in soft tissue sarcomas, by preclinical and clinical studies of rational combinations incorporating pazopanib, and by the identification of clinicopathologic and/or biological predictors of benefit or lack thereof. The identification of pazopanib’s activity in soft tissue sarcomas is an important step forward: it provides proof-of-concept for the use of antiangiogenic activity in soft tissue sarcoma treatment and also brings a new therapy to our current patients. However, more work is needed to address adequately the major unmet need faced by patients with advanced soft tissue sarcomas.
Disclosure
The authors report no conflicts of interest in this work.
References
Mittal K, Ebos J, Rini B. Angiogenesis and the tumor microenvironment: vascular endothelial growth factor and beyond. Semin Cancer Biol. 2014;41(2):235–251. | ||
Zhao HL, Yang F, Huang X, Zhou QH. Overview of fundamental study of pazopanib in cancer. Thorac Cancer. 2014;5(6):487–493. | ||
Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21(2):154–165. | ||
Heldin CH, Lennartsson J. Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb Perspect Biol. 2013;5(8):a009100. | ||
Tornillo L. Gastrointestinal stromal tumor – an evolving concept. Front Med. 2014;1:43. | ||
Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51(15):4632–4640. | ||
Votrient® (pazopanib) [package insert]. London, UK: GlaxoSmithKline; 2015. | ||
Kumar R, Knick VB, Rudolph SK, et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther. 2007;6(7):2012–2021. | ||
Hurwitz HI, Dowlati A, Saini S, et al. Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res. 2009;15(12):4220–4227. | ||
Posadas EM, Limvorasak S, Sharma S, Figlin RA. Targeting angiogenesis in renal cell carcinoma. Expert Opin Pharmacother. 2013;14(16):2221–2236. | ||
Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28(6):1061–1068. | ||
Sternberg CN, Hawkins RE, Wagstaff J, et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: final overall survival results and safety update. Eur J Cancer. 2013;49(6):1287–1296. | ||
Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369(8):722–731. | ||
du Bois A, Floquet A, Kim JW, et al. Incorporation of pazopanib in maintenance therapy of ovarian cancer. J Clin Oncol. 2014;32(30):3374–3382. | ||
O’Brien ME, Gaafar R, Hasan B, et al. Maintenance pazopanib versus placebo in non-small cell lung cancer patients non-progressive after first line chemotherapy: a double blind randomised phase III study of the lung cancer group, EORTC 08092 (EudraCT: 2010-018566-23, NCT01208064). Eur J Cancer. 2015;51(12):1511–1528. | ||
Sleijfer S, van der Graaf WT, Blay JY. Angiogenesis inhibition in non-GIST soft tissue sarcomas. Oncologist. 2008;13(11):1193–1200. | ||
DuBois S, Demetri G. Markers of angiogenesis and clinical features in patients with sarcoma. Cancer. 2007;109(5):813–819. | ||
Balasubramanian L, Evens AM. Targeting angiogenesis for the treatment of sarcoma. Curr Opin Oncol. 2006;18(4):354–359. | ||
Pakos EE, Goussia AC, Tsekeris PG, Papachristou DJ, Stefanou D, Agnantis NJ. Expression of vascular endothelial growth factor and its receptor, KDR/Flk-1, in soft tissue sarcomas. Anticancer Res. 2005;25(5):3591–3596. | ||
Wang J, Coltrera MD, Gown AM. Cell proliferation in human soft tissue tumors correlates with platelet-derived growth factor B chain expression: an immunohistochemical and in situ hybridization study. Cancer Res. 1994;54(2):560–564. | ||
Chao C, Al-Saleem T, Brooks JJ, Rogatko A, Kraybill WG, Eisenberg B. Vascular endothelial growth factor and soft tissue sarcomas: tumor expression correlates with grade. Ann Surg Oncol. 2001;8(3):260–267. | ||
Yudoh K, Kanamori M, Ohmori K, Yasuda T, Aoki M, Kimura T. Concentration of vascular endothelial growth factor in the tumour tissue as a prognostic factor of soft tissue sarcomas. Br J Cancer. 2001;84(12):1610–1615. | ||
Hayes AJ, Mostyn-Jones A, Koban MU, A’Hern R, Burton P, Thomas JM. Serum vascular endothelial growth factor as a tumour marker in soft tissue sarcoma. Br J Surg. 2004;91(2):242–247. | ||
