Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 9
Paraneoplastic pemphigus: a short review
Authors Wieczorek M, Czernik A
Received 6 July 2016
Accepted for publication 30 August 2016
Published 23 September 2016 Volume 2016:9 Pages 291—295
DOI https://doi.org/10.2147/CCID.S100802
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Marta Wieczorek,1 Annette Czernik2
1Clinical Department of Dermatology, Central Clinical Hospital of the Ministry of the Interior and Administration, Warsaw, Poland; 2Department of Dermatology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Abstract: Paraneoplastic pemphigus (PNP) is a fatal autoimmune blistering disease associated with an underlying malignancy. It is a newly recognized blistering disease, which was first recognized in 1990 by Dr Anhalt who described an atypical pemphigus with associated neoplasia. In 2001, Nguyen proposed the term paraneoplastic autoimmune multiorgan syndrome because of the recognition that the condition affects multiple organ systems. PNP presents most frequently between 45 and 70 years old, but it also occurs in children and adolescents. A wide variety of lesions (florid oral mucosal lesions, a generalized polymorphous cutaneous eruption, and pulmonary involvement) may occur in patients with PNP. The earliest and most consistent finding is severe stomatitis. There is a spectrum of at least five clinical variants with different morphology. Similarly, the histological findings are very variable. Investigations to diagnose PNP should include checking for systemic complications (to identify tumor), skin biopsies (for histopathological and immunofluorescence studies), and serum immunological studies. PNP is characterized by the presence of autoantibodies against antigens such as desmoplakin I (250 kD), bullous pemphigoid aniygen I (230 kD), desmoplakin II (210 kD), envoplakin (210 kD), periplakin (190 kD), plectin (500 kD), and a 170 kD protein. Unlike other forms of pemphigus, PNP can affect other types of epithelia, such as gastrointestinal and respiratory tract. Treatment of PNP is difficult, and the best outcomes have been reported with benign neoplasms that have been surgically excised. The first-line treatment is high-dose corticosteroids with the addition of steroid-sparing agents. Treatment failures are often managed with rituximab with or without concomitant intravenous immunoglobulin. In general, the prognosis is poor, not only because of eventual progression of malignant tumors but also because treatment with aggressive immunosuppression therapy often results in infectious complications, which is unfortunately at this time the most common cause of death in PNP.
Keywords: paraneoplastic pemphigus, direct and indirect immunofluorescence, mucositis, rituximab
Introduction
Paraneoplastic pemphigus (PNP) is a rare and often fatal autoimmune blistering disease accompanied by both benign and malignant neoplasms. The most frequently reported associated malignancies are lymphomatoid and hematologic, eg, B-cell lymphoma, chronic lymphocytic leukemia, Castleman’s disease, Waldenstrom’s macroglobulinemia, and thymoma (with or without myasthenia gravis). Interactions between the immune system and concomitant neoplasm seem to be the basis of pathogenesis, with autoantibodies directed against both desmosomal and hemidesmosomal antigens. In PNP, the vast majority of patients have autoantibodies to periplakins and envoplakins.
Prognosis depends on the associated tumor. Some patients experience rapid improvement after excision of a benign tumor, such as Castleman’s disease. However, malignant tumors are frequently accompanied not only by higher mortality from the associated malignancy but also because the PNP can be quite severe and unresponsive to treatment.
PNP is a rare disease whose incidence is not fully recognized. In 1990, Anhalt et al1 first described five cases of patients with a rare form of atypical pemphigus that were all associated with lymphoproliferative diseases. Of 100,000 cases of non-Hodgkin’s lymphoma and chronic lymphocytic leukemia reported to the US Food and Drug Administration, a total of 12 were found to be complicated by PNP.2 PNP mostly affects adults between 45 and 70 years of age, but it may also be found in children, particularly when associated with Castleman disease. There is no known correlation between incidence of the disease and specific sex, race, or place of origin.3
Pathogenesis
Etiopathogenesis of PNP is not fully known.4 Skin lesions are thought to be caused by an autoimmune response generated by antibodies to tumor antigens that cross-react with epithelial antigens. Tumor autoantibodies produce and release cytokines (such as interleukin-6) that favor the differentiation of B-cells5 and foster the development of the humoral branch of the immune system.
