Back to Journals » International Journal of Nanomedicine » Volume 15
Past, Present, and Future of Anticancer Nanomedicine
Received 20 March 2020
Accepted for publication 19 May 2020
Published 6 August 2020 Volume 2020:15 Pages 5719—5743
DOI https://doi.org/10.2147/IJN.S254774
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Kyungeun Kim,1 Dongwoo Khang1– 4
1College of Medicine, Gachon University, Incheon 21999, South Korea; 2Lee Gil Ya Cancer and Diabetes Institute, Gachon University, Incheon 21999, South Korea; 3Gachon Advanced Institute for Health Science & Technology (GAIHST), Gachon University, Incheon 21999, South Korea; 4Department of Physiology, School of Medicine, Gachon University, Incheon 21999, South Korea
Correspondence: Dongwoo Khang
Department of Physiology, School of Medicine, Lee Gil Ya Cancer and Diabetes Institute, Gachon Advanced Institute for Health Science & Technology (GAIHST), Gachon University, Incheon 21999, South Korea
Tel/Fax +82 32 899 1525
Email [email protected]
Abstract: This review aims to summarize the methods that have been used till today, highlight methods that are currently being developed, and predict the future roadmap for anticancer therapy. In the beginning of this review, established approaches for anticancer therapy, such as conventional chemotherapy, hormonal therapy, monoclonal antibodies, and tyrosine kinase inhibitors are summarized. To counteract the side effects of conventional chemotherapy and to increase limited anticancer efficacy, nanodrug- and stem cell-based therapies have been introduced. However, current level of understanding and strategies of nanodrug and stem cell-based therapies have limitations that make them inadequate for clinical application. Subsequently, this manuscript reviews methods with fewer side effects compared to those of the methods mentioned above which are currently being investigated and are already being applied in the clinic. The newer strategies that are already being clinically applied include cancer immunotherapy, especially T cell-mediated therapy and immune checkpoint inhibitors, and strategies that are gaining attention include the manipulation of the tumor microenvironment or the activation of dendritic cells. Tumor-associated macrophage repolarization is another potential strategy for cancer immunotherapy, a method which activates macrophages to immunologically attack malignant cells. At the end of this review, we discuss combination therapies, which are the future of cancer treatment. Nanoparticle-based anticancer immunotherapies seem to be effective, in that they effectively use nanodrugs to elicit a greater immune response. The combination of these therapies with others, such as photothermal or tumor vaccine therapy, can result in a greater anticancer effect. Thus, the future of anticancer therapy aims to increase the effectiveness of therapy using various therapies in a synergistic combination rather than individually.
Keywords: stem cell-based therapy, T cell-mediated therapy, macrophage repolarization, nanodrug-based immunotherapy
Introduction
As research progresses, great success in treating many diseases has achieved what was previously thought to be incurable. The invention of vaccines and the consequent eradication of smallpox, the discovery of insulin and its use for treating diabetic patients, and the use of anesthesia in surgery are some of the major medical developments made to this day.1–3 Unfortunately, the cure for cancer, one of the leading worldwide causes of death, is still a challenge as of today.4
There are three standard strategies for cancer treatment: surgery, radiotherapy, and chemotherapy. Surgery and radiotherapy are very effective for removing tumors, but they are limited to low-stage tumors and are not sufficiently applicable to high-stage tumors.5 While most early-stage tumors can be potentially cured by complete excision, stage III and IV tumors require chemotherapy treatment. However, even with chemotherapy, there is no guarantee of a cure.6 Therefore, the ultimate treatment for malignant tumors requires the novel design of new drugs and combination therapies to maximize anticancer efficacy.
The design of novel target-based anticancer drugs allows to selectively target and completely eradicate cancerous cells. However, most of the strategies that destroy the tumor cell also adverse effects the normal cells during chemotherapy.7 Therefore, the next generation of cancer treatment should not only should target malignant tumor cells, but also inactivate the cancer functions that harm non-tumor cells.8
The rapid growth of target-based anticancer drugs has provided the possibility to selectively destroy tumor cells located deep within tissues,8 even though there are barriers to overcome.
Cancer therapies, including chemotherapy, hormonal therapy, monoclonal antibody therapies, and tyrosine kinase inhibitor therapies are already clinically applied. However, they have critical limitations, such as relative ineffectiveness in certain types of cancers or serious side effects that disqualify them as safe drugs.9–12 Chemotherapy, which has been long-used in cancer treatment, leads to side effects by adversely affecting the function of normal cells in clinical settings.7 Hormonal therapy is effective but can only be applied to a small range of cancers. Monoclonal antibodies and tyrosine kinase inhibitors have been deemed as a great advancement in anticancer therapy, but their effectiveness is limited to a few cancers, which have certain hyperactive receptors.13,14
Newer methods, like selective accumulation of nanodrugs by adjusting their physiochemical properties or the use of human stem cells, have been employed. However, the use of these nanodrugs resulted in toxicity and was not sufficiently effective considering aroused side effects by use of nanoparticles.15,16 Recently, great efforts have been made to develop a novel immunotherapeutic strategy that effectively directs immune cells to react against invasive cancer cells.17–19 The immune system, which includes immune checkpoints, the tumor microenvironment, dendritic cells (DC), or macrophages, can be appropriately manipulated to create the desired anticancer effect.20,21 Furthermore, when nanoparticles are simultaneously used in combination with other technologies, maximum anticancer efficacy can be achieved.22 Specifically, the use of nanoparticles in combination with tumor vaccines and natural killer cells, or photothermal therapy for the ablation of tumor cells is considered a very effective strategy.23 In conclusion, the focus of this review is to summarize previous approaches and current research regarding anticancer therapy, as well as address the future perspective of anticancer therapy (Figure 1).
 |
Figure 1 Past, current, and future anticancer therapies. (A) Previous approaches for anticancer therapy. Even though many of these methods are still effective, they have limitations and are thus not the most effective for attacking cancerous cells. (B) Current research in anticancer therapy. Cancer immunotherapies, including T cell-mediated therapy and macrophage repolarization, are the most promising. (C) Future perspective of anticancer therapy. The future of anticancer therapy lies in nanotechnology based anticancer immunotherapy and other combination therapies. |
Previous Anticancer Therapies
Conventional Chemotherapy
Although the use of conventional anticancer chemotherapeutic drugs causes severe side effects, the use of these drugs is still widespread.24 This is probably because these drugs are effective against all types of cancer due to their non-selective drug biodistribution, unlike other more efficient drugs that usually require specific biomarkers or activated receptors and can thus only be used to treat certain cancers, such as monoclonal antibodies or tyrosine kinase inhibitors.25,26 Thus, for the majority of cancers that do not have these biological traits, conventional chemotherapy is the first treatment of choice.6
Conventional chemotherapy inhibits DNA synthesis and mitosis, thereby preventing the proliferation of cancer cells.27 The most common chemotherapeutic drugs are alkylating agents, antimetabolites, and topoisomerase inhibitors, which stop cancer cell proliferation by different mechanisms (Figure 2).28–30
 |
Figure 2 Conventional chemotherapy. (A) Alkylating agents. A = adenosine, T = tyrosine, G = guanine, C = cytosine, X = alkylating agent. Alkylating agents react with the N7 of guanine, resulting in abnormal base pairing, which leads to miscoding and strand breakage. (B) Antimetabolites. Y = Antimetabolite. Antimetabolites are structurally similar to endogenous compounds and destroy cancer cells by posing as purines or pyrimidines, which are building blocks of DNA. In B, the antimetabolite is masquerading as a purine. (C) Topoisomerase inhibitors. Z = Topoisomerase inhibitor. Topoisomerases normally decrease the torsion of the DNA. When this process is stopped by topoisomerase inhibitors, the torsion of the DNA strand increases and causes DNA breakage. |
Alkylating agents such as nitrogen mustards (eg cyclophosphamide) or nitrosoureas alkylate guanines at N7 (eg carmustine), leading to abnormal base pairing, miscoding, and strand breakage, and thereby stopping cell proliferation and destroying cancer cells.28,31 Antimetabolites, a different type of anticancer drug including folic acid analogs (eg methotrexate), purine analogs (eg azathioprine), and pyrimidine analogs (eg 5-fluorouracil),29 are structurally similar to endogenous genetic products. Thus, they can genetically alter the cancer gene and can inhibit nucleotide synthesis.32 On the contrary, topoisomerase inhibitors, such as camptothecins (eg irinotecan), anthracyclines (eg doxorubicin), and podophyllotoxins (eg etoposide) block the activation of the topoisomerase by relaxing overwound DNA.30 Due to the double helix structure of DNA, during DNA replication and transcription, winding of the DNA occurs. The topoisomerase enzyme appropriately breaks and mends DNA strands to relax the winding and decrease the torsion.33 Topoisomerase inhibitors inhibit the topoisomerase function, resulting in the overwinding of DNA and increase in its torsion, and thus causing DNA injury. Since alkylating agents, antimetabolites, and topoisomerase inhibitors influence the cell cycle, they can significantly affect fast-growing tumors.34
Unfortunately, these drugs are non-selective and thus also detrimental to normal cells. Fast-growing normal cells, such as bone marrow cells, hair follicles, or intestinal epithelial cells, are particularly vulnerable to these drugs.35–37 Furthermore, these drugs are ineffective against some cancer cells,38 for instance slow-growing tumor cells. Some cancer cells have become resistance to chemotherapeutic drugs by increasing DNA repair, inactivating cancer drugs, or promptly pumping the drugs out to prevent their accumulation in the cytosol.9 Due to these drawbacks, researchers have focused on other strategies for curing cancer.
Hormonal Anticancer Therapy
The development of tamoxifen for curing breast cancer has completely changed the landscape of cancer therapy, opening the era of “targeted cancer therapy”.39 Tamoxifen, a selective estrogen receptor modulator, specifically targets tumors that have estrogen receptors. Hormonal therapy utilizes the endocrine system to attack hormone responsive tumor tissues;40 however, it has certain drawbacks.10 Since estrogen receptors are naturally present not only in the breast tissue but in tissues all over the human body, the activation of estrogen receptors in endometrium, bone, and adipose tissues, for example, elicits side effects.41 As the endometrium of the female uterus has a large number of estrogen receptors and tamoxifen is also a partial agonist of endometrial tissue, development of endometrial cancer is possible. The activation of endometrial estrogen receptors causes endometrial cell proliferation and cancerization.42 Although tamoxifen, as a hormonal anticancer therapy, possess many side effects and limitations, it is meaningful that it has started research into the field of targeted therapy. Tamoxifen serves as a cornerstone for following research regarding other cancer receptors and biological markers, which can be targeted with drugs, and thus destroying the cancer cell.39
Monoclonal Antibody Therapy
The limitation of conventional chemotherapy has led researchers to look for alternative methods, and the efficacy of tamoxifen has encouraged the development of better targeted chemotherapies. The use of monoclonal antibodies (mAbs), which are receptor-specific antibodies that detect overexpressed receptors in the tumor cell and selectively attack malignant cells, constitutes one of the earliest, most widely-used, powerful, and effective approaches for targeted tumor therapy.43,44 Monoclonal antibodies are more effective than previous chemotherapies because they selectively bind to overexpressed cancer membrane receptors, and are thus less harmful to normal cells. They also have advantages when compared to hormonal therapy because mAbs target more specific molecular receptors and ligands.45 Tumor signaling takes place when receptors and ligands form a complex that initiates an intracellular response, leading to events, such as cell proliferation and genetic mutation. Therefore, by targeting the overexpressed receptors that are involved in cancer cell signaling, treating a broad range of tumors is possible.11
Blockage of the epidermal growth factor receptor (EGFR) family members is a major function of mAb-based drugs.46,47 EGFRs are often overexpressed in many epithelial malignant tumors, and higher levels of their overexpression correlated with more advanced tumor stages, poorer prognosis, and greater resistance to anticancer therapy.48 HER1 and HER2, which are the major subtypes of EGFR, are frequently utilized in mAb anticancer therapy.49–51 HER1 (Cetuximab; Erbitux) and HER2 (Trastuzumab; Herceptin) inhibitors, are used for treating (HER1-rich) metastatic colorectal52–56 and (HER2-rich) breast cancers,57,58 respectively. However, EGFR mAb application is only limited to EGFR-overexpressed tumors.12 Even in colorectal and breast cancers, the EGFR receptor is overexpressed in only 25–82% of colorectal cancers and 18% of breast cancers,59–63 meaning not all tumors have high EGFR expression.
Tumor progression and growth require metastasis and angiogenesis, and the vascular endothelial growth factor receptor (VEGFR) is a major receptor involved in these processes.64–66 By inhibiting VEGFR with a mAb such as Bevacizumab (Avastin), the tumor is deprived of oxygen and nutrition, thus tumor cell growth is stopped, effectively treating colorectal and lung cancers.55,67-70 Bevacizumab was also approved for use in combination with antimetabolite 5-fluorouracil or carboplatin together with paclitaxel against colorectal and lung cancers (Table 1).68,71 However, like EGFR mAbs, VEGFR mAbs can only be applied to tumors with VEGFR overexpression, which corresponds to 48%, 66.7%, and 72.2% in colorectal cancer and in lung adenocarcinoma and squamous cell carcinoma,72,73 respectively. Therefore, many types of colorectal and lung cancers are untreatable using this therapy.
