Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 6
Pathophysiology and therapeutic options in osteogenesis imperfecta: an update
Authors Brizola E, Felix T, Shapiro J
Received 17 September 2015
Accepted for publication 11 January 2016
Published 30 March 2016 Volume 2016:6 Pages 17—30
DOI https://doi.org/10.2147/RRED.S96578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Mingzhao Xing
Evelise Brizola,1 Temis M Félix,2 Jay R Shapiro3
1Bone and Osteogenesis Imperfecta Department, Kennedy Krieger Institute, Johns Hopkins School of Medicine, Baltimore, MD, USA; 2Medical Genetics Service, Hospital de Clínicas de Porto Alegre, Federal University of Rio Grande do Sul, Porto Alegre, Rio Grande do Sul, Brazil; 3Osteoporosis and Metabolic Bone Disorders Center, Bethesda, MD, USA
Abstract: Osteogenesis imperfecta (OI) is a rare, heritable systemic disorder of bone and connective tissue, which in almost 90% of cases is due to mutations affecting the normal synthesis of type I collagen. In 1979, four OI phenotypes were categorized which were inherited as autosomal dominant characteristics. Individuals with OI present both genetic and phenotypic variabilities. Major characteristics of OI are bone fragility, blue sclerae, dentinogenesis imperfecta, short stature, scoliosis, and joint hyperextensibility. Both autosomal dominant and recessive inheritance are now recognized. Advances in molecular diagnosis have led to a major expansion in our understanding of the genetic basis for different OI phenotypes. To date, sequence variants in 17 genes are described as causative of OI. These genes regulate the synthesis of type I collagen pro-alpha polypeptide chains, proteins involved in type I collagen processing in the endoplasmic reticulum and proteins involved in osteoblast function. These new genetic associations have also led to uncertainty with regard to the current classification of OI phenotypes. Bisphosphonates have been widely used to improve bone mass and decrease fractures in both children and adults with OI. While effective in many but not all children when administered for 2–4 years, bisphosphonates have not proven effective in adults with OI. Studies are limited for treatment of adults with teriparatide and denosumab. Advances have been reported in the surgical management of OI. Although the role of physical therapy in the management of children and adults was previously described, this important treatment modality is significantly underutilized.
Keywords: osteogenesis imperfecta, sequence variants, collagen, bisphosphonates, bone, treatment
Introduction
Osteogenesis imperfecta (OI) is a rare clinically and genetically heterogeneous systemic disorder of bone and connective tissue characterized by bone fragility and physical findings related to the underlying connective tissue disorder. The incidence of OI is approximately 6–7/100,000 births, and approximately 500,000 persons worldwide have OI.1 In USA, there are between 25,000 and 50,000 affected individuals.2 Individuals with OI have a lifelong risk of fracture, which may occur at times spontaneously or following minimal trauma. Other systemic manifestations are blue sclerae, dentinogenesis imperfecta (DI), early-onset hearing loss, short stature, kyphoscoliosis, and joint hyperextensibility. Less frequent are basilar invagination, cardiac valvular and vascular disease, and ocular complications, including glaucoma and decreased corneal thickness.1–5
There has been remarkable progress in understanding the molecular basis for this disease. Over the last decade, advances in molecular biology have greatly expanded the association of previously unrecognized gene sequence variants with phenotypes now categorized as OI. As a consequence, the spectrum of OI phenotypes is now considerably enhanced compared to the four phenotypes first described by Sillence et al.6 That classification employed clinical severity, radiological features, and inheritance, dividing OI into four types: mild (OI type I), lethal (OI type II), severe progressive (OI type III), and moderately deforming (OI type IV).6 These four types were inherited in an autosomal dominant manner and each was subsequently shown to involve mutations affecting type I collagen pro-alpha chains.
The Online Mendelian Inheritance in Man database currently lists OI types I–XVII based on recently defined sequence variants including several proteins not in the classic collagen pathway. Approximately 90% of OI cases involve autosomal dominant inheritance with mutations in the COL1A1 or COL1A2 genes.5 To date, the Human Collagen Mutation Database (http://www.le.ac.uk/genetics/ collagen/) lists the following genes associated with a phenotype within the broad clinical spectrum considered OI: BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, P3H1, P4HB, PLOD2, PLS3, PPIB, SEC24D, SERPINF1, SERPINH1, SP7, TMEM38B, WNT1, and SPARC.
OI classification
A classification of OI can be based on a definitive genotype or on a phenotype that is highly variable and more difficult to categorize in many individuals even within the same family. Furthermore, expression of one gene locus can translate into different skeletal phenotypes. Access to genetic testing is not available for many individuals. Therefore, many individuals with OI do not know what clinical type they represent or the genetic basis for their phenotype. Recognizing the emerging confusion due to the expansion of genetic data, the International Skeletal Dysplasia Society kept the Sillence classification with the inclusion of type V OI and without any direct molecular reference.7 The new causative OI genes have been arranged according to clinical severity into the five OI types (Table 1).5,7 In 2010, Van Dijk et al1 proposed a revised classification of OI with the exclusion of type VII and VIII OI, since these types were added based on genotype but with clinical and radiological characteristics indistinguishable from Sillence types II and IV OI.
 | Table 1 OI nomenclature according clinical severity |
It is important to emphasize that the diagnosis of OI still requires historical, clinical, and radiological evidence of the disorder. Fracture history per se in a child does not establish the diagnosis of OI, and several other skeletal disorders including secondary causes of osteoporosis can mimic OI clinically. These include hypophosphatasia, idiopathic juvenile osteoporosis, celiac disease, Ehlers–Danlos syndrome, vitamin D deficiency, and hypophosphatemic disorders. In addition, child physical abuse is relevant considering that this is more frequent than OI and usually presents a history of multiple fractures, but in a pattern different from OI.8
The recommendation of this review, from a clinical standpoint, is that a clinical classification such as the one suggested by Bonafe et al7 seems to be the best option. Probably, clinical features will continue being the basis for OI diagnosis and newer genetic findings will be adapted into recognizable phenotypes.
Phenotypes and genotypes of OI
The discovery of new genotypes related to OI phenotypes has been significantly expanded with the advent of single-nucleotide polymorphism array technology and the increased application of whole-exome sequencing.9 The genes described as OI-causative regulate the synthesis of type I collagen alpha polypeptide chains, several proteins involved in type I collagen processing, or proteins involved in osteoblast development and function.8 Nonetheless, the correlation between genotype and phenotype in OI is extremely complex due to high variability in the expressivity of the disorder (Table 2).