PNP is often a harbinger of benign and malignant neoplasms, most commonly malignancies of the lymphatic system. On the basis of the review of 163 cases of PNP examined between 1990 and 2003, Table 1 lists the most frequent concomitant cancers.6 Ohzono et al7 described the associated tumors in 104 PNP cases over a period of 16 years (between January 1997 and April 2013). Their clinical and histopathological findings were generally similar to those in previous reports.7
 | Table 1 Relative frequencies of concomitant neoplasms Note: aLiposarcoma, leiomyosarcoma, reticulum cell sarcoma, malignant nerve sheath tumor. |
Some patients have tumors that are difficult to define, such as follicular dendritic cell sarcomas located in the retroperitoneal space.8 Studies of patients with non-Hodgkin lymphoma revealed that most severe lesions in the course of the PNP occur 2–3 years after diagnosis of lymphoma.6 Castleman disease also known as giant lymph node hyperplasia occurs most commonly in children. When it occurs in localized form, it can be simply treated with surgical resection.
There is no consensus regarding the diagnostic criteria for PNP. The first set of criteria were made by Anhalt et al1,5,8,9 and include the following:
- Characteristic clinical appearance and histopathology.
- Detection of tissue bound, circulating autoantibodies via direct immunofluorescence, indirect immunofluorescence (IIF), and immunoprecipitation studies.
To expand on this later, Anhalt1,5,8,9 further developed defining characteristics of PNP such as painful inflammation of the oral mucosa, a polymorphous skin eruption with corresponding histologic findings often showing lichenoid or acantholytic changes, supportive immunofluorescence findings showing intercellular and basement membrane binding, serum antibodies that bind simple, columnar, and transitional epithelium, coexistence of lymphoproliferative disorders, and the presence of anti-dsg, desmoplakin I and II, envoplakin, periplakin, bullous pemphigoid antigen 1, and plectin antibodies.9
Histology
The diagnosis is made based on clinical, histological, and immunofluorescent findings. Often diagnosis of the disease necessitates multiple biopsies.10 Histology varies depending on the type of skin lesions. Acantholysis occurs over the basal layer in blisters. Additionally, there might be vacuolar degeneration of the basal layer associated with band-like infiltrate of lymphocytes in the dermis as one would typically see in lesions that are clinically and histopathologically lichenoid.
Immunology
Immunopathology plays an important role in the diagnosis of PNP. In direct immunofluorescence examination, intercellular deposits of IgG and C3 are ascertained. Unlike pemphigus vulgaris, linear C3 deposits and, to a lesser extent, IgG can be observed at epidermal–dermal junction. This is likely attributed to binding of BP180 and BP230. In fact, IIF and enzyme-linked immunosorbent assay (ELISA) testing found about 40% of patients had circulating autoantibodies to BP180, and these patients were more likely to have skin lesions involving tense bullae as seen in bullous pemphigoid.11 IIF shows that IgG antibodies bind with the stratified epithelium in the esophagus and other tissues of monkeys. In sharp contrast to pemphigus vulgaris and foliaceus, they also bind with the transitional and cylindrical epithelium of the urinary bladder, bronchi, small intestine, and colon, as well as, to a lesser extent, with myocardium and skeletal muscles and thyroid epithelium.
Previously, the highest sensitivity (75%) and specificity were exhibited by rat bladder immunofluorescence testing. Immunoprecipitation and immunoblotting can demonstrate antibodies to all desmosomal proteins: desmoglein 3 (130 kD), desmoplakin 1 (250 kD), BP230, desmoplakin 2 (210 kD), envoplakin (210 kD), plectin (>400 kD), periplakin (190 kD), epiplakin,12 and occasionally desmoglein 1 (160 kD). Also, studies using novel mammalia ELISAs showed that PNP sera reacted with desmocollins 1–3 in various patterns.13 Fluorescence on bladder epithelium (transitional) reflects the binding with intracellular proteins. ELISA may show autoantibodies to desmoglein 3 and, less often, desmoglein 1.14 New, commercially available ELISA testing to envoplakin may increase the sensitivity and specificity of serologic testing to surpass that of current immunofluorescence testing. Using immunoprecipitation and mass spectrometry, the 170 kD antigen target has been identified as a protease inhibitor, α-2 macroglobulin-like protein, a broad range protease inhibitor expressed in stratified epithelia and other tissues damaged in PNP.15
Clinical
Typically, the first symptoms include severe painful oral erosions, frequently hemorrhagic, which spread to involve the entire vermilion and tongue (Figure 1). The lesions are polymorphic, and symptoms such as blisters, erosions, spots, papules, and plaques can occur. These clinical findings may be accompanied by a positive Nikolsky sign. Cutaneous lesions usually appear subsequent to the onset of mucosal lesions and may involve any site, but the upper body is most usually involved.