Graeven U, Andre N, Achilles E, Zornig C, Schmiegel W. Serum levels of vascular endothelial growth factor and basic fibroblast growth factor in patients with soft-tissue sarcoma. J Cancer Res Clin Oncol. 1999;125(10):577–581. | ||
Yoon SS, Segal NH, Park PJ, et al. Angiogenic profile of soft tissue sarcomas based on analysis of circulating factors and microarray gene expression. J Surg Res. 2006;135(2):282–290. | ||
Vacca A, Scavelli C, Montefusco V, et al. Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma. J Clin Oncol. 2005;23(23):5334–5346. | ||
Yi-Shin Kuo D, Timmins P, Blank SV, et al. Phase II trial of thalidomide for advanced and recurrent gynecologic sarcoma: a brief communication from the New York Phase II consortium. Gynecol Oncol. 2006;100(1):160–165. | ||
McMeekin DS, Sill MW, Darcy KM, et al. A phase II trial of thalidomide in patients with refractory uterine carcinosarcoma and correlation with biomarkers of angiogenesis: a Gynecologic Oncology Group study. Gynecol Oncol. 2012;127(2):356–361. | ||
McMeekin DS, Sill MW, Darcy KM, et al. A phase II trial of thalidomide in patients with refractory leiomyosarcoma of the uterus and correlation with biomarkers of angiogenesis: a gynecologic oncology group study. Gynecol Oncol. 2007;106(3):596–603. | ||
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–3140. | ||
Pacey S, Ratain MJ, Flaherty KT, et al. Efficacy and safety of sorafenib in a subset of patients with advanced soft tissue sarcoma from a phase II randomized discontinuation trial. Invest New Drug. 2011;29(3):481–488. | ||
Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27(19):3133–3140. | ||
von Mehren M, Rankin C, Goldblum JR, et al. Phase 2 Southwest Oncology Group-directed intergroup trial (S0505) of sorafenib in advanced soft tissue sarcomas. Cancer. 2012;118(3):770–776. | ||
Bompas E, Le Cesne A, Tresch-Bruneel E, et al. Sorafenib in patients with locally advanced and metastatic chordomas: a phase II trial of the French Sarcoma Group (GSF/GETO). Ann Oncol. 2015;26(10):2168–2173. | ||
Valentin T, Fournier C, Penel N, et al. Sorafenib in patients with progressive malignant solitary fibrous tumors: a subgroup analysis from a phase II study of the French Sarcoma Group (GSF/GETO). Invest New Drug. 2013;31(6):1626–1627. | ||
Ray-Coquard I, Italiano A, Bompas E, et al. Sorafenib for patients with advanced angiosarcoma: a phase II Trial from the French Sarcoma Group (GSF/GETO). Oncologist. 2012;17(2):260–266. | ||
Chevreau C, Le Cesne A, Ray-Coquard I, et al. Sorafenib in patients with progressive epithelioid hemangioendothelioma: a phase 2 study by the French Sarcoma Group (GSF/GETO). Cancer. 2013;119(14):2639–2644. | ||
Grignani G, Palmerini E, Dileo P, et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol. 2012;23(2):508–516. | ||
Papaetis GS, Syrigos KN. Sunitinib: a multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. Biodrugs. 2009;23(6):377–389. | ||
Sutent® (sunitinib) [package insert]. New York, NY: Pfizer; 2015. | ||
George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol. 2009;27(19):3154–3160. | ||
Mahmood ST, Agresta S, Vigil CE, et al. Phase II study of sunitinib malate, a multitargeted tyrosine kinase inhibitor in patients with relapsed or refractory soft tissue sarcomas. Focus on three prevalent histologies: leiomyosarcoma, liposarcoma and malignant fibrous histiocytoma. Int J Cancer. 2011;129(8):1963–1969. | ||
Hensley ML, Sill MW, Scribner DR, et al. Sunitinib malate in the treatment of recurrent or persistent uterine leiomyosarcoma: a Gynecologic Oncology Group phase II study. Gynec Oncol. 2009;115(3):460–465. | ||
Stacchiotti S, Negri T, Zaffaroni N, et al. Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol. 2011;22(7):1682–1690. | ||
Stacchiotti S, Tamborini E, Marrari A, et al. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin Cancer Res. 2009;15(3):1096–1104. | ||
Stopeck A, Sheldon M, Vahedian M, Cropp G, Gosalia R, Hannah A. Results of a phase I dose-escalating study of the antiangiogenic agent, SU5416, in patients with advanced malignancies. Clin Cancer Res. 2002;8(9):2798–2805. | ||