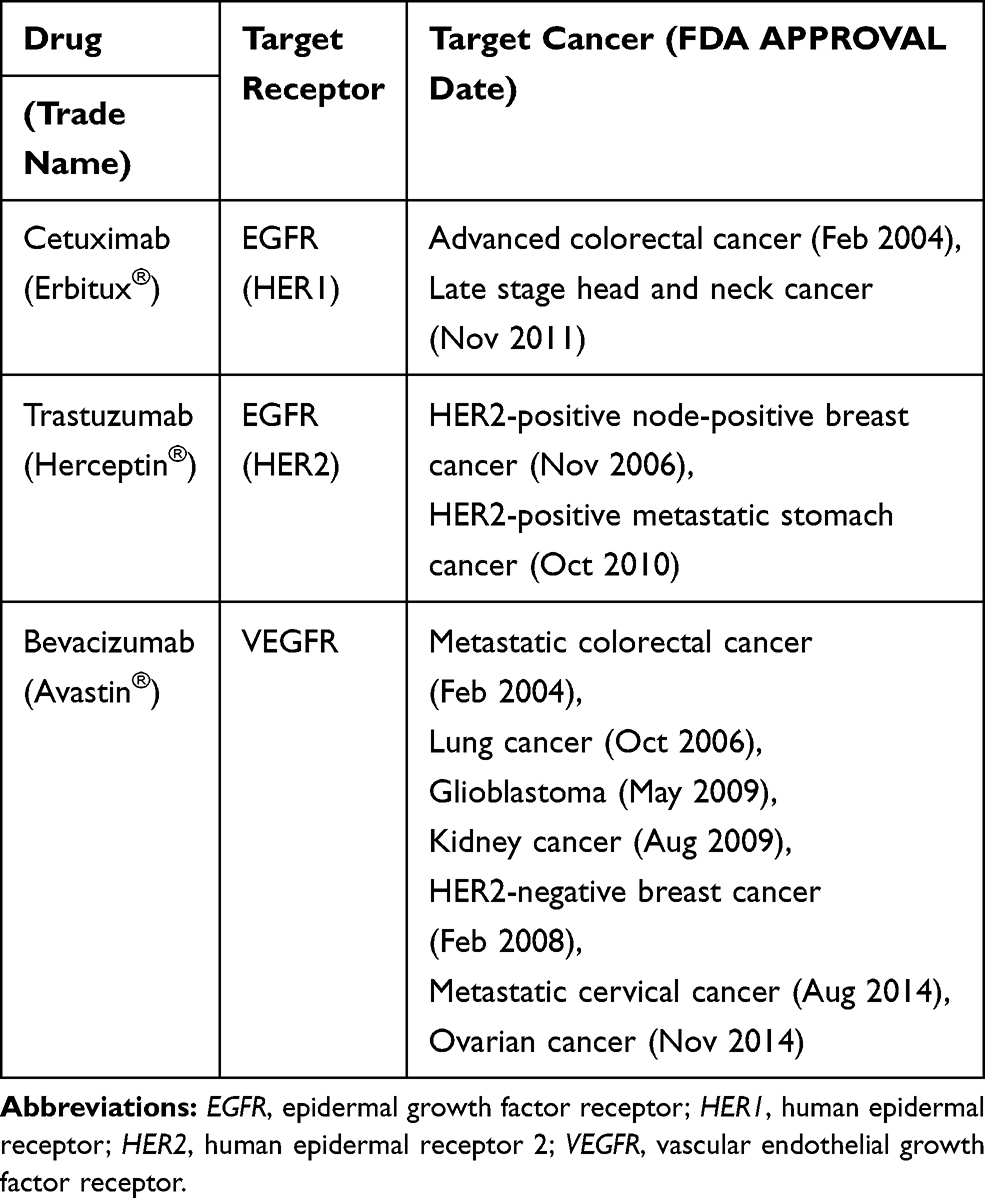 |
Table 1 Major Monoclonal Antibodies in Cancer Treatment |
Tyrosine Kinase Inhibitor Therapy
Tyrosine kinase inhibitors (TKIs) function similarly to mAbs; however, they have a different mechanism of action (Figure 3).74 TKIs target overexpressed receptors, similarly to mAbs. However, unlike mAbs which only block a specific cancer receptor, TKIs also stop the intracellular signaling system that is usually initiated when the receptor and ligand interact.14 Tyrosine kinase is the enzyme responsible for the activation of many proteins in tumor signaling. By blocking this enzyme, TKIs prevent the activation of the signaling systems that lead to tumor formation and proliferation.75,76 Imatinib (Gleevec) is a widely known TKI used for treating acute lymphoblastic leukemia (ALL) and chronic myeloblastic leukemia (CML) by inhibiting the transcription of the BCR/ABL gene and treating gastrointestinal stromal tumor by inhibiting the c-kit gene expression.77–82 Erlotinib and Gefitinib block the signals initiated by the HER1 tumor receptor; however, Lapatinib and Neratinib block the HER2 tumor receptor (Table 2).83–85
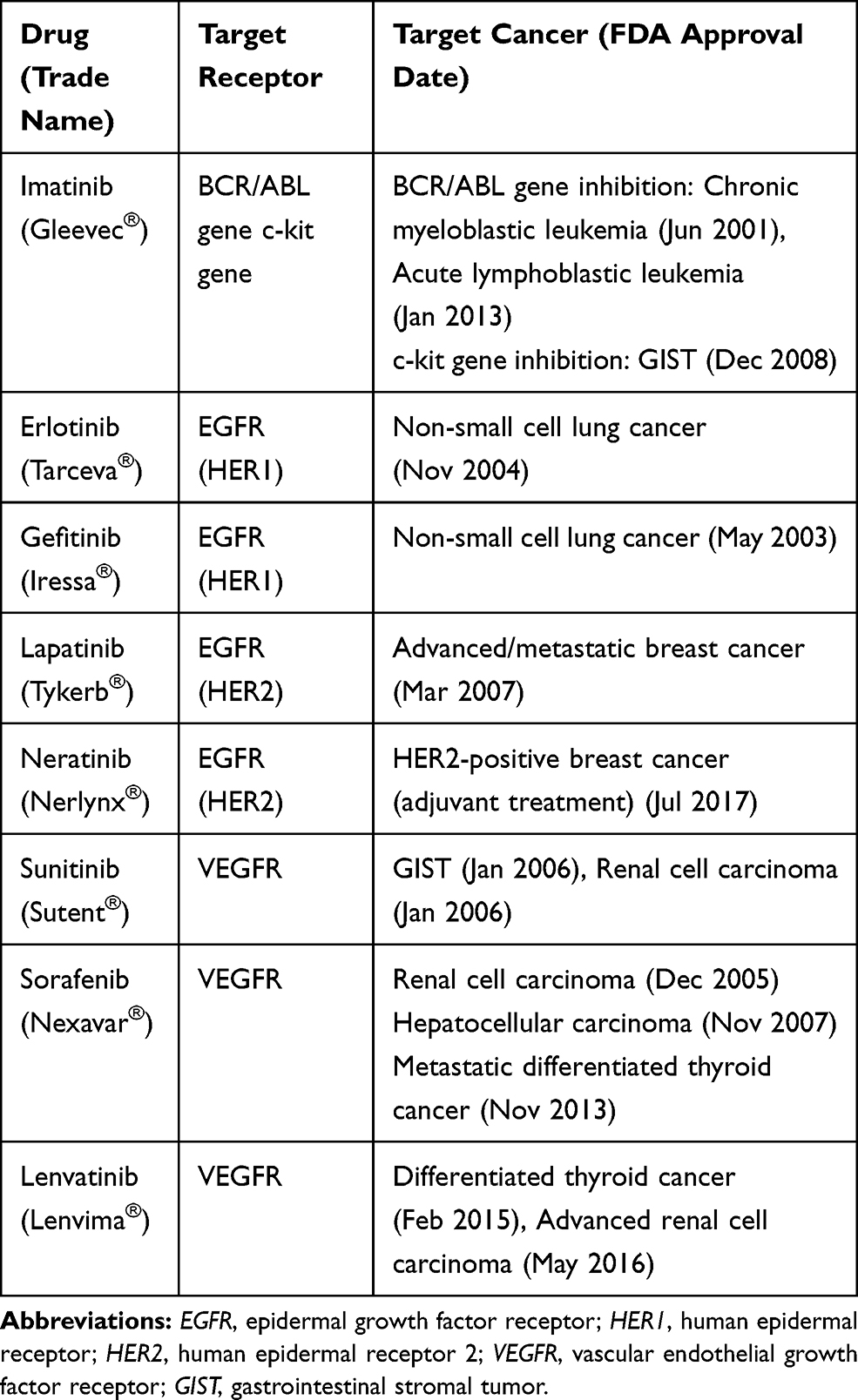 |
Table 2 Major Tyrosine Kinase Inhibitors in Cancer Treatment |
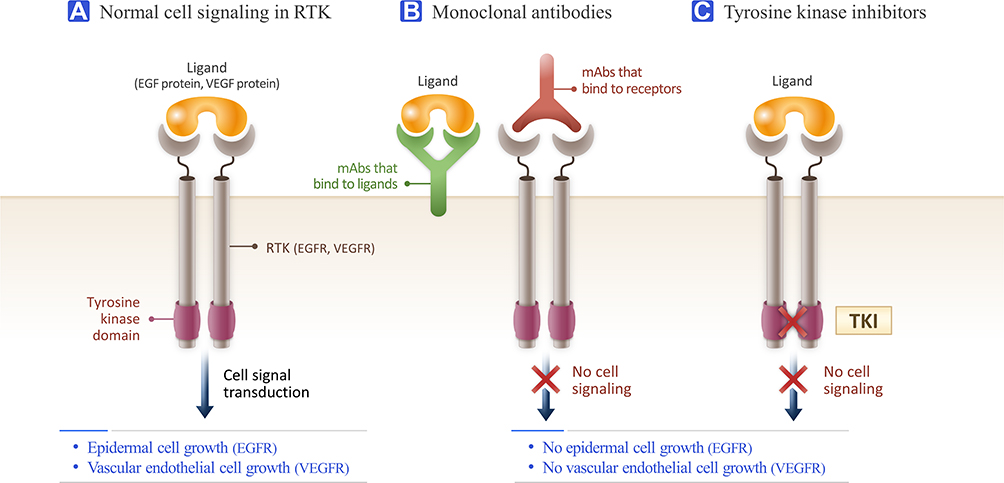 |
Figure 3 Comparison of monoclonal antibodies (mAbs) and tyrosine kinase inhibitor (TKI) activity in receptor tyrosine kinase (RTK). (A) Normal cell signaling of RTK. When ligands bind to RTKs, neighboring RTKs cross-link to each other to form dimers, activating the tyrosine kinase domain in each RTK to phosphorylate the tyrosine kinase domain in the paired RTK. This phosphorylation initiates intracellular cell signaling and thus cellular activities, including proliferation and growth. (B) Monoclonal antibodies. MAbs stop the ligand from binding to RTKs, thus causing no dimerization, no phosphorylation, and no cell signaling. MAbs can bind to ligands or receptors, depending on the type. (C) TKI. TKIs inhibit RTK phosphorylation. The signaling transduction cascade is not started and thus cell signaling and resulting cellular activities do not occur. |
Though monoclonal antibodies and TKIs are both very effective anticancer drugs, they still have some clinical limitations. First of all, they are only effective in treating the specific cancers that express the target proteins. For example, Trastuzumab is effective only against breast cancer and Gleevec is only applicable to ALL and CML.58,79,82 Unfortunately, these drugs are poorly applicable to other tumors, such as lung or pancreatic cancer.86,87 Additionally, these types of drugs can destroy normal cells that have the same type of receptors. Although their cytotoxicity is much more limited when compared with that of conventional anticancer chemotherapy, they still elicit severe side effects. MAbs frequently cause immune-related dermatological problems, and TKIs can induce hematologic problems, such as anemia, macrocytosis, or neutropenia.88–91 Therefore, future cancer therapies should focus on treating a wider range of tumors and overcoming the side effects associated with mAbs and TKIs.
Established Approaches in Nanodrug Therapy
A new type of anticancer drug, nanodrugs, was invented not only to effectively reach the tumor site but also to be easily captured by the tumor cells.92–94 Researchers believed that by carefully designing the unique physiochemical properties of nanodrugs, their anticancer efficacy could be enhanced.15 Additionally, nanodrugs have the ability to evade cancer efflux pumps and optimize intracellular endosomal drug delivery, allowing nanodrugs to selectively destroy the cancer cell.95–97 However, the greatest drawbacks of nanodrug delivery for the clinical application of nanodrugs are the toxicity and clearance issues. Although various efforts have been made to maximize tumor targeting efficiency and minimize the clearance problems, the predicted toxicity of nanodrugs has prevented them from becoming the ultimate cure for cancer.98,99
In previous research on mAb drugs, tumors were targeted via membrane-receptors to increase their targeting efficiency. However, nanotechnology research has shown that changes in nanoparticle (NP) shape, size, and material can influence their biodistribution and improve their cellular uptake efficiency.15,100-104 For example, gold and liposome NPs are preferentially localized in the liver and blood, respectively.105–107 Additionally, nanospheres, nanorods, and single walled carbon nanotubes (swCNTs) show different biodistributions.98 However, biodistribution control by changing the nanoshape is currently not a practical approach for selectively targeting specific tumor tissues.108,109
The major limitation of conventional nanodrugs is their accumulation in the reticuloendothelial system (RES), a group of important immune organs, rather than in the specific tumors they are meant to target.110 However, RES accumulation can be minimized by increasing the circulation time of nanodrugs in the blood, which increases the interaction time between nanodrugs and immune cells. Therefore, to maximize the targeted nanodrug delivery, it is essential to determine the optimal blood circulation time.15 One strategy to adjust the optimal blood circulation time of nanodrugs is to control their polyethylene glycol (PEG)/polylactic acid (PLA) ratio by changing the molecular weight ratios of PEG and PLA.111,112
In some cases, drugs are required to be not only delivered to the targeted tissue, but also into the exact organelle in the tumor cell111,113,114 by using the intracellular uptake pathways.111 A conventional target organelle is the mitochondria, since mitochondria attack can deprive the tumor of its energy. To reach the mitochondria, nanodrugs are released from the early endolysosomes, and PEGylation is often used to prompt the releasing process.111,115,116
As mentioned above, nanodrugs have shown great results for tumor targeting, but have shown clearance issues and immune toxicity that still hamper the use of nanodrugs to destroy cancer cells.112,117 Therefore, to apply these nanodrugs in the clinic, developments must be made to avoid the potential side effects. However, instead of reforming nanodrugs to minimize side effects, other methods of utilizing nanodrugs have been introduced, which is a topic that will be discussed later on.108
Stem Cell-Based Cell Therapy
Among the currently used anticancer therapies, stem cell therapy is one of the promising strategies. Stem cell therapy is mainly based on the innate ability of mesenchymal stem cells to home to cancer cells.118–120
By selecting the appropriate types of mesenchymal stem cells (MSCs) for MSC therapy and loading them with cytotoxic drugs, drugs can be delivered into the targeted cancer cells to efficiently destroy them.121–124 There are many lineages of MSCs, such as bone marrow, adipose, and umbilical cord blood MSCs. Each type of MSC has tumor-associated chemokine receptors which enable them to selectively target different cancer cell lines.121,125-127 Therefore, a certain type of tumor can be targeted by selecting an appropriate MSC.128,129
The various MSC lineages differ in biodistribution and chemokine receptors.123,130 Specific chemokine receptors showed selective responses toward different cluster of differentiation (CD) antigens in the physiological tumor microenvironment (TME).131 Knowing the relationships between different MSC lineages and their homing abilities to different tumor types is important for developing MSC-mediated therapies.132
Among the malignant tumors, brain, pancreatic, and small-cell lung cancers (SCLCs), are neoplasms that are still difficult to treat.133–138 Stem cells are more promising for destroying malignant tumors than conventional targeted chemotherapy because they target tumor cells more effectively.123 For example, previous research indicated that CXCL12 and IL-8 chemokine receptors of bone marrow MSCs (BM-MSCs) reacted more strongly with chemokines released by lung cancer cells, compared to those produced by other cancer cells, such as breast or brain cancer cells, resulting in increased targeting ability for lung cancer cells.130,139,140
However, stem cell therapy still has some drawbacks. The use of genetically engineered MSCs with increased anticancer efficacy mediated by the secretion of therapeutic proteins or expression of suicide-inducing enzymes, resulted in some unexpected long-term side effects, such as a shorter lifespan and lower drug delivery efficacy, and resistance to therapy by some large cancer cell populations.141 On the other hand, there are other two methods that deliver drugs in better and safer ways, namely drug conjugation to the MSC surface membranes and facilitated intracellular drug loading into MSCs. MSCs with surface conjugated drugs had better drug loading ability,122,130,142,143 drug stability, and homing ability, and were able to further induce cancer cell self-apoptosis than MSCs with intracellularly uploaded drugs.16,130 Therefore, surface-conjugated drugs had increased anticancer efficacy, and a reduced dose of injected MSCs was required to provide the same anticancer effect as that of facilitated intracellular drug loading into MSCs.