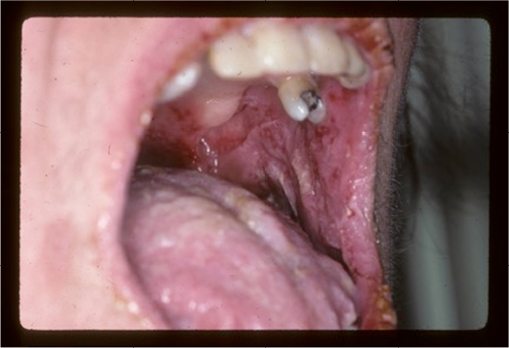 | Figure 1 Oral manifestations of paraneoplastic pemphigus. Massive erosions of the oral mucosa. |
Symptoms can be classified into several groups according to the types of alterations:
- Pemphigus-like: superficial vesicles, flaccid vesicles, erosions, crust, and erythema.
- Bullous pemphigoid-like: scaly erythematous papules, which may or may not be associated with stretched vesicles.
- Erythema multiform-like: polymorphic alterations, mainly erythematous peeling pellets with erosions and sometimes even with hard-to-heal ulcerations.
- Graft-versus-host disease: disseminated dusky red scaly papules.
- Lichen planus-like: small flat scaly papules and intense mucous membrane involvement.
PNP may not only involve the buccal mucosa but also the mucous membranes of the esophagus, stomach, duodenum, and intestines and also the pulmonary epithelium.2,3
Patients with PNP can develop life-threatening restrictive bronchiolitis consistent with bronchiolitis obliterans. The frequency of the involvement of the respiratory system and pathological mechanisms are not known. In a study of 17 patients with PNP, restrictive bronchiolitis was found only in three patients. However, in another analysis where 28 patients with PNP and concomitant Castleman’s disease were examined, the respiratory system was affected in 26 cases.16,17 Pulmonary disease, when present, is irreversible despite aggressive therapy.2,3,18 The recently discovered autoantigen, epiplakin, has demonstrated correlation with development of bronchiolitis obliterans in Japanese patients.12 Epiplakin is present in the respiratory bronchiole, and mice injected with epiplakin autoantibody showed abnormal changes in the histopathology of their pulmonary epithelia. While more research is needed, these early results indicate that epiplakin may represent a specific autoantigen in PNP-related bronchiolitis obliterans.
The differential diagnosis includes pemphigus vulgaris, mucous membrane pemphigoid, erythema multiforme, Stevens–Johnson syndrome, lichen planus, graft-versus-host disease, and herpes simplex virus infection. When PNP is suspected in a patient with no know history of malignancy, an extensive baseline workup should be conducted, including: blood cell count, lactate dehydrogenase, flow cytometry, as well as computed tomography of the chest, abdomen, and pelvis. In up to a third of patients with PNP, discovery of the underlying malignancy occurred subsequent the onset of PNP symptoms.
Treatment
It is vital to define and treat the associated cancer in PNP. In patients with an operable malignancy, a surgical cure is often the best chance of inducing remission of PNP.19
Concurrent to a thorough medical workup in patients without operable malignancies, several nonsurgical treatments have proven effective in reducing symptoms in patients with PNP. Initially, glucocorticosteroid therapy should be implemented – prednisone (0.5–1.0 mg/kg).20 Cutaneous lesions tend to crust over and heal faster than alterations in the mucosa. Steroid-sparing agents can be added to glucocorticoid therapy to reduce the total steroid burden. Immunosuppressants such as cyclosporin, cyclophosphamide, azathioprine, and mycophenolate mofetil are often used in combination with prednisone.21 However, because of the tumorgenic properties of these agents, often new alternative therapies are being used particularly in patients whose malignancy is in remission.