 | Table 2 Expanded classification of OI on molecular basis |
The majority of OI cases are related to autosomal dominant inheritance caused by heterozygosity for mutations in the COL1A1 or COL1A2 genes corresponding to OI types I, II, III, and IV. Individuals with OI type I may experience few fractures associated or not to mild deformities (Figure 1A). OI type II is characterized by severe bone fragility and neonatal mortality. OI type III is a severe form leading to multiple fractures, even in utero, and bone deformities (Figures 1B and 2). OI type IV presents moderate bone involvement with high clinical variability.5,7 De novo mutations are responsible for approximately 60% of mild (OI-I) or moderately deforming OI (OI-IV), almost 100% of severely deforming OI (OI-III), and practically all perinatal lethal cases (OI-II).10 Occasionally, type III and IV OI are the result of autosomal recessive inheritance.1,5
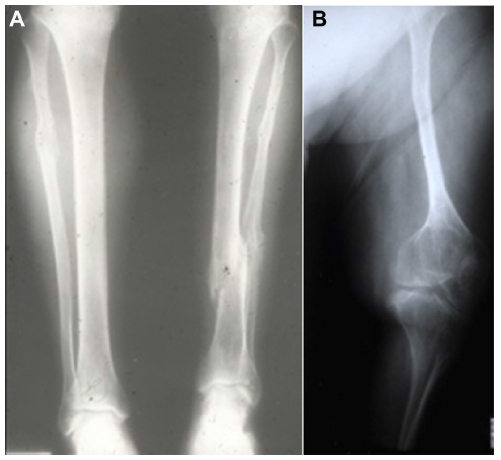 | Figure 1 (A, B) Radiologic demonstration of marked differences in phenotype: OI types I and III. |
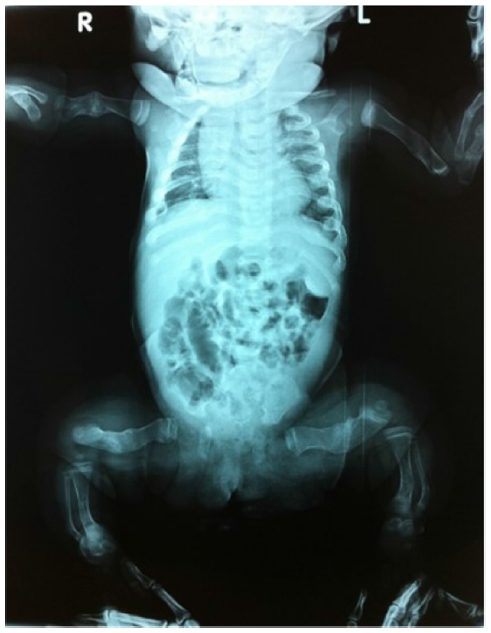 | Figure 2 OI type III, 2-month-old baby girl. |
OI type V is an autosomal dominant disorder with a distinctive phenotype representing 4%–5% of OI cases.5 OI type V is the result of heterozygous mutations (approximately −14C>T) in the 5′UT region of the IFITM5 gene, which encodes interferon-induced transmembrane protein 5.5,11,12 It is characterized by variable degree of severity and specific features such as hyperplastic callus, dislocation of the radial head, and progressive calcification of the interosseous membranes of forearms and legs.12 However, a child with clinical features of type III/IV OI was identified with a de novo IFITM5 mutation (approximately −14C>T) but without the classic features described for OI-V.13
OI types VI–XVI are recessively inherited. Collectively, these account for approximately 3%–5% of individuals with OI and express moderately severe to lethal phenotypes, which in some variants clinically resemble OI types II and III.8
OI-VI is associated with homozygous or compound heterozygous mutations in SERPINF1. This gene encodes pigment epithelium derived factor that is a multifunctional secreted glycoprotein of the serpin family with potent antiangiogenic activity.14,15 It functions in bone homeostasis and osteoid mineralization. Affected individuals have a moderate-to-severe OI, presenting decreased bone mineral density (BMD) with no fractures at birth and without blue sclerae or DI. Bone biopsy shows a characteristic “fish-scale” pattern caused by disorganization of the bone matrix, with large amount of unmineralized osteoid tissue simulating osteomalacia.15–17
Individuals with OI types VII, VIII, and IX have severe to lethal phenotypes caused, respectively, by mutations in the CRTAP, LEPRE1, and PPIB genes. Approximately 1.5% of West Africans and 0.4% of African Americans carry a founder mutation in LEPRE1 gene and three lethal cases of OI-VIII were reported in individuals of these ethnicities.18,19 In addition to features common to severe OI phenotypes, mutations in CRTAP and LEPRE1 genes (OI-VII, OI-VIII) are associated with formation of bulbous epiphysis with popcorn calcifications at the distal femurs.19,20
Type X OI is the result of mutations involving the SERPINH1 gene, which regulates folding and trafficking of the collagen triple helix in the endoplasmic reticulum.5,21 This mutation was reported in a child from Saudi Arabia with triangular facies, blue sclerae, micrognatia, severe bone deformities, and bilateral renal calculi leading to impaired renal function.21
The main features of Bruck syndrome are bone fragility with congenital contractures of large joints. It is classified as types 1 and 2 according to the gene mutated, FKBP10 (OI-XI) and PLOD2, respectively; however, the phenotypes are indistinguishable (Figure 3).22–24 PLOD2 mutation expression in a boy from nonconsaguineous Turkish parents led to congenital join contractures with pterygium and severe OI-like osteopenia including multiple fractures.25 Interestingly, two brothers from a Spanish family also with PLOD2 mutations had phenotypes of different severity, one had a mild form with few fractures and white sclerae and the second had a more severe form with multiple fractures and blue sclerae; however, neither had congenital joint contractures.24
 | Figure 3 Male, 15-years-old, OI type XI (Bruck syndrome – FKBP10 mutation). |
SP7 gene is associated with OI type XII. This moderate form of OI was described in one child from an Egyptian family displaying decreased BMD, recurrent fractures, mild bone deformities, white sclerae, delayed teeth eruption, and absence of DI.26
Mutations in BMP1 gene are categorized as OI type XIII. It was identified in a few individuals with severe but variable phenotypes. Individuals of Egyptian origin had generalized decreased BMD, light blue sclerae, delay of motor development, severe growth deficiency, bone deformities, platispondyly, kyphoscoliosis, umbilical hernia, and several spontaneous fractures.27 Nonetheless, Turkish individuals had increased BMD, white sclerae, and normal motor development, also, multiple fractures after minimal trauma, and not all of them had deformities in lower limbs.28 In addition, a boy with consanguineous parents of Pakistani descent was identified with a homozygous mutation in BMP1 associated with white sclerae, normal bone density, multiple fractures up to age 2, and delay of motor development.29
A homozygous mutation in TMEM38B gene is associated with OI type XIV, which appears to be an ancient mutation in Israeli Bedouin and Saudi families from the Arabian Peninsula.30,31 Affected individuals had bone involvement of variable severity with decreased BMD, multiple fractures ranging from prenatal onset up to 6 years of age, white or blue sclerae, mild-to-moderate short stature, and normal teeth and hearing.30,31
Mutations in WNT1 gene are related to OI-XV. Components of the canonical Wnt/β-catenin signaling pathway are integral for normal osteoblast differentiation as well as the differentiation of several types of stem cells.32 Fahiminiya et al33 have reported WNT1 mutations in four children from three families. These children had short stature, low bone density, and multiple vertebral compression fractures in addition to multiple long-bone fractures during the first years of life.33 A homozygous nonsense mutation was identified in two siblings, this led to brain malformation and severe intellectual disability not associated with major OI phenotypes.34
Keupp et al35 have also reported that hypofunctional alleles of WNT1 (one frameshift mutation, two missense mutations, one splice site mutation, and one nonsense mutation) cause autosomal-recessive OI in consanguineous families. Of note, these authors identified a heterozygous WNT1 missense mutation in a family affected by dominantly inherited early-onset osteoporosis.35 Studies indicated that altered WNT1 proteins fail to activate canonical LRP5-mediated WNT-regulated β-catenin signaling.31,34,35 Pyott et al36 have reported biallelic loss-of-function mutations in WNT1 causing a phenotype consistent with recessively inherited type III OI.