However, when drugs are not properly conjugated in the loading process, the same drugs can diminish the homing ability of MSCs to cancer cells, increase self-apoptosis, and cause unexpected tumorigenesis due to uncontrollable differentiation.144 Additionally, the use of stem cells for cancer therapy is still controversial.145 Unlike human embryonic stem cells, MSCs do not cause ethical problems, since they are obtained from tissues such as adult bone marrow, adipose tissue, or the umbilical cord. However, safety issues related to the use of MSCs are still a concern, especially regarding their long-term fate. Unwanted differentiation of MSCs may occur, potentially leading to tumor growth and metastasis, instead of destroying tumor cells.120,146,147 Therefore, studies that continuously monitor MSCs, even after long-term therapy, are essential to guarantee their effectivity and safety.
To overcome such limitations, the combination of nanomedicine and MSC therapy has shown a huge potential to effectively target tumors.16,144,148 By conjugating nanodrugs onto the MSC membrane, a higher efficiency of drug delivery can be attained, since nanodrugs have better conjugation and tumor-targeting abilities.143,149 Another promising method is the use of nanovesicles (NVs) in MSC therapy. Instead of conjugating nanodrugs onto the surface of MSCs, it is possible to create NVs using MSC membranes, which can induce a more effective anticancer response.
Current Immuno-Anticancer Therapy
Cancer immunotherapy is a promising and novel anticancer therapy that focuses on the immunological aspects of cancer. Through immuno-anticancer therapy, the innate and adaptive immune system can be utilized to attack invasive cancer cells.
T Cell-Mediated Therapy
T lymphocytes, which play a major role in the adaptive immune system, have the ability to naturally destroy cancer cells by recognizing the cancer antigens that tumors express and responding specifically to the tumors.150,151 T cell therapy is also important because while T cells play a major role in adaptive immunity, they simultaneously activate the immune system to amplify the innate immune response against cancer cells. By doing so, the innate and adaptive immune mechanisms synergize to fight against cancer.152,153
However, cancer cells have evolved to avoid the T cell attack. Tumor cells activate immune checkpoints, secrete cytokines that inhibit cytotoxic T and natural killer (NK) cells, and downregulate antigen presentation by decreasing the expression levels of major histocompatibility complex (MHC) II.154,155 The goal of T cell-mediated therapy is to overcome the cancer cells’ evasion of the T cell attack by recognizing and effective killing cancer cells.18 There are three major approaches for T cell-mediated therapy: immune checkpoint therapy, tumor microenvironment targeted-therapy, and dendritic cell interaction-based therapy.19
Immune Checkpoint Therapy
A normal immune response involves the recognition of the foreign antigens via helper T cells, which secrete cytokines that enable the proliferation of cytotoxic T cells. These proliferated cytotoxic T cells directly attack the foreign antigens, resulting in an effective immune response.151 However, cancer cells can avoid T cell immune activation.154,155 Immune checkpoints originally exist to inhibit the activation of CD8 cytotoxic T cells, stopping an autoimmune response. However, cancer cells evade the attack of the immune system by activating these immune checkpoints that stop the T cell attack.20 Although there are many immune checkpoints, the programmed cell death protein (PD) 1/PD-ligand 1 (PD-L1) and the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) checkpoints are most often utilized in immune checkpoint therapy.156,157 The PD-1/PD-L1 checkpoint stops the immune response, resulting in the apoptosis of the cancer cells; meanwhile, the CTLA-4 checkpoint blocks the cytotoxic T cells from recognizing and attacking tumor cells. By modulating these checkpoints, cancer cells can avoid the immune system (Figure 4).158
 |
Figure 4 PD-1 and CTLA-4 immune checkpoints and immune checkpoint inhibitors. (A) CTLA-4 immune checkpoint. The CTLA-4 immune checkpoint stops cytotoxic T cells from recognizing antigens presented by dendritic cells (DC) via major histocompatibility complex (MHC). When CTLA-4 receptors bind to CD80/CD86, tumor antigens are not recognized. (B) Anti-CTLA-4 antibody. When anti-CTLA-4 antibodies, eg ipilimumab, bind to the CTLA-4 receptor, the CTLA-4 immune checkpoint, which was originally activated by the binding of CD80/86 with CTLA-4, is not activated. This leads to the activation of the cytotoxic T cells and the successful recognition of tumor antigens presented by the APC MHC molecules. (C) PD-1 immune checkpoint. The PD-1 immune checkpoint stops cytotoxic T cells from secreting cytokines that induce tumor cell death. The PD-1 receptor of the cytotoxic T cell binds PD-L1, a ligand presented by the tumor cell. (D) Anti-PD-1 and anti-PD-L1 antibodies. When anti-PD-1 antibodies, eg Nivolumab, bind the PD-1 receptor, the PD-1 immune checkpoint is no longer activated. A similar effect can be obtained using anti-PD-L1 antibodies, eg Pembrolizumab, which results in the activation of cytotoxic T cells and thus causes tumor cell death. |
The use of anti-PD-1, anti-PD-L1, and anti-CTLA-4 antibodies has been effective in treating tumor types unresponsive to already established anticancer therapies. Nivolumab, an anti-PD-1 antibody, treated both squamous and non-squamous non-SCLCs (NSCLC) more effectively than conventional chemotherapy.159,160 Pembrolizumab, an anti-PD-L1 antibody, was also effective for treating NSCLCs having the PD-L1 ligand.161–163 Ipilimumab, an anti-CTLA-4 antibody, was used against melanomas.164–166
The main drawback of immune checkpoint inhibition therapy is that only a limited patient population can benefit from it; this is because it is only effective when PD-1, PD-L1, and CTLA-4 play an active role in tumorigenesis.167 For example, Nivolumab and Pembrolizumab are only applicable to cancers that inactivate the PD-1/PD-L1 immune checkpoint.168
There are currently seven FDA-approved immune checkpoint inhibitors: ipilimumab, pembrolizumab, nivolumab, atezolizumab, avelumab, durvalumab, and cemiplimab-rwlc. These drugs treat a wide range of cancers, including advanced melanoma, lymphoma, NSCLC, and renal cell carcinoma.169 Despite advances in the treatment of various cancer types by immune checkpoint inhibition, application of this therapy to treat solid tumors, such as pancreatic and brain tumors, is still unsuccessful (Table 3).168,170
 |
Table 3 FDA Approved Immune Checkpoint Inhibitors |
Tumor Microenvironment-Targeted Therapy
Tumor microenvironment-targeted therapy is another strategy that stops cancer cells from evading the immune system.171–173 Various cellular infiltrates within the tumor microenvironment (TME) (eg blood vessel endothelium, immune cells, signaling molecules, and extracellular matrix) also help cancer cells escape the immune system.174
Regulatory T cells (Tregs) play an important role in the TME.175 They not only activate the immune checkpoints, but also secrete immunosuppressive cytokines, such as TGF-β and IL-10, which stop the antitumor response of T cells and other innate immune cells, such as CD4, CD8, and NK cells.176 In contrast, by activating cells which secrete immune-activating cytokines, such as IL-2, IFN-α, TNF, IL-12, and GM-CSF, an effective anticancer response can be initiated.173,177,178 In particular, IL-2 inhibits metastatic melanoma. Importantly, a limited number of patients respond to this cytokine treatment; but those who do respond to it experience durable, complete, and apparently curative results.179–181
Dendritic Cell Interaction-Based Therapy
DCs are antigen-presenting cells (APCs).182,183 Both foreign and self-antigens are presented on DCs as peptides by MHC class II molecules. This presentation results in the activation of T cells and the stimulation of the adaptive immune defense system.184 Specifically, by activating the adaptive immune response, DCs link the innate and adaptive immune systems.153 As discussed, T cell-mediated therapy relies in the activation of T cells in order to effectively attack cancer cells. Therefore, by properly activating DCs, one can stimulate T cells to attack the cancer cells.185–188
The most important question is how to manipulate the DCs to initiate T cell activation. T cells are activated through the immunological response of cell membrane-bound molecules, such as BTLA-HVEM, CD40-CD40L, CTLA4-CD80/CD-86, and CD70-CD27, and various secreted cytokines including, IL-6, IL-12, IL-23, IL-27, and TNF-β1. By understanding that the immunomodulatory mechanism involving DCs and T cells is a “functional partnership,” it is possible to develop novel immunotherapies to target tumors (Figure 5).189–196
 |
Figure 5 Immunomodulatory mechanisms between dendritic cells (DC) and T cells. The activity of T cells is regulated by the membrane molecules presented by the dendritic cells. (A) T cells are activated by interactions such as the MHC antigen recognition by T cell receptors, CD40-CD40L, and CD70-CD27. Cytokines such as IL-6, IL-12, IL-23, and IL-27 also activate T cell immunity. (B) T cell activity is suppressed by BTLA-HVEM and CD80/86-CTLA-4. Cytokines such as TGF-β and IL-27 also suppress T cell activity. |
Macrophage Repolarization Therapy
Like T cells, macrophages also play a vital role in the TME and can destroy cancer cells.197–200 Macrophages located in the TME, named tumor-associated macrophages (TAM), can release immunosuppressive cytokines, including IL-1β, IL-6, and TNF-α. TAMs are usually differentiated into M2-type macrophages, which are anti-inflammatory, unlike M1-type macrophages which show pro-inflammatory abilities. M2-type macrophages suppress antitumor immune responses and promote tumor growth, whereas M1-type macrophages do the opposite. Due to these traits, researchers have tried to repolarize macrophages from M2- to M1-type.197,201,202
To repolarize macrophages from the M2- to the M1-type, the use of nanovesicles (NV) was proposed.22 NVs derived from M1-type macrophages (M1NVs) contain approximately three times more protein than that in non-nanosized exosome vesicles derived from the same number of M1-type macrophages. M1NVs are also engulfed by M2-type macrophages, and possess homing abilities towards tumor sites, guided by leukocyte-derived adhesion molecules with lymphocyte function-associated antigen 1(LFA-1), and are not toxic or immunogenic.22,203 To summarize, the use of NVs is more effective and safer than the direct injection of M1-type macrophages into the TME, because the latter approach adversely repolarized M1- to M2-type macrophages.22
Importantly, M1NV also boosts the antitumor efficacy when combined with the PD-L1 antibody, by significantly decreasing the tumor volume. This synergistic effect originates because M1NV and PD-L1 have completely different mechanisms of action. M1NV inhibits M2-type macrophages, stopping angiogenesis and immunosuppression, while PD-L1 antibody only induces the apoptosis of tumor cells by the cytotoxic T cell attack.22,203
However, the TAM strategy is still at a very early developmental stage, and differences between human and mouse macrophages make the clinical application of laboratory findings difficult. Therefore, to effectively design a macrophage-repolarization strategy, the interaction between the human immune system and the TME needs to be further investigated and elucidated.22
Future Anticancer Therapy
The future of anticancer therapy will involve the combination of existing strategies. Therefore, to obtain the greatest anticancer effect, it is essential to determine which strategies work together well in a combined manner.204,205 By understanding the exact mechanisms by which drugs destroy tumors, the ultimate combination therapy to treat cancer can be designed. Until now, the most promising anticancer strategy is NP immunotherapy. Indeed, the field of nanotechnology can be exploited to boost the anticancer efficacy of conventional immunotherapy.206–208
Nanodrug-Based Anticancer Immunotherapy
Although the clinical application of conventional anticancer nanodrugs was unsuccessful, nanomedical cancer treatment is still considered a promising approach.112,117,206 This approach includes the use of NPs to stimulate the immune system rather than to directly release cytotoxic anticancer drugs to the cancer cells.