Kosmider O, Denis N, Dubreuil P, Moreau-Gachelin F. Semaxinib (SU5416) as a therapeutic agent targeting oncogenic Kit mutants resistant to imatinib mesylate. Oncogene. 2007;26(26):3904–3908. | ||
Kuenen BC, Tabernero J, Baselga J, et al. Efficacy and toxicity of the angiogenesis inhibitor SU5416 as a single agent in patients with advanced renal cell carcinoma, melanoma, and soft tissue sarcoma. Clin Cancer Res. 2003;9(5):1648–1655. | ||
Heymach JV, Desai J, Manola J, et al. Phase II study of the antiangiogenic agent SU5416 in patients with advanced soft tissue sarcomas. Clin Cancer Res. 2004;10(17):5732–5740. | ||
Sahade M, Caparelli F, Hoff PM. Cediranib: a VEGF receptor tyrosine kinase inhibitor. Future Oncol. 2012;8(7):775–781. | ||
Fox E, Aplenc R, Bagatell R, et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J Clin Oncol. 2010;28(35):5174–5181. | ||
Kummar S, Allen D, Monks A, et al. Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol. 2013;31(18):2296–2302. | ||
Judson I, Scurr M, Gardner K, et al. Phase II study of cediranib in patients with advanced gastrointestinal stromal tumors or soft-tissue sarcoma. Clin Cancer Res. 2014;20(13):3603–3612. | ||
Avastin® (bevacizumab) [package insert]. South San Francisco, CA: Genentech; 2015. | ||
D’Adamo DR, Anderson SE, Albritton K, et al. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. J Clin Oncol. 2005;23(28):7135–7142. | ||
Yang SH, Lin JK, Chen WS, et al. Pneumothorax after bevacizumab-containing chemotherapy: a case report. Jpn J Clin Oncol. 2011;41(2):269–271. | ||
Zhang Y, Yang H, Zhao M, He J. Bilateral pneumothorax after bevacizumab-containing chemotherapy in fibrosarcoma. J Thorac Dis. 2012;4(2):229–231. | ||
Dickson MA, D’Adamo DR, Keohan ML, et al. Phase II trial of gemcitabine and docetaxel with bevacizumab in soft tissue sarcoma. Sarcoma. 2015;2015:532478. | ||
Hingorani P, Eshun F, White-Collins A, Watanabe M. Gemcitabine, docetaxel, and bevacizumab in relapsed and refractory pediatric sarcomas. J Pediatr Hematol Oncol. 2012;34(7):524–527. | ||
Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23(3):785–790. | ||
Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002. [Erratum appears in J Clin Oncol. 2007;25(24):3790]. J Clin Oncol. 2007;25(19):2755–2763. | ||
Glade Bender JL, Lee A, Reid JM, et al. Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children’s oncology group phase I consortium report. J Clin Oncol. 2013;31(24):3034–3043. | ||
Plummer R, Madi A, Jeffels M, et al. A phase I study of pazopanib in combination with gemcitabine in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71(1):93–101. | ||
Munhoz RR, D’Angelo SP, Gounder MM, et al. A phase IB/II study of gemcitabine and docetaxel in combination with pazopanib for the neoadjuvant treatment of soft tissue sarcomas. Oncologist. 2015;20(11):1245–1246. | ||
Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European Organisation for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC study 62043). J Clin Oncol. 2009;27(19):3126–3132. | ||
van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. | ||
Kasper B, Sleijfer S, Litiere S, et al. Long-term responders and survivors on pazopanib for advanced soft tissue sarcomas: subanalysis of two European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62043 and 62072. Ann Oncol. 2014;25(3):719–724. | ||
Coens C, van der Graaf WT, Blay JY, et al. Health-related quality-of-life results from PALETTE: a randomized, double-blind, phase 3 trial of pazopanib versus placebo in patients with soft tissue sarcoma whose disease has progressed during or after prior chemotherapy – a European Organization for Research and Treatment of Cancer–Soft Tissue and Bone Sarcoma Group Global Network Study (EORTC 62072). Cancer. 2015;121(17):2933–2941. | ||
Amdahl J, Manson SC, Isbell R, et al. Cost-effectiveness of pazopanib in advanced soft tissue sarcoma in the United Kingdom. Sarcoma. 2014;2014:481071. | ||