OI-XVI is associated with mutations in CREB3L1 gene. It was reported in a Turkish family with consanguinity. One affected was a fetus from a pregnancy medically interrupted at 19 weeks where postmortem exam showed thin ribs and long-bone fractures. The second child showed intrauterine fractures, pulmonary infections, and cardiac insufficiency leading to death in the first year of life.37
Mutations in P4HB and SEC24D genes are causative of Cole–Carpenter syndrome 1 and 2 (CCS-1 and CCS-2), respectively.38,39 CCS-1 is an OI-like disorder that is characterized by severe bone fragility, fractures, craniosynostosis, ocular proptosis, hydrocephalus, and dysmorphic facial features.38 Individuals initially described with CCS-2 were from a German family and displayed severe pre- and postnatal bone fragility with multiple fractures, diminished ossification of the skull, craniofacial dysmorphism, and short stature.39 A study with the zebrafish model showed that Sec24D plays an important role in the extracellular matrix secretion in cartilage. With reference to facial dysmorphism, Sec24D deficiency is shown to affect craniofacial morphogenesis in late stages of chondroblast development.40
In 2013, X-linked osteoporosis with early-onset fractures occurring in childhood was described in five families associated with a mutation in PLS3, which encodes the actin-binding protein, plastin 3. The rare heterozygous sequence variant (rs140121121) was associated with decreased BMD and an increased risk of fracture among women in the general population.41
SPARC (OI-XVII) is the gene most recently associated with an OI phenotype. Two distinctive homozygous missense mutations in SPARC were reported from two unrelated girls with severe bone fragility. Besides severe bone fragility, one had severe early-onset scoliosis requiring spinal fusion at age 6 years. The second girl was born prematurely of a consanguineous couple, with hypotonia and gross motor developmental delay. Histomorphometric study showed matrix hypermineralization.42
Current pharmacological management options
There is no “optimal treatment” for OI if the primary object of treatment is to limit or prevent fractures in children or adults. Relief from musculoskeletal pain and improvement of compressed vertebral bodies and mobility can also be sought as secondary outcomes. Critical assessment of treatment outcomes is limited by the small numbers of participants in clinical trials and the short duration of many trials, which is frequently limited to 1 or 2 years of observation.
Bisphosphonates
For over 2 decades, bisphosphonates (BPs) have been considered the first-line drug for treating children and adults with moderate or severe forms of OI. Individuals with type I OI may be treated with BPs depending on the frequency of fractures. As will be discussed, the use of BPs in OI has recently come under critical review. Third-generation BPs are nitrogen-containing analogs of pyrophosphate, which inhibit osteoclast bone resorption by interfering with the mevalonic acid pathway and protein prenelation.43 Bone formation is subsequently increased as osteoblastic bone formation completes mineralization at sites of active bone formation (transients). BPs have strong affinity for bone mineral, particularly at sites of increased bone turnover.43 In OI, the response to BP therapy differs between children and adults. BP treatment is generally efficient during childhood due to high bone turnover; however, after puberty there is a decrease in bone turnover, which limits the effectiveness of BP in decreasing fracture rate.2,44
Currently, individual centers in the USA and Europe may use different BPs. Nonetheless, alendronate and risedronate by the oral route and pamidronate and zoledronate by intravenous (IV) route are the most frequently used. It is important to note that there are significant differences with regard to schedules, doses, and duration of treatment among various centers and published studies (Table 3).45
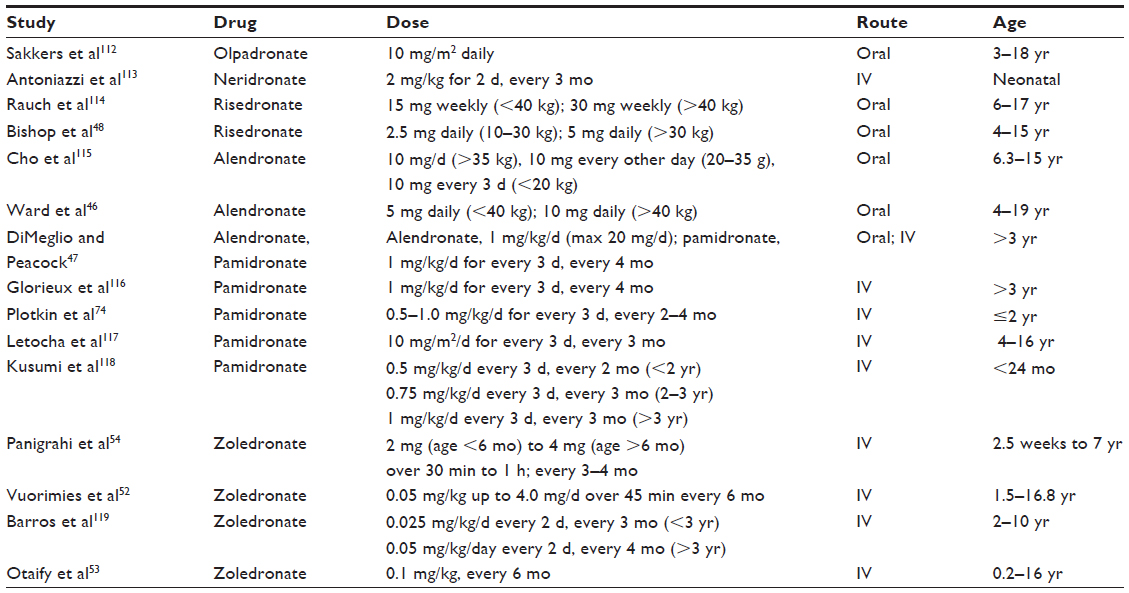 | Table 3 Bisphosphonate treatment protocols for pediatric OI |
Oral BPs are advantageous as they do not require IV access; however, the bioavailability is low and compliance may be poor. Several studies have reported the treatment effects of oral BPs in children and adults, but to date there is no conclusive evidence of its effect on sustained reduction of fracture rate. Two years of alendronate treatment in children with OI was associated with improved lumbar spine areal BMD (LS-aBMD) and decreased bone turnover; however, there was no significant difference in regard to the height of the vertebral bodies, cortical thickness, mobility, bone pain, and fracture incidence.46
Equally beneficial effects following 2 years of BP therapy including increase in BMD, decrease in bone turnover, and a trend to decrease fracture incidence were observed comparing alendronate to pamidronate in a sample of 18 children with OI.47 In a randomized, double-blind, placebo-controlled trial, a daily dose of risedronate was administered for 1 year to children with OI.48 Approximately 81% had a mild form of OI. After 12 months, risedronate treatment increased LS-aBMD and reduced the risk of recurrent fractures by 42%. However, during an extension open-label phase, 2–3 years after the placebo-controlled phase, there was no difference in lumbar spine and total body aBMD between the groups. In addition, 53% of patients in the treated group versus 65% of patients receiving placebo reported vertebral or nonvertebral fractures.48
Palomo et al49 reviewed the results of pamidronate treatment in 37 children with OI who started BP therapy before 5 years of age, and who had a subsequent follow-up period of at least 10 years. At baseline, 35% of vertebrae were affected by compression fractures, whereas only 6% appeared compressed at the final evaluation, indicating vertebral reshaping during growth. Although long-term pamidronate therapy was associated with higher Z-scores for LS-aBMD and vertebral reshaping, the long-bone fracture rate remained high and the majority of patients developed scoliosis.49
Bradbury et al50 treated 37 adults with type I OI with risedronate (35 mg/weekly) for 24 months following which there was an increase on lumbar spine BMD and a decrease in bone biomarkers; however, posttreatment, there was no difference in bone pain or fracture incidence. In addition, Shapiro et al51 reported a marginal decreased fracture incidence associated with IV BP therapy in adult patients with moderate to severe OI but not in individuals with mild OI, and no significant change was observed with oral BP in any type of OI.