Historically, previous approaches of nanodrug therapy focused on direct attacking cancer cells or transporting cytotoxic drugs into cancer cells.209 However, due to advantages in preventing metastasis and recurrence, immunotherapy using NPs is not only considered as a way of transporting anticancer drugs into the cancer cells but also regarded as activators of the innate immune response.210–213 Additionally, it is expected that nanodrug anticancer immunotherapy will be able to overcome the limitations of the current immunotherapies in terms of efficacy and cytotoxic side effects.97
The cancer-immunity response is identical to the conventional immune response to antigens.158,214 When cancer cells enter apoptosis, cell death as the result of a genetically programmed process, or necrosis, cell death caused by external cellular injury, antigens from tumor cells are released and captured by APCs and presented through MHC class II molecules to T cells.215,216 The MHC class II molecules trigger immature T cells to differentiate into tumor-specific cytotoxic T lymphocytes (Tc). Tcs then infiltrate the TME, recognize the tumor cells, and induce their apoptosis.217
According to this scenario, NPs can boost the efficacy of the tumor immune response through three main mechanisms (Figure 6).211
 |
Figure 6 Nanoparticles boost the anti-tumor immune response. (A) Efficiency of nanoparticles for tumor-antigen delivery. Nanoparticles (NPs) aid tumor antigen delivery by protecting tumor antigens from degradation. Tumor cell-derived artificial NP membranes encapsulate and thus protect tumor antigens from degradation. As NP-coated tumor antigens are less degraded than tumor antigens not coated with NPs, more antigens are able to reach the APCs, thus resulting in higher immunogenicity. The physiochemical properties of NPs also aid the selectivity of delivery of tumor antigens to lymph nodes, where tumor antigens are more easily transferred to APCs. (B) Adjuvant delivery. NPs can deliver adjuvants along with the tumor antigen. Adjuvants are molecules that increase the immunogenicity of tumor antigens; they include MPLA, LPS, and STING. When tumor antigens are delivered with adjuvants, a greater immune response of APCs is initiated. (C) Tumor microenvironment (TME) modulation. NPs can suppress the activity of regulatory T cells (Treg) through various mechanisms, eg NPs conjugated to anti-CTLA inhibitors. Tregs are immunosuppressive cells that inhibit the activity of cytotoxic T cells, which inhibit tumor cell activity. Another mechanism is the suppression of tumor-associated macrophages (TAM) by NPs, eg liposomal doxorubicin NPs. TAMs suppress helper T cells (Th) through the TGF-β signaling pathway, and Th cells facilitate cytotoxic T cell attack of tumor cells. TAMs also directly attack tumor cells. To summarize, NPs can decrease the number of tumor cells in the TME through Tregs and TAMs. |
First, NPs can aid the delivery of cancer antigens to APCs. Tumor antigens must be expressed on the membrane of APCs to induce tumor immunity. However, these native tumor antigens are easily degraded by enzymes in the body and have low immunogenicity because they are not easily transferred to APCs. NPs facilitate cancer antigen delivery to APCs by minimizing tumor antigen degradation in the body via encapsulation and transfer of cancer antigens to the APCs.213,218 NPs also induce delivery of the tumor proteins to lymph nodes, where cancer antigens are more easily transferred to APCs.218,219 This selective delivery to the lymph nodes is determined by the physiochemical properties of the NP, such as particle size, surface charge, shape, and hydrophobicity.220–222 Among them, particle size is the most important factor that determines the delivery to the lymph nodes. While small NPs (<5 nm) easily leak out of the blood vessels, larger NPs (>100 nm) remain in the circulatory system and eventually reach in the lymph nodes.223 However, when NPs of 25 and 100 nm were intradermally injected, NPs of 25 nm were more significantly delivered to the draining lymph nodes than 100 nm NPs.208,221 Therefore, medium-size NPs (5–100 nm) were optimal for the selective delivery of the tumor proteins to the lymph nodes, resulting in higher expression of tumor proteins on APCs.224
Second, nanoparticles can deliver adjuvants to promote an anticancer response.225,226 Adjuvants are molecules that increase immunogenicity, such as 3-O-desacyl-4ʹ-monophosphoryl lipid A (MPLA), lipopolysaccharides (LPS), and agonists of the stimulator of interferon (IFN) genes.227–229 When tumor antigens are presented alone, they often fail to elicit an immune response. However, when NPs deliver these adjuvants to the cytoplasm of APCs, a more sensitive antigen-specific T cell immune response is generated.225
Third, NPs can modulate TME to increase the activation of the immune system.230,231 NPs can control and reduce the activation regulatory T cells (Treg) in the TME. NPs can also inhibit tumor cytokines, like TGFβ, that are present in the TME,232–234 inducing an immune response against the tumor.235
Although many types of NPs are investigated in anticancer immunotherapy, polylactic-co-glycolic acid (PLGA) NPs, liposomes, micelles, and gold NPs were widely used (Table 4).236–239 Among them, PLGA NPs are considered excellent biomaterials in immunotherapy due to their low systemic toxicity and high biodegradability.236,240 PLGA NPs are nonspecifically uptaked by DCs without any immunological recognition process and thus, PLGA NPs are suitable for targeting DCs for delivering antigens, vaccines, and other therapeutic molecules.240–242
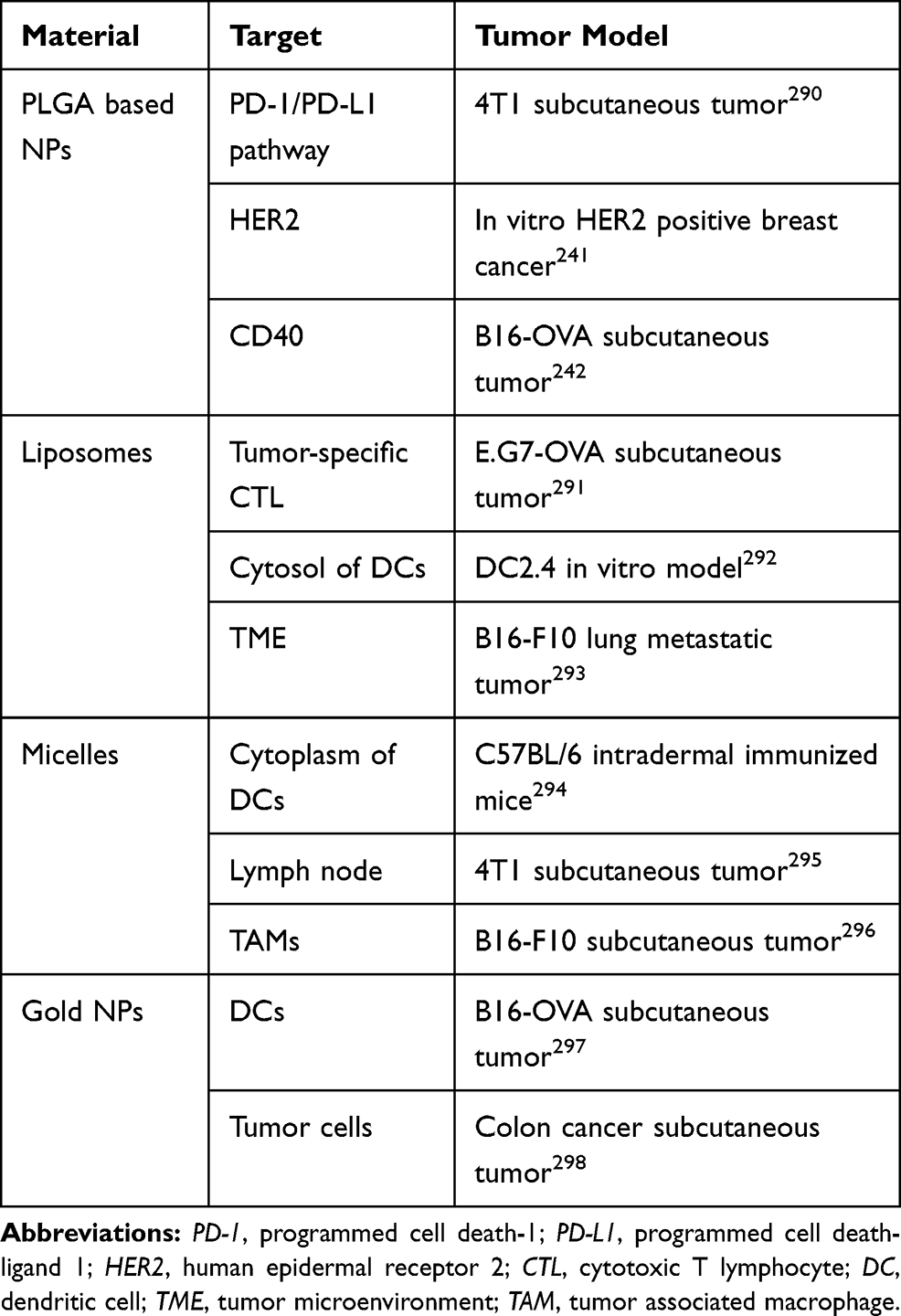 |
Table 4 Nanoparticle (NP) Systems Used for Immunotherapy |
Among the clinically approved anticancer nanodrugs, Abraxane is one of the most successful drugs. Abraxane, an FDA-approved drug, is applied for the treatment of non-small cell lung cancer (NSCLC), is regarded as a more effective drug than paclitaxel (PTX), one of conventional chemotherapy for treating NSCLC.243 Abraxane is an albumin bounded PTX, and the albumin formulation allows PTX to more easily penetrate tumors than PTX alone. Albumin particles extravasate from the blood through albumin receptors, increasing the concentration of the drug in the local tumor site.244–246
Although the use of NPs in immunotherapy has increased the effectiveness of the tumor immune response, there are still some limitations. Since the NPs are usually intravenously (IV) rather than locally administrated, they are sequestered and accumulated in RES organs, such as the liver or kidney, instead of reaching the tumor site.213,247
A solution to this problem was discovered in the field of interventional radiology.248 By injecting NPs locally into the tumor rather than systemically, with the help of imaging guidance, drugs are directly and precisely injected into the tumor. The local administration of the NPs has shown greater anticancer effect and less toxicity to other non-target cells, compared with systemic IV administration.249,250
Another method that resulted in highly specific delivery of NPs to tumor cells and low toxicity to non-tumor cells involved coating NPs on NK cells.149 NK cell membranes can elicit tumor-specific immune responses by regulating the tumor immune response with cytokines such as TNF-α, activating T cells to attack tumor cells, and inducing M1-type macrophage repolarization.251–253 NK cell membrane-decorated NPs (NK-NPs) can improve NK cell membrane immunotherapy. An example of NPs that are coated on NK cell membranes is the photosensitizer 4,4ʹ,4”,4”’ –(porphine-5,10,15,20-tetrayl) tetrakis (benzoic acid) (TCPP)-loaded mPEG-PGLA polymeric NP (T-NP). TCPPs that were loaded on T-NPs not only directly eradicated primary tumor cells through photodynamic therapy (PDT) but also triggered the generation of damage-associated molecular patterns (DAMP) caused by PDT-induced immunogenic cell death of tumor cells. The DAMPs activated APCs, consequently enhanced the antitumor immunity efficiency of the NK cell membranes. Additionally, NK-NPs reduced primary tumor growth, and induced an abscopal effect, which means an inhibition of proliferation in distant tumors caused by the elimination of primary tumors (Figure 7).149,254
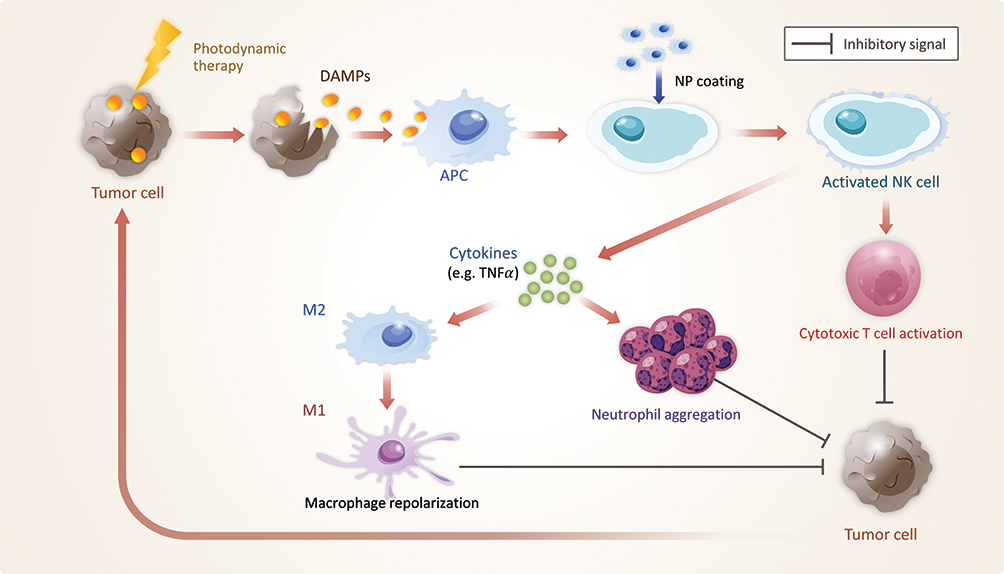 |
Figure 7 NP coating on NK cells. When photodynamic therapy (PDT) is applied, tumor cells release damage-associated molecular patterns; APCs captured these to activate NK cells. NK cells trigger antitumor immune responses that attack tumor cells, and this process is upregulated by NP coating. Tumor immune responses controlled by TNFα are increased. M2 macrophages are repolarized into M1 macrophages to create a pro-inflammatory effect; meanwhile, neutrophils aggregate, causing inflammation. Cytotoxic T cells also become activated. Through these immune responses, tumor cells are more efficiently attacked. NPs also play an important role in PDT. NPs, such as T-NPs, serve as photosensitizers in PDT. |
In addition, the conjugating NPs with small molecules, such as peptides, has shown versatile functionalities.255 In addition, conjugating a chemotherapeutic drug with a functional peptide, peptides are allowed to form an autocatalytic and transformable state in drug delivery system. Specifically, by attaching a short PEG chain with a tumor targeting peptide and by using a hydrogen-bonding peptide in scaffold, peptide-nanodrug selectively accumulated into the tumor site and drugs sustained released the chemotherapeutic drug.256
However, the use of NPs in cancer nanomedicine is still hampered by the development of side effects. For instance, TiO2, SiO2, Au, and Ag NPs can create gaps in the endothelium of tens to hundreds micrometers in width,257 a process called endothelial leakiness (NanoEL).258,259 These gaps can be exploited by cancer cells for intravasation into circulation and extravasation to a new tumor site, thus accelerating the metastasis process.260–262 Therefore, the potential of NanoEL to stimulate metastasis needs to be considered during the design of drugs to treat cancer, especially in nanomedicine (Figure 8).257
 |
Figure 8 NP-induced endothelial leakiness (NanoEL). (A) Intravasation. Some NPs form sizable gaps in the tumor vasculature. Through these gaps, tumor cells can migrate from the primary tumor site into the blood stream. (B) Extravasation. After the entrance of tumor cells into the circulatory system, the tumor cells extravasate out of the bloodstream to a new tumor site through a process known as metastasis. |
Other Combination Therapies
As previously mentioned, the future of cancer therapy lies in the combination of various existing methods to effectively kill cancer cells. The use of monoclonal antibodies has been more effective in treating cancer when combined with conventional chemotherapy, rather than alone.263 Likewise, immune checkpoint inhibitors are also more effective in attacking tumor cells when used in combination with other types of anticancer therapies, such as the combination of M1NV with the PD-L1 antibody (Table 5).22,264
 |
Table 5 Major FDA Approved Combination Therapies |
Nanomedicine in Photothermal Therapy (PTT)
The use of NPs in photothermal therapy (PTT) is advantageous.23,265 PTT is a type of PDT that utilizes heat created by electromagnetic radiation. The thermal ablation of tumor cells using PTT is a possible clinical approach for the treatment of local, solid tumors.266–268 By treating the tumors with photosensitizers, which are molecules that can be activated by light to produce chemical responses that can damage cellular structures, and near-infrared wavelength radiation, it is possible to attack tumor cells via hyperthermia.269,270
However, PTT still has some limitations. PTT can only be applied as a secondary treatment after the surgical removal of solid tumors and is not applicable to metastatic tumors. Additionally, hyperthermia can induce the tumor cells to release antigens and pro-inflammatory cytokines, which could adversely increase the tumor cell action.271 These limitations can be overcome with the help of NPs.