Delea TE, Amdahl J, Nakhaipour HR, et al. Cost-effectiveness of pazopanib in advanced soft-tissue sarcoma in Canada. Curr Oncol. 2014;21(6):e748–e759. | ||
Villa G, Hernandez-Pastor LJ, Guix M, Lavernia J, Cuesta M. Cost-effectiveness analysis of pazopanib in second-line treatment of advanced soft tissue sarcoma in Spain. Clin Transl Oncol. 2015;17(1):24–33. | ||
Alcindor T. Response of refractory Ewing sarcoma to pazopanib. Acta Oncol. 2015;54(7):1063–1064. | ||
Attia S, Okuno SH, Robinson SI, et al. Clinical activity of pazopanib in metastatic extraosseous Ewing sarcoma. Rare Tumors. 2015;7(2):5992. | ||
Yamamoto Y, Nozawa M, Shimizu N, Minami T, Yoshimura K, Uemura H. Pazopanib for recurrent extraosseous Ewing’s sarcoma of the retroperitoneum. Int J Urol. 2014;21(11):1183–1184. | ||
Bally O, Tassy L, Richioud B, Decouvelaere AV, Blay JY, Derbel O. Eight years tumor control with pazopanib for a metastatic resistant epithelioid hemangioendothelioma. Clin Sarcoma Res. 2015;5:12. | ||
Schallier D, Berendes BJ, Lefesvre P, Everaert H. Effective treatment of pleural epitheloid hemangio-endothelioma with pazopanib: a case report. Anticancer Res. 2016;36(1):351–354. | ||
Sugitani A, Asai K, Kojima K, et al. Primary pleural synovial sarcoma treated with pazopanib. Int Med. 2015;54(16):2051–2055. | ||
Maruzzo M, Martin-Liberal J, Messiou C, et al. Pazopanib as first line treatment for solitary fibrous tumours: the Royal Marsden Hospital experience. Clin Sarcoma Res. 2015;5:5. | ||
Hashimoto A, Takada K, Takimoto R, et al. [Effective treatment of metastatic rhabdomyosarcoma with pazopanib]. Gan to Kagaku Ryoho. 2014;41(8):1041–1044. Japanese. | ||
Irimura S, Nishimoto K, Kikuta K, et al. Successful treatment with pazopanib for multiple lung metastases of inguinal epithelioid sarcoma: a case report. Case Rep Oncol. 2015;8(3):378–384. | ||
Koizumi A, Inoue T, Takayama K, et al. [Pazopanib for three patients with recurrence of retroperitoneal liposarcoma: initial clinical experience]. Hinyokika Kiyo. 2015;61(4):153–158. Japanese. | ||
Lee SJ, Kim ST, Park SH, et al. Successful use of pazopanib for treatment of refractory metastatic hemangiopericytoma. Clin Sarcoma Res. 2014;4:13. | ||
Duffaud F, Sleijfer S, Litiere S, et al. Hypertension (HTN) as a potential biomarker of efficacy in pazopanib-treated patients with advanced non-adipocytic soft tissue sarcoma. A retrospective study based on European Organisation for Research and Treatment of Cancer (EORTC) 62043 and 62072 trials. Eur J Cancer. 2015;51(17):2615–2623. | ||
Ronellenfitsch U, Dimitrakopoulou-Strauss A, Jakob J, et al. Preoperative therapy with pazopanib in high-risk soft tissue sarcoma: a phase II window-of-opportunity study by the German Interdisciplinary Sarcoma Group (GISG-04/NOPASS). BMJ Open. 2016;6(1):e009558. | ||
Nakano K, Motoi N, Inagaki L, et al. Differences in the responses to pazopanib and the prognoses of soft tissue sarcomas by their histological eligibility for the PALETTE study. Jpn J Clin Oncol. 2015;45(5):449–455. | ||
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. | ||
Fu S, Hou MM, Naing A, et al. Phase I study of pazopanib and vorinostat: a therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Ann Oncol. 2015;26(5):1012–1018. | ||
Halaven® (eribulin) [package insert]. Woodcliff Lake, NJ: Eisai; 2016. | ||
Yondelis® (trabectedin) [package insert]. Horsham, PA: Janssen; 2015. | ||
Morgan SS, Cranmer LD. Systematic therapy for unresectable or metastatic soft-tissue sarcomas: past, present, and future. Curr Oncol Rep. 2011;13(4):331–349. | ||
Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415–423. | ||
Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al. Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens – a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol. 1999;17(1):150–157. | ||
Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. [Erratum appears in N Engl J Med. 2007;356(3):318]. N Engl J Med. 2006;355(24):2542–2550. | ||
Tewari KS, Sill MW, Long HJ, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370(8):734–743. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.