Pamidronate is the most used BP for children with OI; however, more recently zoledronate also has been adopted as a treatment option for this population. Zoledronic acid given intravenously is 200-fold more potent than pamidronate and longer-lasting, possibly up to 2 years in children, in suppressing bone turnover. To date, information is limited as regards efficacy and safety in OI patients. However, recent publications indicate the relative short-term safety of zolendronate administered at 6-month intervals in children with OI.52,53 The advantage of this drug compared to pamidronate is the schedule of infusion, that is, a shorter infusion requires short time and is less frequent. Zoledronate is given for 30 minutes every 6 months and pamidronate is infused for 4 hours every 3–4 months.53,54 There is no data indicating that zoledronate is more effective in decreasing fracture rate compared to pamidronate. On the basis of this, zoledronate was designed to be administered yearly in adults with osteoporosis; the current practice of zoledronate therapy in children with OI remains to be assessed.
Positive effects have been described following BP therapy such as increase of BMD, reduced bone turnover, reduction of fractures in children, increase in vertebral body height, relief of musculoskeletal pain, and improvement in mobility.51,54–57 Nonetheless, in contrast to the generally positive effect on fracture incidence in children, the effect on prevention of fractures in adults still requires further investigation.57,58
Recent Cochrane reports assessed the results of BP treatment from meta-analyses of randomized and quasi-randomized controlled clinical trials.59,60 Including eight and 14 trials (403 and 819 participants), respectively, both reports stated an increase of BMD for children and adults with OI following either oral or IV BP treatment, but the effect on decreasing fracture rate was inconclusive. Moreover, the Dwan et al60 review found no significant evidence of positive effect of BP on improvement of clinical status such as reduction of pain and functional mobility.
Rijks et al61 reviewed ten randomized controlled trials (519 participants) with regard to effectiveness and safety of BP in children with OI. LS-aBMD increased and bone resorption biomarkers decreased following either oral or IV BP. Significant decrease in fracture incidence was reported in 70% of the studies following BP therapy. However, the authors highlighted a common problem in interpreting these treatment reports: 1) fracture incidence was reported in several ways, 2) differentiation between vertebral fractures and fractures of extremities required definition, and 3) confounding factors need to be considered, for example, age, pubertal status, and physical activity.
In 2015, Hald et al62 conducted a meta-analysis specifically to assess the effects of BP therapy on the prevention of fractures. Six randomized, placebo-controlled trials (424 subjects) were included. The results of BP therapy on fracture prevention were considered “inconclusive,” and the proportion of participants who experienced a fracture was not significantly reduced.62 The meta-analysis by Shi et al63 reported the efficacy of BP treatment on BMD and fracture rate in patients with OI. In children, BPs were efficacious in reducing fractures (relative risk [RR] =0.80; 95% confidence interval [CI] =0.66–0.97), whereas in adults, BP seemed equivalent to placebo (RR =0.82; 95% CI =0.42–1.59). There was also no significant difference in fracture reduction when comparing oral and IV BPs.
BPs are stored in the skeleton for years, although more rapid turnover in children may shorten residence in bone compared to adults.64 Although data regarding the safety of BP is reported, issues such as bone turnover after several years of BP treatment go unreported. Delayed eruption of primary teeth following BP therapy is reported in children with OI, and Kamoun-Goldrat and Le Merrer65 described a mean delay of 1.67 years in tooth eruption associated to BP treatment. Atypical femoral fracture following prolonged BP therapy previously described in adults with osteoporosis is now reported in a small number of children and adults with OI.66–68 It raises questions about oversuppression of bone remodeling and its contribution to microdamage accumulation and increased bone fragility.64,69 Hegazy et al70 reported the occurrence of unusual femoral stress fractures in six children with OI on long-term pamidronate therapy. These fractures occurred around intramedullary rods in the subtrochanteric or diaphyseal regions of the femur and were caused by no or minimal trauma, similar to the atypical femoral fractures previously described in adults with osteoporosis on long-term BP therapy and with suppression of bone turnover.
Delayed healing of osteotomy sites secondary to BP therapy was previously reported in children and adolescents with OI.71 However, the same group recently published new data based on a protocol focused on patients treated with zoledronate, interval of 4 months without BP infusion after osteotomy, and a different surgical technique using a manual osteotomy rather than a high-powered saw.72 It was observed that, with the newer approach, delayed healing of the osteotomies over 12 months occurred in 42% of the osteotomies performed versus 72% with the previous approach (P=0.001).
In a review published in 2014 of four retrospective studies and one case report involving mean duration of BP from 4.5 to 6.8 years, the authors concluded that there was no evidence to support a causal relationship between BP and osteonecrosis of the jaw in children and adolescents with OI.73
Other effects also associated with BP therapy in OI patients are gastrointestinal pain associated with oral BPs, flu-like symptoms after the first BP infusion, transient asymptomatic hypocalcemia, and fracture nonunion.42,46,74,75
Teriparatide
Teriparatide (human recombinant parathyroid hormone) is a parathyroid hormone analog, which increases bone mass by increasing osteoblast bone formation. Highly effective in the treatment of age-related osteoporosis, teriparatide decreases the risk of fracture by increasing trabecular number and trabecular thickness.76,77 A randomized, double-blind placebo-controlled trial assessed the effect of teriparatide treatment on 79 adults with OI.77 Positive effects on bone markers were observed with a significant increase in hip and spine aBMD, vertebral volumetric BMD, and estimated vertebral strength. This response was more pronounced in type I OI than in individuals with OI types IV and III. There was no significant difference in self-reported fractures when treated and control groups were compared.77
Thirteen postmenopausal women with type I OI previously treated with BPs received daily injections of teriparatide for 18 months.78 Lumbar spine BMD increased significantly up to 3.5% but no difference was observed for total hip BMD. As in the Orwoll et al77 study, teriparatide significantly increased markers of bone formation and bone resorption suggesting a normal osteoblastic response to therapy. Fracture response was not reported.78 Teriparatide is reported to promote healing of an atypical femoral fracture in an adult patient with OI.79 This may be related to the ability of this drug to stimulate chondrocyte proliferation and differentiation and cartilage production at the site of fracture.
Denosumab (anti-RANK-ligand antibody)
The RANK, RANKL complex regulates bone-remodeling cycles by regulating osteoblast/osteoclast coupling and osteoclast differentiation. RANK is present on the osteoclast precursor, and RANKL produced by the osteoblast is part of the TNF superfamily and, along with the soluble decoy receptor osteoprotegerin, are essential regulators of osteoclast development and function.80 Denosumab is a human monoclonal antibody to RANKL; studies involving age-related osteoporosis have showed the efficacy of denosumab in reducing signaling via RANK, leading clinically to prevention of bone loss.80,81 Due to poor response to BP treatment in OI-VI, Semler et al81 treated four children with denosumab who had shown continuously elevated urinary bone resorption markers during a previous treatment with BP.82 Treatment was well tolerated and bone resorption markers decreased to the normal range. However, this report did not address change in BMD or the impact on fracture rate in these children.81 Subsequently, these authors described long-term denosumab effects over 8–12 cycles of treatment on these same patients.83 There was a BMD increase, normalization of vertebral shape, and decrease in fracture rate. Denosumab treatment also improved BMD and longitudinal bone growth in two children with COL1A1/A2 mutations previously treated with BPs.84
New therapeutic approaches
Antisclerostin antibody
Sclerostin is a negative regulator of bone formation released from osteocytes that modulates osteoblast activity acting through Wnt/β-catenin pathway. Preclinical studies have demonstrated that treatment with antisclerostin monoclonal antibody improves bone mass and bone strength, and enhances repair of fractures in animal models.85,86 Interestingly, at the tissue level, different mechanisms are involved when comparing osteoanabolic therapy (teriparatide) and sclerostin antibody (Scl-Ab). While osteoanabolic therapy increases bone remodeling through increase in both bone formation and resorption, Scl-Ab increases bone formation while decreasing bone resorption.87
In humans, anti-sclerostin antibody (romozumab) was administrated to postmenopausal women and healthy men (aged 45–59 years), and the results showed an increase of bone formation markers and a decrease of bone resorption markers. In addition, there was a significant increase in lumbar spine and hip aBMD.88 In patients with OI, Palomo et al89 did not find a relationship between circulating serum levels of sclerostin and LS-aBMD.