When light-responsive heating NPs (L-HNPs) are injected into tumors, they selectively accumulate in the local tumor site, allowing transfer heat to tumor cells without damaging surrounding healthy tissue.268,272 The L-HNPs effectively absorb energy from a light beam (wavelength of 700–980 nm and 1000–1400 nm) and have a large light-to-heat conversion efficiency.268,273-275 Among metallic NPs, gold were widely used as L-HNPs.267,276-278 Some carbon nanotube based materials also have shown high light-to-heat conversion efficiency and biocompatibility.279,280 In addition, light-absorbing organic NPs, such as polyaniline conductive polymers and indocyanine green (ICG) are also attractive materials as photothermal agents since those materials are biodegradable unlike other inorganic NPs.281–285
The combined use of NPs and PTT has shown therapeutic efficacy for untreated and distant CT26 colon carcinoma and metastasized TC-1 submucosa-lung cancer, which were originally inaccessible by conventional PTT. In these cases, PTT can attack metastatic tumors without performing surgical resection. Therefore, the combined use of NPs and PTT therapies offers more advantages for cancer treatment (Figure 9).23,265
 |
Figure 9 The use of NPs in PTT. (A) Metastatic cancer prior to treatment. When PTT is applied without the use of NPs, it requires prior surgical removal of the solid tumor. (B) The application of PTT without NPs. (C) Results after PTT without NPs. This method shows limited effectiveness in removing tumor cells that remain after surgical excision. It is not effective against diffuse, metastatic tumors. (D, E) The application of PTT with NPs. Polydopamine-coated gold NPs are injected in the tumor vasculature for this purpose. This method does not need surgical excision prior to PTT. (F) Results of PTT with NPs. This method is effective for treating solid tumors even without their surgical removal, and it is also effective for treating diffuse, metastatic tumors. |
Nanomedicine in Tumor Vaccines
Tumor vaccines that can prevent long-term recurrence are also under development.286,287 To prevent the long-term recurrence of tumors, immune checkpoint inhibitor-modified NPs are used to activate the immune system in the TME. Adjuvant-loaded NPs were made by entrapping Imiquimod (IQ), an immune response adjuvant, into photoresponsive polydopamine NPs (IQ/PNs), and modifying them with the anti-PD-L1 antibody (PD-L1Ab-IQ/PNs). Anti-PD-L1 antibodies on IQ/PNs increased the binding of NPs to CT26 cancer cells overexpressing PD-L1, attacking them, and preventing tumor recurrence (Figure 10).288
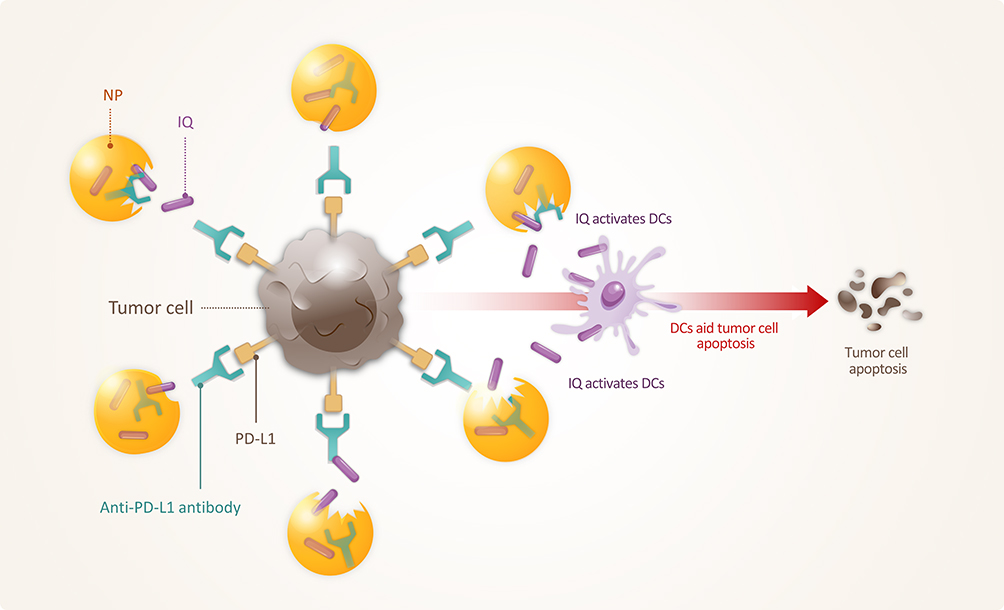 |
Figure 10 PD-L1Ab-IQ/PN. Entrapment of Imiquimod (IQ) in polydopamine NPs represents adjuvant-loaded NPs (IQ/PNs), and with the help of anti-PD-L1 antibodies, IQ/PNs are able to efficiently bind tumor cells overexpressing PD-L1, resulting in the apoptosis of the tumor cells. Through this method, tumor recurrence can be prevented. |
Therefore, the next generation of cancer therapeutics will probably rely on the combination of nanomedicine and immunotherapy to control the immune response against cancer cells and thus destroy them more effectively. By using this approach in combination with other proven therapies mentioned above, the effectiveness of the treatment can be boosted to even higher levels.16,130
Discussion
The ultimate goal of anticancer research is its application in the clinic; significant research in this direction is still being conducted because the current anticancer strategies are neither complete nor ideal for patient treatment.
The beginning of this review describes established and widely employed anticancer approaches. Even though some of them have shown limitations, they are still widely used and some of them even constitute an important stepping-stone to developing the cancer therapies of the future. Conventional chemotherapy has clear application limits, thus more effective cancer drugs are under investigation.289 For instance, hormonal therapy introduced the concept of targeted therapy, which was further developed using monoclonal antibodies and TKIs because they target certain receptors and ligands on tumor cells.14,39,74 However, target receptor-based approaches are only applicable to specific cancer cells that overexpress those corresponding receptors, thus, they are ineffective for treatment many types of tumors.12 The modification of nanodrug physiochemical properties has also been studied for two decades but this approach has been quickly discarded for cancer treatment due to its toxicity and relative ineffectiveness.15 Among the established approaches discussed above, stem cells, especially MSCs, have great targeting efficacy due to their unique homing abilities, but their genetic modification to enhance their attack of cancer cells has raised some concerns regarding long-term toxicity in clinical applications.16
The next part of this review summarizes some more practical methods that are currently under investigation and in the early stages of clinical application. The newest strategy is cancer immunotherapy. T cells, one of the most important immune cells, have the ability to attack cancer cells.19 Thus, by modulating the immune system to initiate the T cell attack, we can achieve a desired level of cancer treatment. Immune checkpoint inhibition could affect the inhibitory factors that prevent T cell attack of cancer cells, thus using our own immune system to attack tumors.157 Additionally, by utilizing anticancer cytokines in the TME, such as IL-2, IFN-α, TNF, IL-12, and GM-CSF, along with the repolarization of M2-type macrophages, is possible to attack a wider range of cancers.21,197 Lastly, DCs play a significant role in connecting the innate and adaptive immune systems, and if the proper stimulation is provided via DCs, T cells could be induced to destroy cancer cells.185 Combination immunotherapy strategies may be more effective compared with single immunotherapy strategies.210
The last part of this review provides information about combination therapy, which is the future of cancer treatment. The use of NPs in immunotherapy has already shown encouraging results.130 The use of NPs in combination with photothermal therapy and tumor vaccines has been shown to be effective in attacking tumor cells.267
In conclusion, the two main keywords of future anticancer strategies would be the combinational therapy combining cancer-immunology and nanodrug delivery system. Reciprocally, the current anticancer nano-therapeutic efficacy can dramatically enhance with the combination of established immunotherapy.
Acknowledgment
This research was supported by grants from Gachon University of Funds (2017-0601) and the National Research Foundation of Korea (2019R1A2C1085986).
Disclosure
The authors declare that there is no conflict of interest in this work.
References
1. Simmons BJ, Falto-Aizpurua LA, Griffith RD, Nouri K. Smallpox: 12,000 years from plagues to eradication: a dermatologic ailment shaping the face of society. JAMA Dermatol. 2015;151(5):521. doi:10.1001/jamadermatol.2014.4812
2. Moroder L, Musiol HJ. Insulin-from its discovery to the industrial synthesis of modern insulin analogues. Angew Chem Int Ed Engl. 2017;56(36):10656–10669. doi:10.1002/anie.201702493
3. O’Neill D. A global perspective on the history of anaesthesia. Lancet. 2017;390(10111):2434–2435. doi:10.1016/S0140-6736(17)33048-9
4. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
5. Dans M, Smith T, Back A, et al. NCCN guidelines insights: palliative care, version 2.2017. J Natl Compr Canc Netw. 2017;15(8):989–997. doi:10.6004/jnccn.2017.0132
6. NCCN. NCCN clinical practice guidelines in oncology. Available from: https://www.nccn.org/professionals/physician_gls/default.aspx#detection.
7. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21(1):15–25.
8. Sawyers C. Targeted cancer therapy. Nature. 2004;432(7015):294–297.
9. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627.
10. Yang G, Nowsheen S, Aziz K, Georgakilas AG. Toxicity and adverse effects of Tamoxifen and other anti-estrogen drugs. Pharmacol Ther. 2013;139(3):392–404. doi:10.1016/j.pharmthera.2013.05.005
11. Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15(6):361–370. doi:10.1038/nrc3930
12. Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1(2):118–129. doi:10.1038/35101072
13. Li GN, Wang SP, Xue X, Qu XJ, Liu HP. Monoclonal antibody-related drugs for cancer therapy. Drug Discov Ther. 2013;7(5):178–184.
14. van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35(8):692–706. doi:10.1016/j.ctrv.2009.08.004
15. Kim SW, Khang D. Multiple cues on the physiochemical, mesenchymal, and intracellular trafficking interactions with nanocarriers to maximize tumor target efficiency. Int J Nanomedicine. 2015;10:3989–4008. doi:10.2147/IJN.S83951
16. Kwon S, Yoo KH, Sym SJ, Khang D. Mesenchymal stem cell therapy assisted by nanotechnology: a possible combinational treatment for brain tumor and central nerve regeneration. Int J Nanomedicine. 2019;14:5925–5942. doi:10.2147/IJN.S217923
17. Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi:10.1186/s12916-016-0623-5
18. Bender E. Cancer immunotherapy. Nature. 2017;552(7685):S61. doi:10.1038/d41586-017-08699-z
19. Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell. 2016;164(6):1233–1247. doi:10.1016/j.cell.2016.01.049
20. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):165. doi:10.1038/s12276-018-0191-1
21. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting Tumor Microenvironment for Cancer Therapy. Int J Mol Sci. 2019;20(4):840. doi:10.3390/ijms20040840
22. Choo YW, Kang M, Kim HY, et al. M1 macrophage-derived nanovesicles potentiate the anticancer efficacy of immune checkpoint inhibitors. ACS Nano. 2018;12(9):8977–8993. doi:10.1021/acsnano.8b02446
23. Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat Commun. 2016;7:13193. doi:10.1038/ncomms13193
24. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108(2):153–164. doi:10.1016/S0092-8674(02)00625-6
25. Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;11(24 Pt 1):8782–8788. doi:10.1158/1078-0432.CCR-05-1664
26. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–1454. doi:10.1093/jnci/djm135
27. Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24(9):1161–1180. doi:10.1016/j.chembiol.2017.08.028
28. Kondo N, Takahashi A, Ono K, Ohnishi T. DNA damage induced by alkylating agents and repair pathways. J Nucleic Acids. 2010;2010:543531. doi:10.4061/2010/543531
29. Kaye SB. New antimetabolites in cancer chemotherapy and their clinical impact. Br J Cancer. 1998;78 Suppl 3:1–7. doi:10.1038/bjc.1998.747
30. Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109(7):2894–2902. doi:10.1021/cr900097c
31. Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12(2):104–120. doi:10.1038/nrc3185
32. Sato T, Issa JJ, Kropf P. DNA hypomethylating drugs in cancer therapy. Cold Spring Harb Perspect Med. 2017;7(5):a026948. doi:10.1101/cshperspect.a026948
33. Pommier Y, Sun Y, Huang SN, Nitiss JL. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol. 2016;17(11):703–721. doi:10.1038/nrm.2016.111
34. Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138(2):255–271. doi:10.1016/j.pharmthera.2013.01.011
35. Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616–1634. doi:10.1093/jnci/91.19.1616
36. West HJ. Chemotherapy-induced hair loss (Alopecia). JAMA Oncol. 2017;3(8):1147. doi:10.1001/jamaoncol.2017.1026
37. Andreyev J, Ross P, Donnellan C, et al. Guidance on the management of diarrhoea during cancer chemotherapy. Lancet Oncol. 2014;15(10):e447–e460. doi:10.1016/S1470-2045(14)70006-3
38. Mohammad RM, Muqbil I, Lowe L, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35 Suppl:S78–S103. doi:10.1016/j.semcancer.2015.03.001
39. Jordan VC. Tamoxifen: catalyst for the change to targeted therapy. Eur J Cancer. 2008;44(1):30–38. doi:10.1016/j.ejca.2007.11.002
40. Pritchard KI. Combining endocrine agents with chemotherapy: which patients and what sequence? Cancer. 2008;112(3 Suppl):718–722. doi:10.1002/cncr.23189
41. Jordan VC, Assikis VJ. Endometrial carcinoma and tamoxifen: clearing up a controversy. Clin Cancer Res. 1995;1(5):467–472.