Sinder et al90 treated rapidly growing 3-week-old Brtl/+ mice model of OI for 5 weeks with Scl-Ab. Scl-Ab had anabolic effects in Brtl/+ and led to new cortical bone formation and increased cortical bone mass. This anabolic action resulted in improved mechanical strength to wild-type levels without altering the underlying brittle nature of the material.90
However, Roschger et al91 reported somewhat different results using male Col1a1 (Jrt)/+ mice. Once-weekly IV Scl-Ab injections for growing and adult mice had no effect on weight or femur length. No significant treatment-associated differences occurred in alkaline phosphatase activity, procollagen type I N-propeptide, or C-telopeptide. Micro-CT analyses at the femur showed that Scl-Ab treatment was associated with higher trabecular bone volume and higher cortical thickness in wild-type mice at both ages and in growing OI mice, but not in adult OI mice.91
Cathepsin K antibody
Cathepsin K is highly expressed in osteoclasts and is an essential enzyme involved in the degradation of type I collagen in the organic bone matrix.43 In an animal model, the cathepsin K monoclonal antibody (Odanacatib) effectively suppressed bone resorption.92 A Phase III randomized, placebo-controlled trial assessed the effect of Odanacatib on fracture risk over 5 years of treatment in 16,713 women with osteoporosis aged 65 years or older.93 There was an increase in lumbar spine and total hip BMD over the 5 years and a significant reduction in the risk of fractures of hip (47% reduction, P<0.001), spine (23% reduction, P<0.001), and nonvertebral (23% risk reduction, P<0.001).43,93 Applicability to the collagen defect in OI remains to be determined.
Transforming growth factor-β
TGF-β is produced by osteoblasts and acts to coordinate bone remodeling by coupling osteoblasts and osteoclasts in the process of bone remodeling. TGF-β is secreted predominantly in an inactive latent form and is deposited into the bone matrix.94 Several studies have reported on the effect of TGF-β overexpression in bone cells and bone. In 1996, Erlebacher et al95 used transgenic mice to evaluate the role of TGF-β2 in bone development and observed that increased expression of TGF-β2 in osteoblasts resulted in an osteoporosis-like phenotype.
In 2000, Gebken et al96 studied the cell-surface expression and functional properties of TGF-β receptors I, II, and III on osteoblasts from a group of OI patients compared to healthy controls. The number of TGF-β receptors was significantly higher on OI osteoblasts than on age-matched control osteoblasts in spite of similar steady-state levels for TGF-β receptor II mRNA in OI and control cells. Furthermore, receptor affinity was not significantly diminished. Thus, osteoblasts from OI patients were shown to have an elevated number of cell-surface receptors for TGF-β, without any evidence for a transcriptional regulation of TGF-β receptor II.96 Excessive TGF-β signaling has been identified in Marfan syndrome involving abnormalities in both lung and aorta.97
Grafe et al94 have reported that excessive TGF-β signaling is a mechanism of OI in both recessive (Crtap–/–) and dominant (Col1a2tm1.1Mcbr) OI mouse models. Higher expression of TGF-β target genes, a higher ratio of phosphorylated Smad2 to total Smad2 protein, and higher in vivo Smad2 reporter activity were observed in these models. The binding of type I collagen of Crtap (–/–) mice showed reduced binding to decorin, a known regulator of matrix TGF-β activity. Treatment of Crtap (–/–) with the anti-TGF-β neutralizing antibody 1D11 corrected the bone phenotype and improved lung abnormalities in both recessive and dominant forms of OI.94
Prenatal and postnatal transplantation of mesenchymal stem cells
Severe to lethal forms of OI may be diagnosed in utero by ultrasonography starting at the 16th week. Signs include diffuse osteopenia, poor ossification of the skull, increased nuchal transluceny, fractures, delay of growth, micromelia, ventriculomegaly, and bowing of long bones.98 In the oim/oim mouse, allogenic transplanted wild-type donor mesenchymal stem cells (MSCs) homed to bone, improving collagen content and mineralization.99 In humans, improvement of linear growth and reduction of fracture rate followed prenatal and postnatal cell transplantation in OI.100,101 Additionally, prenatal transplantation of allogeneic MSCs in three OI pregnancies indicated that it has appeared to be safe.100 A clinical trial in human pregnancy is currently in progress.101
Multidisciplinary management
Orthopedic treatment
Orthopedic management and/or surgical treatment may be necessary in cases of severe bone deformity impairing function, with recurrent fractures, nonunion of fracture, severe scoliosis, and basilar invagination where neurosurgical intervention may be required. Long periods of immobilization secondary to recurrent fractures or surgery may lead to reduction of bone mass and loss of muscle strength, thereby increasing the risk of further fractures.102 Straightening, realignment, and fixation of long bones by intramedullary rodding is a classic technique for limb stabilization in OI patients. The purpose of rodding is to provide fracture protection and to improve alignment of long bone, thus improving function in growing children.103 The main complication following intramedullary rod fixation is proximal migration of the rod and breakage or disassembly of the rod creating an unprotected segment in the bone.102–104
The need for rodding is not limited to severe types of OI. Lower limb deformities were treated with bone splint technique (osteotomies and internal plating combined with cortical strut allografts) in nine children with type I OI (aged 5–12 years).102 Bone healing occurred within 12–16 weeks after surgery in all patients, and significant improvement on mobility was observed in eight patients. No participant experienced further fractures, deformity, or nonunion.
Nonunion of fractures occurs in approximately 20% of OI patients (Figure 4). Surgical repair can be difficult due to poor bone quality.105 Recently, stabilization of long-bone nonunion fractures was performed through compressed sandwich allograft cortical struts method in 12 OI patients (aged 11–78 years) at 13 sites of nonunion fractures.106 All nonunions healed with allograft incorporation to the bone diaphysis, with all patients recovering to the prefracture level of function, thus reinforcing this approach as durable and safe.106
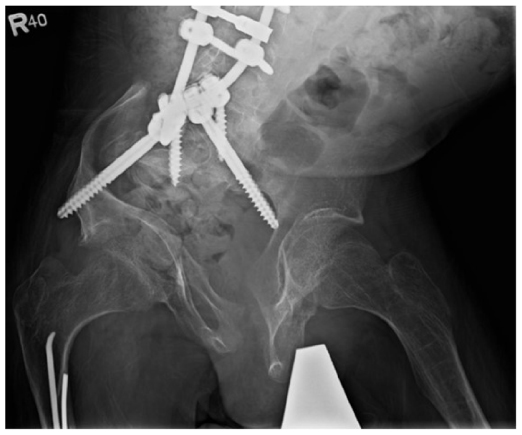
 | Figure 4 Male, 18-years-old, OI type III. |
Rehabilitation
An increased incidence of long-bone fractures is the defining feature in individuals with OI. The majority of these fractures occur in early childhood, directly affecting the child’s motor development.106–108 The clinical type and severity of OI have a major bearing on functional status, including motor development, range of joint motion, and muscle strength.107 Children with moderate to severe forms of OI have greater functional limitation, which negatively affects the development of motor function and the level of ambulation when compared to children with mild forms of OI. In addition, gait acquisition is directly affected by joint range of motion and muscle strength.108 Young adults with type I OI demonstrate full independence, while limitations in self-care, mobility, and domestic life activities were observed in some individuals with OI types IV and V.109
To date, there are no physiotherapeutic treatment protocols available for children and adults with OI. A recent study investigated a rehabilitation approach combining resistance training, body-weight-supported treadmill training, and neurodevelopmental treatment associated with side-alternating whole-body vibration in 53 individuals with OI (ages 2.5–24.8 years) for 6 months within a period of 12 months of treatment.110 There was improvement of mobility between 0 and 6 months of treatment, and also an increase in lean mass and aBMD was observed. However, 46 patients had received BP treatment for years, and the program involved different approaches to treatment.