42. Jordan VC. Tamoxifen and endometrial cancer. Lancet. 1989;1(8640):733–734. doi:10.1016/S0140-6736(89)92255-1
43. Rajewsky K. The advent and rise of monoclonal antibodies. Nature. 2019;575(7781):47–49. doi:10.1038/d41586-019-02840-w
44. Harris M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol. 2004;5(5):292–302. doi:10.1016/S1470-2045(04)01467-6
45. Firer MA, Gellerman G. Targeted drug delivery for cancer therapy: the other side of antibodies. J Hematol Oncol. 2012;5:70. doi:10.1186/1756-8722-5-70
46. Xu MJ, Johnson DE, Grandis JR. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017;36(3):463–473. doi:10.1007/s10555-017-9687-8
47. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi:10.1126/science.1099314
48. Chakraborty S, Li L, Puliyappadamba VT, et al. Constitutive and ligand-induced EGFR signalling triggers distinct and mutually exclusive downstream signalling networks. Nat Commun. 2014;5:5811. doi:10.1038/ncomms6811
49. Ferrari A, Vincent-Salomon A, Pivot X, et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat Commun. 2016;7:12222. doi:10.1038/ncomms12222
50. Xia Y, Fan C, Hoadley KA, Parker JS, Perou CM. Genetic determinants of the molecular portraits of epithelial cancers. Nat Commun. 2019;10(1):5666. doi:10.1038/s41467-019-13588-2
51. Fry EA, Taneja P, Inoue K. Oncogenic and tumor-suppressive mouse models for breast cancer engaging HER2/neu. Int J Cancer. 2017;140(3):495–503. doi:10.1002/ijc.30399
52. van Helden EJ, Menke-van der Houven van Oordt CW, Heymans MW, Ket JCF, van den Oord R, Verheul HMW. Optimal use of anti-EGFR monoclonal antibodies for patients with advanced colorectal cancer: a meta-analysis. Cancer Metastasis Rev. 2017;36(2):395–406. doi:10.1007/s10555-017-9668-y
53. Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28(8):1713–1729. doi:10.1093/annonc/mdx175
54. Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–1643.
55. Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360(6):563–572.
56. Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417.
57. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med. 2017;377(2):122–131.
58. Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2017;389(10087):2415–2429.
59. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137.
60. Radinsky R, Risin S, Fan D, et al. Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin Cancer Res. 1995;1(1):19–31.
61. Goldstein NS, Armin M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on Cancer Stage IV colon adenocarcinoma: implications for a standardized scoring system. Cancer. 2001;92(5):1331–1346.
62. Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345.
63. Rimawi MF, Shetty PB, Weiss HL, et al. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer. 2010;116(5):1234–1242.
64. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13(12):871–882.
65. Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. 2009;6(10):569–579.
66. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
67. Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371(17):1609–1618.
68. Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, Phase 3 TRIBE study. Lancet Oncol. 2015;16(13):1306–1315.
69. Kato T, Seto T, Nishio M, et al. Erlotinib plus bevacizumab phase ll study in patients with advanced non-small-cell lung cancer (JO25567): updated safety results. Drug Saf. 2018;41(2):229–237.
70. Hsu WH, Yang JC, Mok TS, Loong HH. Overview of current systemic management of EGFR-mutant NSCLC. Ann Oncol. 2018;29(suppl_1):i3–i9.
71. Rosell R, Dafni U, Felip E, et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): an international, multicentre, single-arm, Phase 2 trial. Lancet Respir Med. 2017;5(5):435–444.
72. Lee JC, Chow NH, Wang ST, Huang SM. Prognostic value of vascular endothelial growth factor expression in colorectal cancer patients. Eur J Cancer. 2000;36(6):748–753.
73. Ohta Y, Endo Y, Tanaka M, et al. Significance of vascular endothelial growth factor messenger RNA expression in primary lung cancer. Clin Cancer Res. 1996;2(8):1411–1416.
74. Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341(6151):1192–1198.
75. Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17(1):36.
76. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365.
77. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–1583.
78. Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–566.
79. Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a Phase II study. Blood. 2002;99(10):3530–3539.
80. Hughes TP, Kaeda J, Branford S, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349(15):1423–1432.
81. Holtz MS, Slovak ML, Zhang F, Sawyers CL, Forman SJ, Bhatia R. Imatinib mesylate (STI571) inhibits growth of primitive malignant progenitors in chronic myelogenous leukemia through reversal of abnormally increased proliferation. Blood. 2002;99(10):3792–3800.
82. Brown JB, Pai RK, Burgess MA, Chennat J, Zureikat AH. Pathologic complete response in a large gastric GIST: using molecular markers to achieve maximal response to neoadjuvant imatinib. J Natl Compr Canc Netw. 2018;16(12):1424–1428.
83. Lee JK, Hahn S, Kim DW, et al. Epidermal growth factor receptor tyrosine kinase inhibitors vs conventional chemotherapy in non-small cell lung cancer harboring wild-type epidermal growth factor receptor: a meta-analysis. JAMA. 2014;311(14):1430–1437.
84. Cataldo VD, Gibbons DL, Perez-Soler R, Quintas-Cardama A. Treatment of non-small-cell lung cancer with erlotinib or gefitinib. N Engl J Med. 2011;364(10):947–955.
85. Park JW, Liu MC, Yee D, et al. Adaptive randomization of neratinib in early breast cancer. N Engl J Med. 2016;375(1):11–22.
86. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311.
87. Xu JW, Wang L, Cheng YG, et al. Immunotherapy for pancreatic cancer: a long and hopeful journey. Cancer Lett. 2018;425:143–151.
88. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454.
89. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465.
90. Picard M, Galvao VR. Current knowledge and management of hypersensitivity reactions to monoclonal antibodies. J Allergy Clin Immunol Pract. 2017;5(3):600–609.
91. Kantarjian H, Sawyers C, Hochhaus A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346(9):645–652.
92. Bjornmalm M, Thurecht KJ, Michael M, Scott AM, Caruso F. Bridging bio-nano science and cancer nanomedicine. ACS Nano. 2017;11(10):9594–9613.
93. Pelaz B, Alexiou C, Alvarez-Puebla RA, et al. Diverse applications of nanomedicine. ACS Nano. 2017;11(3):2313–2381.
94. Hartshorn CM, Bradbury MS, Lanza GM, et al. Nanotechnology strategies to advance outcomes in clinical cancer care. ACS Nano. 2018;12(1):24–43.
95. Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1866–1879.
96. Wang N, Feng Y, Zeng L, Zhao Z, Chen T. Functionalized multiwalled carbon nanotubes as carriers of ruthenium complexes to antagonize cancer multidrug resistance and radioresistance. ACS Appl Mater Interfaces. 2015;7(27):14933–14945.
97. Zang X, Zhao X, Hu H, Qiao M, Deng Y, Chen D. Nanoparticles for tumor immunotherapy. Eur J Pharm Biopharm. 2017;115:243–256.
98. Kim SW, Park JY, Lee S, Kim SH, Khang D. Destroying deep lung tumor tissue through lung-selective accumulation and by activation of caveolin uptake channels using a specific width of carbon nanodrug. ACS Appl Mater Interfaces. 2018;10(5):4419–4428.
99. Zhao J, Castranova V. Toxicology of nanomaterials used in nanomedicine. J Toxicol Environ Health B Crit Rev. 2011;14(8):593–632.
100. Toy R, Peiris PM, Ghaghada KB, Karathanasis E. Shaping cancer nanomedicine: the effect of particle shape on the in vivo journey of nanoparticles. Nanomedicine (Lond). 2014;9(1):121–134. doi:10.2217/nnm.13.191
101. Hoshyar N, Gray S, Han H, Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (Lond). 2016;11(6):673–692. doi:10.2217/nnm.16.5
102. Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. doi:10.1021/mp800049w
103. Verma SK, Jha E, Panda PK, et al. Mechanistic insight into size-dependent enhanced cytotoxicity of industrial antibacterial titanium oxide nanoparticles on colon cells because of reactive oxygen species quenching and neutral lipid alteration. ACS Omega. 2018;3(1):1244–1262. doi:10.1021/acsomega.7b01522
104. Verma SK, Jha E, Sahoo B, et al. Mechanistic insight into the rapid one-step facile biofabrication of antibacterial silver nanoparticles from bacterial release and their biogenicity and concentration-dependent in vitro cytotoxicity to colon cells. RSC Adv. 2017;7(64):40034–40045. doi:10.1039/C7RA05943D
105. Sonavane G, Tomoda K, Makino K. Biodistribution of colloidal gold nanoparticles after intravenous administration: effect of particle size. Colloids Surf B Biointerfaces. 2008;66(2):274–280. doi:10.1016/j.colsurfb.2008.07.004
106. Arvizo R, Bhattacharya R, Mukherjee P. Gold nanoparticles: opportunities and challenges in nanomedicine. Expert Opin Drug Deliv. 2010;7(6):753–763. doi:10.1517/17425241003777010
107. Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–794. doi:10.1016/j.jconrel.2013.09.013
108. Manzoor AA, Lindner LH, Landon CD, et al. Overcoming limitations in nanoparticle drug delivery: triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012;72(21):5566–5575. doi:10.1158/0008-5472.CAN-12-1683
109. Park K. Facing the truth about nanotechnology in drug delivery. ACS Nano. 2013;7(9):7442–7447. doi:10.1021/nn404501g
110. Nie S. Understanding and overcoming major barriers in cancer nanomedicine. Nanomedicine (Lond). 2010;5(4):523–528. doi:10.2217/nnm.10.23
111. Kim SW, Kyung Lee Y, Yeon Lee J, Hee Hong J, Khang D. PEGylated anticancer-carbon nanotubes complex targeting mitochondria of lung cancer cells. Nanotechnology. 2017;28(46):465102. doi:10.1088/1361-6528/aa8c31
112. Pandey G, Mittapelly N, Banala VT, Mishra PR. Multifunctional glycoconjugate assisted nanocrystalline drug delivery for tumor targeting and permeabilization of lysosomal-mitochondrial membrane. ACS Appl Mater Interfaces. 2018;10(20):16964–16976. doi:10.1021/acsami.7b18699
113. Gao P, Pan W, Li N, Tang B. Boosting cancer therapy with organelle-targeted nanomaterials. ACS Appl Mater Interfaces. 2019;11(30):26529–26558. doi:10.1021/acsami.9b01370
114. Fortuni B, Inose T, Ricci M, et al. Polymeric engineering of nanoparticles for highly efficient multifunctional drug delivery systems. Sci Rep. 2019;9(1):2666. doi:10.1038/s41598-019-39107-3
115. Morimoto N, Takei R, Wakamura M, et al. Fast and effective mitochondrial delivery of omega-Rhodamine-B-polysulfobetaine-PEG copolymers. Sci Rep. 2018;8(1):1128. doi:10.1038/s41598-018-19598-2
116. Liu Z, Robinson JT, Sun X, Dai H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008;130(33):10876–10877. doi:10.1021/ja803688x
117. Wang B, He X, Zhang Z, Zhao Y, Feng W. Metabolism of nanomaterials in vivo: blood circulation and organ clearance. Acc Chem Res. 2013;46(3):761–769. doi:10.1021/ar2003336
118. Blau HM, Daley GQ. Stem cells in the treatment of disease. N Engl J Med. 2019;380(18):1748–1760. doi:10.1056/NEJMra1716145
119. Clarke MF. Clinical and therapeutic implications of cancer stem cells. N Engl J Med. 2019;380(23):2237–2245. doi:10.1056/NEJMra1804280
120. Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. 2017;16(1):31. doi:10.1186/s12943-017-0597-8
121. Cihova M, Altanerova V, Altaner C. Stem cell based cancer gene therapy. Mol Pharm. 2011;8(5):1480–1487. doi:10.1021/mp200151a
122. Wu HH, Zhou Y, Tabata Y, Gao JQ. Mesenchymal stem cell-based drug delivery strategy: from cells to biomimetic. J Control Release. 2019;294:102–113. doi:10.1016/j.jconrel.2018.12.019
123. Shi Y, Du L, Lin L, Wang Y. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017;16(1):35–52. doi:10.1038/nrd.2016.193
124. Yeo RW, Lai RC, Zhang B, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65(3):336–341. doi:10.1016/j.addr.2012.07.001
125. Yang KQ, Liu Y, Huang QH, et al. Bone marrow-derived mesenchymal stem cells induced by inflammatory cytokines produce angiogenetic factors and promote prostate cancer growth. BMC Cancer. 2017;17(1):878. doi:10.1186/s12885-017-3879-z
126. Tran TT, Kahn CR. Transplantation of adipose tissue and stem cells: role in metabolism and disease. Nat Rev Endocrinol. 2010;6(4):195–213. doi:10.1038/nrendo.2010.20
127. Medeiros Tavares Marques JC, Cornelio DA, Nogueira Silbiger V, Ducati Luchessi A, de Souza S, Batistuzzo de Medeiros SR. Identification of new genes associated to senescent and tumorigenic phenotypes in mesenchymal stem cells. Sci Rep. 2017;7(1):17837. doi:10.1038/s41598-017-16224-5
128. Dwyer RM, Potter-Beirne SM, Harrington KA, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007;13(17):5020–5027. doi:10.1158/1078-0432.CCR-07-0731
129. Menon LG, Picinich S, Koneru R, et al. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. 2007;25(2):520–528. doi:10.1634/stemcells.2006-0257
130. Kim SW, Lee YK, Hong JH, et al. Mutual destruction of deep lung tumor tissues by nanodrug-conjugated stealth mesenchymal stem cells. Adv Sci (Weinh). 2018;5(5):1700860. doi:10.1016/S1470-2045(16)30030-4
131. Whiteside TL. Exosome and mesenchymal stem cell cross-talk in the tumor microenvironment. Semin Immunol. 2018;35:69–79. doi:10.1016/j.smim.2017.12.003
132. Ullah M, Liu DD, Thakor AS. Mesenchymal stromal cell homing: mechanisms and strategies for improvement. iScience. 2019;15:421–438. doi:10.1016/j.isci.2019.05.004
133. Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366(9494):1385–1396. doi:10.1016/S0140-6736(05)67569-1
134. Jett JR, Schild SE, Kesler KA, Kalemkerian GP. Treatment of small cell lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e400S–e419S. doi:10.1378/chest.12-2363
135. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388(10039):73–85. doi:10.1016/S0140-6736(16)00141-0
136. Notta F, Hahn SA, Real FX. A genetic roadmap of pancreatic cancer: still evolving. Gut. 2017;66(12):2170–2178. doi:10.1136/gutjnl-2016-313317
137. Zhu H, Li T, Du Y, Li M. Pancreatic cancer: challenges and opportunities. BMC Med. 2018;16(1):214. doi:10.1186/s12916-018-1215-3
138. Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392(10145):432–446. doi:10.1016/S0140-6736(18)30990-5
139. Huang J, Zhang Z, Guo J, et al. Genetic modification of mesenchymal stem cells overexpressing CCR1 increases cell viability, migration, engraftment, and capillary density in the injured myocardium. Circ Res. 2010;106(11):1753–1762. doi:10.1161/CIRCRESAHA.109.196030
140. Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256(2):137–165. doi:10.1016/j.canlet.2007.05.013
141. Stuckey DW, Shah K. TRAIL on trial: preclinical advances in cancer therapy. Trends Mol Med. 2013;19(11):685–694. doi:10.1016/j.molmed.2013.08.007
142. Krueger TEG, Thorek DLJ, Denmeade SR, Isaacs JT, Brennen WN. Concise review: mesenchymal stem cell-based drug delivery: the good, the bad, the ugly, and the promise. Stem Cells Transl Med. 2018;7(9):651–663. doi:10.1002/sctm.18-0024
143. Suryaprakash S, Lao YH, Cho HY, et al. Engineered Mesenchymal stem cell/nanomedicine spheroid as an active drug delivery platform for combinational glioblastoma therapy. Nano Lett. 2019;19(3):1701–1705. doi:10.1021/acs.nanolett.8b04697
144. Galipeau J, Sensebe L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. 2018;22(6):824–833. doi:10.1016/j.stem.2018.05.004
145. Lukomska B, Stanaszek L, Zuba-Surma E, Legosz P, Sarzynska S, Drela K. Challenges and controversies in human mesenchymal stem cell therapy. Stem Cells Int. 2019;2019:9628536. doi:10.1155/2019/9628536
146. Lazennec G, Jorgensen C. Concise review: adult multipotent stromal cells and cancer: risk or benefit? Stem Cells. 2008;26(6):1387–1394. doi:10.1634/stemcells.2007-1006
147. Papaccio F, Paino F, Regad T, Papaccio G, Desiderio V, Tirino V. Concise REVIEW: CANCER CELLS, CANCER STEM CELLS, AND MESENCHYMAL STEM CELLS: INFLUENCE IN CANCER DEVELOPMent. Stem Cells Transl Med. 2017;6(12):2115–2125. doi:10.1002/sctm.17-0138
148. Layek B, Shetty M, Nethi SK, Sehgal D, Starr TK, Prabha S. Mesenchymal stem cells as guideposts for nanoparticle-mediated targeted drug delivery in ovarian cancer. Cancers (Basel). 2020;12:4. doi:10.3390/cancers12040965
149. Deng G, Sun Z, Li S, et al. Cell-membrane immunotherapy based on natural killer cell membrane coated nanoparticles for the effective inhibition of primary and abscopal tumor growth. ACS Nano. 2018;12(12):12096–12108. doi:10.1021/acsnano.8b05292
150. Nurieva R, Thomas S, Nguyen T, et al. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006;25(11):2623–2633. doi:10.1038/sj.emboj.7601146