Further investigation is required, including targeted therapy, the acquisition of specific clinical data, and the evaluation of physiotherapy over short- and long-term courses of intervention. The overall approach to effective physiotherapy for the OI patient will improve when experience is enlarged through the publication of more case reports and cohort studies aimed at outcome assessment.
Conclusion
Research in clinical and genetic fields has expanded speedily. Multiple new genes are now recognized as OI causative, reinforcing the need for extensive clinical investigation focused on improving pharmacological and nonpharmacogical therapy linked to these new genetic discoveries. After 20 years of BP therapy in children and adults with OI, questions have arisen about the long-term efficacy of this mode of treatment. In addition, new therapeutic approaches and surgical techniques have been investigated in OI. Efforts are required to improve techniques for gene replacement therapy, which to date has shown only limited success.111 Nevertheless, still there is no cure for OI and the treatment remains based on drug therapy, orthopedic care, rehabilitation, and nutrition.
Disclosure
The authors report no conflicts of interest in this work.
References
Van Dijk FS, Pals G, Van Rijn RR, Nikkels PG, Cobben JM. Classification of osteogenesis imperfecta revisited. Eur J Med Genet. 2010;53(1):1–5. | |
Martin E, Shapiro JR. Osteogenesis imperfecta: epidemiology and pathophysiology. Curr Osteoporos Rep. 2007;5(3):91–97. | |
Basel D, Steiner RD. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet Med. 2009;11(6):375–385. | |
Ben Amor IM, Glorieux FH, Rauch F. Genotype-phenotype correlations in autosomal dominant osteogenesis imperfecta. J Osteoporos. Epub September 6, 2011. | |
Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014;164A(6):1470–1481. | |
Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. | |
Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167(12):2869–2892. | |
Harrington J, Sochett E, Howard A. Update on the evaluation and treatment of osteogenesis imperfecta. Pediatr Clin North Am. 2014; 61(6):1243–1257. | |
Maasalu K, Nikopensius T, Kõks S, et al. Whole-exome sequencing identifies de novo mutation in the COL1A1 gene to underlie the severe osteogenesis imperfecta. Hum Genomics. 2015;9:6. | |
Steiner RD, Adsit J, Basel D. COL1A1/2-related osteogenesis imperfecta. GeneReviews® [serial on the Internet]. 2005. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1295. Accessed March 24, 2015. | |
Semler O, Garbes L, Keupp K, et al. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012;91(2):349–357. | |
Cho TJ, Lee KE, Lee SK, et al. A single recurrent mutation in the 5′UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012;91(2):343–348. | |
Grover M, Campeau PM, Lietman CD, et al. Osteogenesis imperfecta without features of type V caused by a mutation in the IFITM5 gene. J Bone Miner Res. 2013;28(11):2333–2337. | |
Becker J, Semler O, Gilissen C, et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011;88(3):362–371. | |
Cho SY, Ki CS, Sohn YB, Kim SJ, Maeng SH, Jin DK. Osteogenesis imperfecta type VI with severe bony deformities caused by novel compound heterozygous mutations in SERPINF1. J Korean Med Sci. 2013;28(7):1107–1110. | |
Homan EP, Rauch F, Grafe I, et al. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J Bone Miner Res. 2011;26(12):2798–2803. | |
Fratzl-Zelman N, Schmidt I, Roschger P, et al. Unique micro- and nano-scale mineralization pattern of human osteogenesis imperfecta type VI bone. Bone. 2015;73:233–241. | |
Cabral WA, Barnes AM, Adeyemo A, et al. A founder mutation in LEPRE1 carried by 1.5% of West Africans and 0.4% of African Americans causes lethal recessive osteogenesis imperfecta. Genet Med. 2012;14(5):543–551. | |
Cabral WA, Chang W, Barnes AM, et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39(3):359–365. | |
Baldridge D, Schwarze U, Morello R, et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum Mutat. 2008;29(12):1435–1442. | |
Christiansen HE, Schwarze U, Pyott SM, et al. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86(3):389–398. | |
Alanay Y, Avaygan H, Camacho N, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:551–559. | |
Kelley BP, Malfait F, Bonafe L, et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res. 2011;26:666–672. | |
Puig-Hervás MT, Temtamy S, Aglan M, et al. Mutations in PLOD2 cause autosomal-recessive connective tissue disorders within the Bruck syndrome – osteogenesis imperfecta phenotypic spectrum. Hum Mutat. 2012;33(10):1444–1449. | |
Ha-Vinh R, Alanay Y, Bank RA, et al. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet A. 2004;131(2):115–120. | |
Lapunzina P, Aglan M, Temtamy S, et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;9;87(1):110–114. | |
Martínez-Glez V, Valencia M, Caparrós-Martín JA, et al. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012;33(2):343–350. | |
Asharani PV, Keupp K, Semler O, et al. Attenuated BMP1 function compromises osteogenesis, leading to bone fragility in humans and zebrafish. J Hum Genet. 2012;90(4):661–674. | |
Valencia M, Caparrós-Martin J, Sirerol-Piquer MS, et al. Report of a newly identified patient with mutations in BMP1 and underlying pathogenetic aspects. Am J Med Genet A. 2014;164A(5):1143–1150. | |
Shaheen R, Alazami AM, Alshammari MJ, et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet. 2012;49(10):630–635. | |
Volodarsky M, Markus B, Cohen I, et al. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum Mutat. 2013;34(4):582–586. | |
Ahmadzadeh A, Norozi F, Shahrabi S, Shahjahani M, Saki N. Wnt/β-catenin signaling in bone marrow niche. Cell Tissue Res. Epub October 16, 2015. doi: 10.1007/s00441-015-2300-y. | |
Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013;50(5):345–348. | |
Laine CM, Joeng KS, Campeau PM, et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368(19):1809–1816. | |
Keupp K, Beleggia F, Kayserili H, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92(4):565–574. | |
Pyott SM, Tran TT, Leistritz DF, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. 2013;92(4):590–597. | |
Symoens S, Malfait F, D’hondt S, et al. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis. 2013;8:154. | |
Rauch F, Fahiminiya S, Majewski J, et al. Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am J Hum Genet. 2015;96(3):425–431. | |
Garbes L, Kim K, Rieß A, et al. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet. 2015;96(3):432–439. | |
Sarmah S, Barrallo-Gimeno A, Melville DB, Topczewski J, Solnica-Krezel L, Knapik EW. Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS One. 2010;5(4):e10367. | |
Van Dijk FS, Zillikens MC, Micha D, et al. PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med. 2013;369(16):1529–1536. | |
Mendoza-Londono R, Fahiminiya S, Majewski J; et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet. 2015;96(6):979–985. | |
Russell RG. Pharmacological diversity among drugs that inhibit bone resorption. Curr Opin Pharmacol. 2015;22:115–130 | |
Hoyer-Kuhn H, Netzer C, Semler O. Osteogenesis imperfecta: pathophysiology and treatment. Wien Med Wochenschr. 2015; 165(13–14):278–284. | |
Bachrach LK, Ward LM. Clinical review 1: bisphosphonate use in childhood osteoporosis. J Clin Endocrinol Metab. 2009;94(2):400–409. | |
Ward LM, Rauch F, Whyte MP, et al. Alendronate for the treatment of pediatric osteogenesis imperfecta: a randomized placebo-controlled study. J Clin Endocrinol Metab. 2011;96(2):355–364. | |
DiMeglio LA, Peacock M. Two-year clinical trial of oral alendronate versus intravenous pamidronate in children with osteogenesis imperfecta. J Bone Miner Res. 2006;21(1):132–140. | |
Bishop N, Adami S, Ahmed SF, et al. Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382(9902):1424–1432. | |
Palomo T, Fassier F, Ouellet J, et al. Intravenous bisphosphonate therapy of young children with osteogenesis imperfecta: skeletal findings during follow up throughout the growing years. J Bone Miner Res. Epub June 30, 2015. doi: 10.1002/jbmr.2567. | |
Bradbury LA, Barlow S, Geoghegan F, et al. Risedronate in adults with osteogenesis imperfecta type I: increased bone mineral density and decreased bone turnover, but high fracture rate persists. Osteoporos Int. 2012;23(1):285–294. | |
Shapiro JR, Thompson CB, Wu Y, Nunes M, Gillen C. Bone mineral density and fracture rate in response to intravenous and oral bisphosphonates in adult osteogenesis imperfecta. Calcif Tissue Int. 2010;87(2):120–129. | |
Vuorimies I, Toiviainen-Salo S, Hero M, Mäkitie O. Zoledronic acid treatment in children with osteogenesis imperfecta. Horm Res Paediatr. 2011;75(5):346–353. | |
Otaify GA, Aglan MS, Ibrahim MM, Elnashar M, El Banna RA, Temtamy SA. Zoledronic acid in children with osteogenesis imperfecta and Bruck syndrome: a 2-year prospective observational study. Osteoporos Int. Epub July 3, 2015. | |
Panigrahi I, Das RR, Sharda S, Marwaha RK, Khandelwal N. Response to zolendronic acid in children with type III osteogenesis imperfecta. J Bone Miner Metab. 2010;28(4):451–455. | |
Land C, Rauch F, Munns CF, Sahebjam S, Glorieux FH. Vertebral morphometry in children and adolescents with osteogenesis imperfecta: effect of intravenous pamidronate treatment. Bone. 2006;39(4):901–906. | |
Shapiro JR, Sponseller PD. Osteogenesis imperfecta: questions and answers. Curr Opin Pediatr. 2009;21(6):709–716. | |
Lindahl K, Langdahl B, Ljunggren Ö, Kindmark A. Treatment of osteogenesis imperfecta in adults. Eur J Endocrinol. 2014;171(2):R79–R90. | |
Brizola E, Shapiro JR. Bisphosphonate treatment of children and adults with osteogenesis imperfecta: unanswered questions. Calcif Tissue Int. 2015;97(2):101–103. | |
Phillipi CA, Remmington T, Steiner RD. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2008;(4):CD005088. | |
Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2014;7:CD005088. | |
Rijks EB, Bongers BC, Vlemmix MJ, et al. Efficacy and safety of bisphosphonate therapy in children with osteogenesis imperfecta: a systematic review. Horm Res Paediatr. 2015;84(1):26–42. | |
Hald JD, Evangelou E, Langdahl BL, Ralston SH. Bisphosphonates for the prevention of fractures in osteogenesis imperfecta: meta-analysis of placebo-controlled trials. J Bone Miner Res. 2015;30(5):929–933. | |
Shi CG, Zhang Y, Yuan W. Efficacy of bisphosphonates on bone mineral density and fracture rate in patients with osteogenesis imperfecta: a systematic review and meta-analysis. Am J Ther. Epub 2015 April 3, 2015. | |
Papapoulos SE, Cremers SC. Prolonged bisphosphonate release after treatment in children. N Engl J Med. 2007;356:1075–1076. | |
Kamoun-Goldrat AS, Le Merrer MF. Osteogenesis imperfecta and dentinogenesis imperfecta: diagnostic frontiers and importance in dentofacial orthopedics. Orthod Fr. 2007;78(2):89–99. | |
Schilcher J, Michaelsson K, Aspenberg P. Bisphosphonate use and atypical fractures of the femoral shaft. N Engl J Med. 2011;364:1728–1737. | |
Carpintero P, Del Fresno JA, Ruiz-Sanz J, Jaenal P. Atypical fracture in a child with osteogenesis imperfecta. Joint Bone Spine. 2015;82(4):287–288. | |
Meier RP, IngLorenzini K, Uebelhart B, Stern R, Peter RE, Rizzoli R. Atypical femoral fracture following bisphosphonate treatment in a woman with osteogenesis imperfecta – a case report. Acta Orthop. 2012; 83(5):548–550. | |
Li J, Mashiba T, Burr DB. Bisphosphonate treatment suppresses not only stochastic remodeling but also the targeted repair of microdamage. Calcif Tissue Int. 2001;69:281–286. | |
Hegazy A, Kenawey M, Sochett E, Tile L, Cheung AM, Howard AW. Unusual femur stress fractures in children with osteogenesis imperfecta and intramedullary rods on long-term intravenous pamidronate therapy. J Pediatr Orthop. Epub Jun 8, 2015. | |
Munns CF, Rauch F, Zeitlin L, Fassier F, Glorieux FH. Delayed osteotomy but not fracture healing in pediatric osteogenesis imperfecta patients receiving pamidronate. J Bone Miner Res. 2004;19(11):1779–1786. | |
Anam EA, Rauch F, Glorieux FH, Fassier F, Hamdy R. Osteotomy healing in children with osteogenesis imperfecta receiving bisphosphonate treatment. J Bone Miner Res. 2015;30(8):1362–1368. | |
Hennedige AA, Jayasinghe J, Khajeh J, Macfarlane TV. Systematic review on the incidence of bisphosphonate related osteonecrosis of the jaw in children diagnosed with osteogenesis imperfecta. J Oral Maxillofac Res. 2013;4(4):e1. | |
Plotkin H, Rauch F, Bishop NJ, et al. Pamidronate treatment of severe osteogenesis imperfecta in children under 3 years of age. J Clin Endocrinol Metab. 2000;85(5):1846–1850. | |
Falk MJ, Heeger S, Lynch KA, et al. Intravenous bisphosphonate therapy in children with osteogenesis imperfecta. Pediatrics. 2003;111(3):573–578. | |
Uihlein AV, Leder BZ. Anabolic therapies for osteoporosis. Endocrinol Metab Clin North Am. 2012;41(3):507–525. | |
Orwoll ES, Shapiro J, Veith S, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124(2):491–498. | |
Gatti D, Rossini M, Viapiana O, et al. Teriparatide treatment in adult patients with osteogenesis imperfecta type I. Calcif Tissue Int. 2013;93(5):448–452. | |
Holm J, Eiken P, Hyldstrup L, Jensen JE. Atypical femoral fracture in an osteogenesis imperfecta patient successfully treated with teriparatide. Endocr Pract. 2014;20(10):e187–e190. | |
George S, Brenner A, Sarantopoulos J, Bukowski RM. RANK ligand: effects of inhibition. Curr Oncol Rep. 2010;12(2):80–86. | |
Semler O, Netzer C, Hoyer-Kuhn H, Becker J, Eysel P, Schoenau E. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. 2012;12(3):183–188. | |