151. Dearnley G, et al. T-lymphocytes. Lancet. 1982;1(8275):778–779.
152. Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2(12):933–944. doi:10.1038/nri954
153. Paul WE. Bridging innate and adaptive immunity. Cell. 2011;147(6):1212–1215. doi:10.1016/j.cell.2011.11.036
154. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi:10.1016/j.cell.2017.01.017
155. Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 2015;5(9):915–919. doi:10.1158/2159-8290.CD-15-0563
156. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17(12):e542–e551. doi:10.1016/S1470-2045(16)30406-5
157. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
158. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349
159. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627
160. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
161. Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
162. Peters S, Kerr KM, Stahel R. PD-1 blockade in advanced NSCLC: A focus on pembrolizumab. Cancer Treat Rev. 2018;62:39–49. doi:10.1016/j.ctrv.2017.10.002
163. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi:10.1056/NEJMoa1501824
164. Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377(19):1824–1835. doi:10.1056/NEJMoa1709030
165. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390(10105):1853–1862. doi:10.1016/S0140-6736(17)31601-X
166. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi:10.1056/NEJMoa1003466
167. Lagos GG, Izar B, Rizvi NA. Beyond tumor PD-L1: emerging Genomic biomarkers for checkpoint inhibitor immunotherapy. Am Soc Clin Oncol Educ Book. 2020;40:1–11. doi:10.1200/EDBK_289967
168. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11(1):39. doi:10.1186/s13045-018-0582-8
169. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33(17):1974–1982. doi:10.1200/JCO.2014.59.4358
170. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi:10.1126/science.aan6733
171. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi:10.1038/s41591-018-0014-x
172. Altorki NK, Markowitz GJ, Gao D, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer. 2019;19(1):9–31. doi:10.1038/s41568-018-0081-9
173. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68. doi:10.1016/j.canlet.2016.01.043
174. Spill F, Reynolds DS, Kamm RD, Zaman MH. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol. 2016;40:41–48. doi:10.1016/j.copbio.2016.02.007
175. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16(10):599–611. doi:10.1038/nri.2016.80
176. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523–532. doi:10.1038/nri2343
177. Rusk N. Antitumor T cells. Nat Methods. 2019;16(1):19. doi:10.1038/s41592-018-0271-0
178. Zhou Y, Chen X, Cao J, Gao H. Overcoming the biological barriers in the tumor microenvironment for improving drug delivery and efficacy. J Mater Chem B. 2020.
179. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–5458. doi:10.4049/jimmunol.1490019
180. Andersen R, Donia M, Ellebaek E, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res. 2016;22(15):3734–3745. doi:10.1158/1078-0432.CCR-15-1879
181. Khammari A, Nguyen JM, Leccia MT, et al. Tumor infiltrating lymphocytes as adjuvant treatment in stage III melanoma patients with only one invaded lymph node after complete resection: results from a multicentre, randomized clinical Phase III trial. Cancer Immunol Immunother. 2020. doi:10.1007/s00262-020-02572-1
182. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. 2018;18(2):121–133. doi:10.1038/nri.2017.118
183. Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445–474. doi:10.1146/annurev-immunol-032414-112043
184. Watts C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol. 2004;5(7):685–692. doi:10.1038/ni1088
185. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. 2016;37(12):855–865. doi:10.1016/j.it.2016.09.006
186. Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27(1):74–95. doi:10.1038/cr.2016.157
187. Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22(8):1897–1906. doi:10.1158/1078-0432.CCR-15-1399
188. Rodriguez Perez A, Campillo-Davo D, Van Tendeloo VFI, Benitez-Ribas D. Cellular immunotherapy: a clinical state-of-the-art of a new paradigm for cancer treatment. Clin Transl Oncol. 2020. doi:10.1007/s12094-020-02344-4
189. Jones A, Bourque J, Kuehm L, et al. Immunomodulatory functions of BTLA and HVEM govern induction of extrathymic regulatory T cells and tolerance by dendritic cells. Immunity. 2016;45(5):1066–1077. doi:10.1016/j.immuni.2016.10.008
190. Morel Y, Truneh A, Sweet RW, Olive D, Costello RT. The TNF superfamily members LIGHT and CD154 (CD40 ligand) costimulate induction of dendritic cell maturation and elicit specific CTL activity. J Immunol. 2001;167(5):2479–2486. doi:10.4049/jimmunol.167.5.2479
191. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. doi:10.1084/jem.174.3.561
192. Nolte MA, van Olffen RW, van Gisbergen KP, van Lier RA. Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol Rev. 2009;229(1):216–231. doi:10.1111/j.1600-065X.2009.00774.x
193. Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005;6(3):280–286. doi:10.1038/ni1165
194. van Oosterwijk MF, Juwana H, Arens R, et al. CD27-CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int Immunol. 2007;19(6):713–718. doi:10.1093/intimm/dxm033
195. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi:10.1016/j.immuni.2006.01.001
196. Bourque J, Hawiger D. Immunomodulatory bonds of the partnership between dendritic cells and t cells. Crit Rev Immunol. 2018;38(5):379–401. doi:10.1615/CritRevImmunol.2018026790
197. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40(4):310–327. doi:10.1016/j.it.2019.02.003
198. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. doi:10.1038/nrclinonc.2016.217
199. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev. 2017;114:206–221. doi:10.1016/j.addr.2017.04.010
200. Dancsok AR, Gao D, Lee AF, et al. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology. 2020;9(1):1747340. doi:10.1080/2162402X.2020.1747340
201. Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79:541–566. doi:10.1146/annurev-physiol-022516-034339
202. Brown JM, Recht L, Strober S. The promise of targeting macrophages in cancer therapy. Clin Cancer Res. 2017;23(13):3241–3250. doi:10.1158/1078-0432.CCR-16-3122
203. Cheng L, Wang Y, Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol Ther. 2017;25(7):1665–1675. doi:10.1016/j.ymthe.2017.02.007
204. Gotwals P, Cameron S, Cipolletta D, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17(5):286–301. doi:10.1038/nrc.2017.17
205. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14(8):561–584. doi:10.1038/nrd4591
206. Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. Nanoparticle-based immunotherapy for cancer. ACS Nano. 2015;9(1):16–30. doi:10.1021/nn5062029
207. Musetti S, Huang L. Nanoparticle-mediated remodeling of the tumor microenvironment to enhance immunotherapy. ACS Nano. 2018;12(12):11740–11755. doi:10.1021/acsnano.8b05893
208. Yoon HY, Selvan ST, Yang Y, et al. Engineering nanoparticle strategies for effective cancer immunotherapy. Biomaterials. 2018;178:597–607. doi:10.1016/j.biomaterials.2018.03.036
209. Fontana F, Liu D, Hirvonen J, Santos HA. Delivery of therapeutics with nanoparticles: what’s new in cancer immunotherapy? Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(1). doi:10.1002/wnan.1421
210. Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012;17(17–18):1044–1052. doi:10.1016/j.drudis.2012.05.010
211. Zhuang J, Holay M, Park JH, Fang RH, Zhang J, Zhang L. Nanoparticle delivery of immunostimulatory agents for cancer immunotherapy. Theranostics. 2019;9(25):7826–7848. doi:10.7150/thno.37216
212. Fang RH, Zhang L. Nanoparticle-based modulation of the immune system. Annu Rev Chem Biomol Eng. 2016;7:305–326. doi:10.1146/annurev-chembioeng-080615-034446
213. Park W, Heo YJ, Han DK. New opportunities for nanoparticles in cancer immunotherapy. Biomater Res. 2018;22:24. doi:10.1186/s40824-018-0133-y
214. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012
215. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541.