Land C, Rauch F, Travers R, Glorieux FH. Osteogenesis imperfecta type VI in childhood and adolescence: effects of cyclical intravenous pamidronate treatment. Bone. 2007;40:638–644. | |
Hoyer-Kuhn H, Netzer C, Koerber F, Schoenau E, Semler O. Two years’ experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J Rare Dis. 2014;9:145. | |
Hoyer-Kuhn H, Semler O, Schoenau E. Effect of denosumab on the growing skeleton in osteogenesis imperfecta. J Clin Endocrinol Metab. 2014;99(11):3954–3955. | |
Li X, Warmington KS, Niu QT, et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res. 2010;25:2647–2656. | |
Ominsky MS, Li C, Li X, et al. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res. 2011;26:1012–1021. | |
Ominsky MS, Niu QT, Li C, Li X, Ke HZ. Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. J Bone Miner Res. 2014;29(6):1424–1430. | |
Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. | |
Palomo T, Glorieux FH, Rauch F. Circulating sclerostin in children and young adults with heritable bone disorders. J Clin Endocrinol Metab. 2014;99(5):E920–E925. | |
Sinder BP, Salemi JD, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Rapidly growing Brtl/+ mouse model of osteogenesis imperfecta improves bone mass and strength with sclerostin antibody treatment. Bone. 2015;71:115–123. | |
Roschger A, Roschger P, Keplingter P, et al. Effect of sclerostin antibody treatment in a mouse model of severe osteogenesis imperfecta. Bone. 2014;66:182–188. | |
Stroup GB, Lark MW, Veber DF, et al. Potent and selective inhibition of human cathepsin K leads to inhibition of bone resorption in vivo in a nonhuman primate. J Bone Miner Res. 2001;16:1739–1746. | |
Bone HG, Dempster DW, Eisman JA, et al. Odanacatib for the treatment of postmenopausal osteoporosis: development history and design and participant characteristics of LOFT, the long-term odanacatib fracture trial. Osteoporos Int. 2015;26(2):699–712. | |
Grafe I, Yang T, Alexander S, et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20(6):670–675. | |
Erlebacher A, Derynck R. Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol. 1996;132(1–2):195–210. | |
Gebken J, Brenner R, Feydt A, et al. Increased cell surface expression of receptors for transforming growth factor-beta on osteoblasts from patients with osteogenesis imperfecta. Pathobiology. 2000;68(3):106–112. | |
Wheeler JB, Ikonomidis JS, Jones JA. Connective tissue disorders and cardiovascular complications: the indomitable role of transforming growth factor-beta signaling. Adv Exp Med Biol. 2014;802:107–127. | |
Krakow D. Skeletal dysplasias. Clin Perinatol. 2015;42(2):301–319. | |
Pereira RF, O’Hara MD, Laptev AV, et al. Marrow stromal cells as a source of progenitor cells for nonhematopoietic tissues in transgenic mice with a phenotype of osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1998;95(3):1142–1147. | |
Götherström C, Westgren M, Shaw SW, et al. Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: a two-center experience. Stem Cells Transl Med. 2014;3(2):255–264. | |
Westgren M, Götherström C. Stem cell transplantation before birth – a realistic option for treatment of osteogenesis imperfecta? Prenat Diagn. 2015;35(9):827–832. | |
Lin D, Zhai W, Lian K, Ding Z. Results of a bone splint technique for the treatment of lower limb deformities in children with type I osteogenesis imperfecta. Indian J Orthop. 2013;47(4):377–381. | |
Lee K, Park MS, Yoo WJ, Chung CY, Choi IH, Cho TJ. Proximal migration of femoral telescopic rod in children with osteogenesis imperfecta. J Pediatr Orthop. 2015;35(2):178–184. | |
Nicolaou N, Bowe JD, Wilkinson JM, Fernandes JA, Bell MJ. Use of the Sheffield telescopic intramedullary rod system for the management of osteogenesis imperfecta: clinical outcomes at an average follow-up of nineteen years. J Bone Joint Surg Am. 2011;93:1994–2000. | |
Agarwal V, Joseph B. Non-union in osteogenesis imperfecta. J Pediatr Orthop B. 2005;14(6):451–455. | |
Puvanesarajah V, Shapiro JR, Sponseller PD. Sandwich allografts for long-bone nonunions in patients with osteogenesis imperfecta: a retrospective study. J Bone Joint Surg Am. 2015;97(4):318–325. | |
Engelbert RHH, Pruijs HEH, Beemer FA, Helders PJM. Osteogenesis imperfecta in childhood: treatment strategies. Arch Phys Med Rehabil. 1998;79(12):1590–1594. | |
Brizola E, Staub AL, Félix TM. Muscle strength, joint range of motion, and gait in children and adolescents with osteogenesis imperfecta. Pediatr Phys Ther. 2014;26(2):245–252. | |
Montpetit K, Dahan-Oliel N, Ruck-Gibis J, Fassier F, Rauch F, Glorieux F. Activities and participation in young adults with osteogenesis imperfecta. J Pediatr Rehabil Med. 2011;4(1):13–22. | |
Hoyer-Kuhn H, Semler O, Stark C, Struebing N, Goebel O, Schoenau E. A specialized rehabilitation approach improves mobility in children with osteogenesis imperfecta. J Musculoskelet Neuronal Interact. 2014;14(4):445–453. | |
Niyibizi C, Li F. Potential implications of cell therapy for osteogenesis imperfecta. Int J Clin Rheumtol. 2009;4(1):57–66. | |
Sakkers R, Kok D, Engelbert R, et al. Skeletal effects and functional outcome with olpadronate in children with osteogenesis imperfecta: a 2-year randomized placebo-controlled study. Lancet. 2004;363:1427–1431. | |
Antoniazzi F, Zamboni G, Lauriola S, Donadi L, Adami S, Tatò L. Early bisphosphonate treatment in infants with severe osteogenesis imperfecta. J Pediatr. 2006;149(2):174–179. | |
Rauch F, Munns CF, Land C, Cheung M, Glorieux FH. Risedronate in the treatment of mild pediatric osteogenesis imperfecta: a randomized placebo-controlled study. J Bone Miner Res. 2009;24(7):1282–1289. | |
Cho TJ, Choi IH, Chung CY, Yoo WJ, Park MS, Park YK. Efficacy of oral alendronate in children with osteogenesis imperfecta. J Pediatr Orthop. 2005;25(5):607–612. | |
Glorieux FH, Bishop N, Plotkin H, Chabot G, Lanoue G, Travers RT. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339:947–952. | |
Letocha AD, Cintas HL, Troendle JF, et al. Controlled trial of pamidronate in children with types II and IV osteogenesis imperfecta confirms vertebral gains but not short-term functional improvement. J Bone Miner Res. 2005;20:977–986. | |
Kusumi K, Ayoob R, Bowden SA, Ingraham S, Mahan JD. Beneficial effects of intravenous pamidronate treatment in children with osteogenesis imperfecta under 24 months of age. J Bone Miner Metab. 2015;33(5):560–568. | |
Barros ER, Saraiva GL, de Oliveira TP, Lazaretti-Castro M. Safety and efficacy of a 1-year treatment with zoledronic acid compared with pamidronate in children with osteogenesis imperfecta. J Pediatr Endocrinol Metab. 2012;25(5–6):485–491. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.