216. Garg AD, Agostinis P. Cell death and immunity in cancer: from danger signals to mimicry of pathogen defense responses. Immunol Rev. 2017;280(1):126–148. doi:10.1111/imr.12574
217. Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol. 2016;28(1):22–27. doi:10.1016/j.smim.2015.11.002
218. Tran TH, Tran TTP, Nguyen HT, et al. Nanoparticles for dendritic cell-based immunotherapy. Int J Pharm. 2018;542(1–2):253–265. doi:10.1016/j.ijpharm.2018.03.029
219. Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35(2):814–824. doi:10.1016/j.biomaterials.2013.10.003
220. Zhu G, Zhang F, Ni Q, Niu G, Chen X. Efficient nanovaccine delivery in cancer immunotherapy. ACS Nano. 2017;11(3):2387–2392. doi:10.1021/acsnano.7b00978
221. Reddy ST, van der Vlies AJ, Simeoni E, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25(10):1159–1164. doi:10.1038/nbt1332
222. Kang S, Ahn S, Lee J, et al. Effects of gold nanoparticle-based vaccine size on lymph node delivery and cytotoxic T-lymphocyte responses. J Control Release. 2017;256:56–67. doi:10.1016/j.jconrel.2017.04.024
223. Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Control Release. 2006;112(1):26–34. doi:10.1016/j.jconrel.2006.01.006
224. Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38(5):1404–1413. doi:10.1002/eji.200737984
225. Shekarian T, Valsesia-Wittmann S, Brody J, et al. Pattern recognition receptors: immune targets to enhance cancer immunotherapy. Ann Oncol. 2017;28(8):1756–1766. doi:10.1093/annonc/mdx179
226. Yang R, Xu J, Xu L, et al. Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano. 2018;12(6):5121–5129. doi:10.1021/acsnano.7b09041
227. Zhou Z, Mandal SS, Liao G, Guo J, Guo Z. Synthesis and evaluation of GM2-monophosphoryl lipid A conjugate as a fully synthetic self-adjuvant cancer vaccine. Sci Rep. 2017;7(1):11403. doi:10.1038/s41598-017-11500-w
228. McAleer JP, Zammit DJ, Lefrancois L, Rossi RJ, Vella AT. The lipopolysaccharide adjuvant effect on T cells relies on nonoverlapping contributions from the MyD88 pathway and CD11c+ cells. J Immunol. 2007;179(10):6524–6535. doi:10.4049/jimmunol.179.10.6524
229. Hanson MC, Crespo MP, Abraham W, et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125(6):2532–2546. doi:10.1172/JCI79915
230. Jeanbart L, Swartz MA. Engineering opportunities in cancer immunotherapy. Proc Natl Acad Sci U S A. 2015;112(47):14467–14472. doi:10.1073/pnas.1508516112
231. Overchuk M, Zheng G. Overcoming obstacles in the tumor microenvironment: recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials. 2018;156:217–237. doi:10.1016/j.biomaterials.2017.10.024
232. Ou W, Thapa RK, Jiang L, et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J Control Release. 2018;281:84–96. doi:10.1016/j.jconrel.2018.05.018
233. Xu Z, Wang Y, Zhang L, Huang L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8(4):3636–3645. doi:10.1021/nn500216y
234. Choi J, Kim HY, Ju EJ, et al. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials. 2012;33(16):4195–4203. doi:10.1016/j.biomaterials.2012.02.022
235. Li R, He Y, Zhu Y, et al. Route to rheumatoid arthritis by macrophage-derived microvesicle-coated nanoparticles. Nano Lett. 2019;19(1):124–134. doi:10.1021/acs.nanolett.8b03439
236. Surendran SP, Moon MJ, Park R, Jeong YY. Bioactive nanoparticles for cancer immunotherapy. Int J Mol Sci. 2018;19(12):3877. doi:10.3390/ijms19123877
237. Singh MS, Bhaskar S. Nanocarrier-based immunotherapy in cancer management and research. Immunotargets Ther. 2014;3:121–134. doi:10.2147/ITT.S62471
238. Bookstaver ML, Tsai SJ, Bromberg JS, Jewell CM. Improving vaccine and immunotherapy design using biomaterials. Trends Immunol. 2018;39(2):135–150. doi:10.1016/j.it.2017.10.002
239. Mukherjee B. Nanosize drug delivery system. Curr Pharm Biotechnol. 2013;14(15):1221. doi:10.2174/138920101415140804121008
240. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. doi:10.1016/j.jconrel.2012.01.043
241. Colzani B, Pandolfi L, Hoti A, et al. Investigation of antitumor activities of trastuzumab delivered by PLGA nanoparticles. Int J Nanomedicine. 2018;13:957–973. doi:10.2147/IJN.S152742
242. Rosalia RA, Cruz LJ, van Duikeren S, et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials. 2015;40:88–97. doi:10.1016/j.biomaterials.2014.10.053
243. Gianni L, Mansutti M, Anton A, et al. Comparing neoadjuvant nab-paclitaxel vs paclitaxel both followed by anthracycline regimens in women with ERBB2/HER2-negative breast cancer-the evaluating treatment with neoadjuvant abraxane (ETNA) trial: a randomized phase 3 clinical trial. JAMA Oncol. 2018;4(3):302–308. doi:10.1001/jamaoncol.2017.4612
244. Zhao M, Li H, Ma Y, et al. Nanoparticle abraxane possesses impaired proliferation in A549 cells due to the underexpression of glucosamine 6-phosphate N-acetyltransferase 1 (GNPNAT1/GNA1). Int J Nanomedicine. 2017;12:1685–1697. doi:10.2147/IJN.S129976
245. Bernabeu E, Cagel M, Lagomarsino E, Moretton M, Chiappetta DA. Paclitaxel: what has been done and the challenges remain ahead. Int J Pharm. 2017;526(1–2):474–495. doi:10.1016/j.ijpharm.2017.05.016
246. Zhao M, Lei C, Yang Y, et al. Abraxane, the nanoparticle formulation of paclitaxel can induce drug resistance by up-regulation of P-gp. PLoS One. 2015;10(7):e0131429. doi:10.1371/journal.pone.0131429
247. Yang L, Kuang H, Zhang W, Aguilar ZP, Wei H, Xu H. Comparisons of the biodistribution and toxicological examinations after repeated intravenous administration of silver and gold nanoparticles in mice. Sci Rep. 2017;7(1):3303. doi:10.1038/s41598-017-03015-1
248. Farokhzad OC, Langer R. Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv Drug Deliv Rev. 2006;58(14):1456–1459. doi:10.1016/j.addr.2006.09.011
249. Phillips WT, Bao A, Brenner AJ, Goins BA. Image-guided interventional therapy for cancer with radiotherapeutic nanoparticles. Adv Drug Deliv Rev. 2014;76:39–59. doi:10.1016/j.addr.2014.07.001
250. Hu J, Dong Y, Ding L, et al. Local delivery of arsenic trioxide nanoparticles for hepatocellular carcinoma treatment. Signal Transduct Target Ther. 2019;4:28. doi:10.1038/s41392-019-0062-9
251. Arnon TI, Markel G, Mandelboim O. Tumor and viral recognition by natural killer cells receptors. Semin Cancer Biol. 2006;16(5):348–358. doi:10.1016/j.semcancer.2006.07.005
252. Sivori S, Falco M, Della Chiesa M, et al. CpG and double-stranded RNA trigger human NK cells by Toll-like receptors: induction of cytokine release and cytotoxicity against tumors and dendritic cells. Proc Natl Acad Sci U S A. 2004;101(27):10116–10121. doi:10.1073/pnas.0403744101
253. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi:10.1038/nrc3245
254. Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18(5):313–322. doi:10.1038/nrc.2018.6
255. Chen DB, Zhao YJ, Wang XY, et al. Regulatory factor X5 promotes hepatocellular carcinoma progression by transactivating tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein theta and suppressing apoptosis. Chin Med J (Engl). 2019;132(13):1572–1581. doi:10.1097/CM9.0000000000000296
256. Xue X, Lindstrom A, Qu H, Li Y. Recent advances on small-molecule nanomedicines for cancer treatment. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2020;12(3):e1607. doi:10.1002/wnan.1607
257. Peng F, Setyawati MI, Tee JK, et al. Nanoparticles promote in vivo breast cancer cell intravasation and extravasation by inducing endothelial leakiness. Nat Nanotechnol. 2019;14(3):279–286. doi:10.1038/s41565-018-0356-z
258. Setyawati MI, Tay CY, Bay BH, Leong DT. Gold nanoparticles induced endothelial leakiness depends on particle size and endothelial cell origin. ACS Nano. 2017;11(5):5020–5030. doi:10.1021/acsnano.7b01744
259. Tay CY, Setyawati MI, Leong DT. Nanoparticle density: a critical biophysical regulator of endothelial permeability. ACS Nano. 2017;11(3):2764–2772. doi:10.1021/acsnano.6b07806
260. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi:10.1038/nm1469
261. Schroeder A, Heller DA, Winslow MM, et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2011;12(1):39–50. doi:10.1038/nrc3180
262. Anderberg C, Cunha SI, Zhai Z, et al. Deficiency for endoglin in tumor vasculature weakens the endothelial barrier to metastatic dissemination. J Exp Med. 2013;210(3):563–579. doi:10.1084/jem.20120662
263. Thomas A, Teicher BA, Hassan R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016;17(6):e254–e262.
264. Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48(3):417–433. doi:10.1016/j.immuni.2018.03.007
265. Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat Commun. 2018;9(1):1074. doi:10.1038/s41467-018-03473-9
266. Chu KF, Dupuy DE. Thermal ablation of tumours: biological mechanisms and advances in therapy. Nat Rev Cancer. 2014;14(3):199–208. doi:10.1038/nrc3672
267. Hwang S, Nam J, Jung S, Song J, Doh H, Kim S. Gold nanoparticle-mediated photothermal therapy: current status and future perspective. Nanomedicine (Lond). 2014;9(13):2003–2022. doi:10.2217/nnm.14.147
268. Jaque D, Martinez Maestro L, Del Rosal B, et al. Nanoparticles for photothermal therapies. Nanoscale. 2014;6(16):9494–9530. doi:10.1039/C4NR00708E
269. Yu M, Guo F, Tan F, Li N. Dual-targeting nanocarrier system based on thermosensitive liposomes and gold nanorods for cancer thermo-chemotherapy. J Control Release. 2015;215:91–100. doi:10.1016/j.jconrel.2015.08.003
270. Wang D, Niu L, Qiao ZY, et al. Synthesis of self-assembled porphyrin nanoparticle photosensitizers. ACS Nano. 2018;12(4):3796–3803. doi:10.1021/acsnano.8b01010
271. Chen WR, Singhal AK, Liu H, Nordquist RE. Antitumor immunity induced by laser immunotherapy and its adoptive transfer. Cancer Res. 2001;61(2):459–461.
272. Doughty ACV, Hoover AR, Layton E, Murray CK, Howard EW, Chen WR. Nanomaterial applications in photothermal therapy for cancer. Materials (Basel). 2019;12(5):779. doi:10.3390/ma12050779
273. Pissuwan D, Valenzuela SM, Cortie MB. Therapeutic possibilities of plasmonically heated gold nanoparticles. Trends Biotechnol. 2006;24(2):62–67. doi:10.1016/j.tibtech.2005.12.004
274. Dreaden EC, Mackey MA, Huang X, Kang B, El-Sayed MA. Beating cancer in multiple ways using nanogold. Chem Soc Rev. 2011;40(7):3391–3404. doi:10.1039/c0cs00180e
275. Jain PK, Huang X, El-Sayed IH, El-Sayed MA. Noble metals on the nanoscale: optical and photothermal properties and some applications in imaging, sensing, biology, and medicine. Acc Chem Res. 2008;41(12):1578–1586. doi:10.1021/ar7002804
276. Liu Y, Crawford BM, Vo-Dinh T. Gold nanoparticles-mediated photothermal therapy and immunotherapy. Immunotherapy. 2018;10(13):1175–1188. doi:10.2217/imt-2018-0029
277. Riley RS, Day ES. Gold nanoparticle-mediated photothermal therapy: applications and opportunities for multimodal cancer treatment. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(4):e1449. doi:10.1002/wnan.1449
278. Almeida JP, Figueroa ER, Drezek RA. Gold nanoparticle mediated cancer immunotherapy. Nanomedicine. 2014;10(3):503–514. doi:10.1016/j.nano.2013.09.011
279. Chen YW, Su YL, Hu SH, Chen SY. Functionalized graphene nanocomposites for enhancing photothermal therapy in tumor treatment. Adv Drug Deliv Rev. 2016;105(Pt B):190–204. doi:10.1016/j.addr.2016.05.022
280. Akinoglu EM, Ozbilgin K, Sonmez PK, et al. Biocompatibility of vertically aligned multi-walled carbon nanotube scaffolds for human breast cancer cell line MDA-MB-231. Prog Biomater. 2017;6(4):189–196. doi:10.1007/s40204-017-0078-6
281. Wang J, Wu X, Shen P, et al. Applications of inorganic nanomaterials in photothermal therapy based on combinational cancer treatment. Int J Nanomedicine. 2020;15:1903–1914. doi:10.2147/IJN.S239751
282. Khafaji M, Zamani M, Golizadeh M, Bavi O. Inorganic nanomaterials for chemo/photothermal therapy: a promising horizon on effective cancer treatment. Biophys Rev. 2019;11(3):335–352. doi:10.1007/s12551-019-00532-3
283. Yu J, Javier D, Yaseen MA, et al. Self-assembly synthesis, tumor cell targeting, and photothermal capabilities of antibody-coated indocyanine green nanocapsules. J Am Chem Soc. 2010;132(6):1929–1938. doi:10.1021/ja908139y
284. Yang J, Choi J, Bang D, et al. Convertible organic nanoparticles for near-infrared photothermal ablation of cancer cells. Angew Chem Int Ed Engl. 2011;50(2):441–444. doi:10.1002/anie.201005075
285. Vines JB, Lim DJ, Park H. Contemporary polymer-based nanoparticle systems for photothermal therapy. Polymers (Basel). 2018;10(12):1357. doi:10.3390/polym10121357
286. Le QV, Suh J, Choi JJ, et al. In situ nanoadjuvant-assembled tumor vaccine for preventing long-term recurrence. ACS Nano. 2019;13(7):7442–7462. doi:10.1021/acsnano.9b02071
287. Banchereau J, Palucka K. Immunotherapy: cancer vaccines on the move. Nat Rev Clin Oncol. 2018;15(1):9–10. doi:10.1038/nrclinonc.2017.149
288. Liu WL, Zou MZ, Liu T, et al. Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells. Nat Commun. 2019;10(1):3199. doi:10.1038/s41467-019-11157-1
289. Tannock IF. Conventional cancer therapy: promise broken or promise delayed? Lancet. 1998;351 Suppl 2:SII9–16. doi:10.1016/S0140-6736(98)90327-0
290. Luo L, Zhu C, Yin H, et al. Laser immunotherapy in combination with perdurable PD-1 blocking for treatment of metastatic tumor. ACS Nano. 2018;12:7647–7662. doi:10.1021/acsnano.8b00204
291. Yuba E, Uesugi S, Yoshizaki Y, Harada A, Kono K. Potentiation of cancer immunity-inducing effect by pH-sensitive polysaccharide-modified liposomes with combination of TGF-β type I receptor inhibitor-embedded liposomes. Med Res Arch. 2017;5:1–16. doi:10.18103/mra.v5i5.1243
292. Yuba E, Yamaguchi A, Yoshizaki Y, Harada A, Kono K. Bioactive polysaccharide-based pH-sensitive polymers for cytoplasmic delivery of antigen and activation of antigen-specific immunity. Biomaterials. 2017;120:32–45. doi:10.1016/j.biomaterials.2016.12.021
293. Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal delivery enhances immune activation by STING agonists for cancer immunotherapy. Adv Biosyst. 2017;1:1600013. doi:10.1002/adbi.201600013
294. Yuba E, Sakaguchi N, Kanda Y, Miyazaki M, Koiwai K. pH-responsive micelle-based cytoplasmic delivery system for induction of cellular immunity. Vaccines. 2017;5:41. doi:10.3390/vaccines5040041
295. Peng J, Xiao Y, Li W, et al. Photosensitizer micelles together with IDO inhibitor enhance cancer photothermal therapy and immunotherapy. Adv Sci. 2018;5:1700891. doi:10.1002/advs.201700891
296. Li H, Li Y, Wang X, et al. Rational design of polymeric hybrid micelles to overcome lymphatic and intracellular delivery barriers in cancer immunotherapy. Theranostics. 2017;7:4383–4398. doi:10.7150/thno.20745
297. Lin AY, Almeida JP, Bear A, et al. Gold nanoparticle delivery of modified CpG stimulates macrophages and inhibits tumor growth for enhanced immunotherapy. PLoS One. 2013;8:e63550. doi:10.1371/journal.pone.0063550
298. Meir R, Shamalov K, Sadan T, et al. Fast image-guided stratification using anti-programmed death ligand 1 gold nanoparticles for cancer immunotherapy. ACS Nano. 2017;11:11127–11134. doi:10.1021/acsnano.7b05299
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.