Rituximab – a chimeric anti-CD20 monoclonal antibody – is being used more often in patients with PNP because it appears to be more effective than alternative steroid sparing agents with perhaps reduced tumorgenicity risk. Patients can be treated with the lymphoma protocol at a dose of 375 mg/m weekly for 4 weeks, or the rheumatologic protocol of 1 g once and repeated in 2 weeks. Additional cycles may be administered every 6–12 months depending on clinical response and recovery of the B-cell (CD-20) population. Rituximab is usually very well tolerated, but notable adverse effects of treatment include infusion reactions and an allergic reaction. Severe, life-threatening anaphylactic reactions have occurred. For this reason, rituximab is infused in a monitored setting such as an infusion center where an allergy can be rapidly identified and treated. In addition, progressive multifocal leukoencephalopathy, a fatal and untreatable reactivation of JC virus in the brain, has been reported in association with rituximab. This reactivation occurs only in the setting of severe immunosuppression. This is of concern for PNP patients, who are often substantially immunosuppressed given the concomitant malignancy with or without associated chemotherapy. The first successful treatment of a patient with PNP was in one who had concomitant non-Hodgkin lymphoma that was being treated with rituximab.22 Like many options for PNP, rituximab also has limited effectiveness, and often treatment success depends on controlling the patients underlying malignancy.23
The concomitant use of rituximab with intravenous immunoglobulin (IVIG) has proven successful in those patients who do not respond to conventional therapy or use of rituximab alone. IVIG is dosed at 2 g/kg per cycle, and cycles are repeated on a monthly basis. IVIG is well tolerated and shown to be quite effective in reducing pathogenic autoantibodies rapidly in patients with autoimmune bullous diseases. Another benefit is that IVIG can be added into the patient’s existing treatment regimen without added concern of additional immunosuppression, making it a popular approach among clinicians who treat PNP. IVIG’s favorable safety profile makes it an obvious choice in patients who are often on complicated treatments for PNP and an underlying malignancy. However, the affordability and its inconvenience have limited its use.24
Unfortunately, PNP is often resistant to treatment. The mortality rate ranges from 75% to 90%, with the main cause of death in these patients being respiratory failure.25 Prompt diagnosis and early initiation of treatment are of paramount importance.
Disclosure
The authors report no conflicts of interest in this work.
References
Anhalt GJ, Kim SC, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med. 1990;323:1729–1735. | ||
Nguyen VT, Ndoye A, Bassler KD, et al. Classification, clinical manifestations, and immunopathological mechanisms of the epithelial variant of paraneoplastic autoimmune multiorgan syndrome: a reappraisal of paraneoplastic pemphigus. Arch Dermatol. 2001;137(2):193–206. | ||
Czernik A, Camilleri M, Pittelkow MR, Grando SA. Paraneoplastic autoimmune multiorgan syndrome: 20 years after. Int J Dermatol. 2011;50:905–914. | ||
Zimmermann J, Bahmer F, Rose C, Zillikens D, Schmidt E. Clinical and immunopathologica spectrum of paraneoplastic pemphigus. J Dtsch Dermatol Ges. 2010;8:598–606. German. | ||
Anhalt GJ, Mimouni D. Paraneoplastic pemphigus. In: Goldsmith LA, Katz SI, Gilchrest, et al, editors. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New Yory, NY: McGraw Hill; 2012;1:600. | ||
Kaplan I, Hodak E, Ackerman L, Mimouni D, Anhalt GJ, Calderon S. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol. 2004;40:553. | ||
Ohzono A, Sogame R, Li X, et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol. 2015;173:1447. | ||
Anhalt GJ. Paraneoplastic pemphigus. Adv Dermatol. 1997;12:77. | ||
Anhalt GJ. Paraneoplastic pemphigus. J Investig Dermatol Symp Proc. 2004;9:29–33. | ||
Horn TD, Anhalt GJ. Histologic features of paraneoplastic pemphigus. Arch Dermatol. 1992;128:1091–1095. | ||
Tsuchisaka A, Kawano H, Yasukochi A, et al. Immunological and statistical studies of anti-BP180 antibodies in paraneoplastic pemphigus. J Invest Dermatol. 2014;134(8):2283. | ||
Tsuchisaka A, Numata S, Teye K, et al. Epiplakin is a paraneoplastic pemphigus autoantigen and related to bronchiolitis obliterans in Japanese patients. J Invest Dermatol. 2016;136(2):399–408. | ||
Ishii N, Teye K, Fukuda S, et al. Anti-desmocollin autoantibodies in nonclassical pemphigus. Br J Dermatol. 2015;173(1):59–68. | ||
Hashimoto T. Production of numerous autoantibodies in paraneoplastic pemphigus. Br J Dermatol. 2015;172(4):849–850. | ||
Schepens I, Jaunin F, Begre N, et al. The protease inhibitor alpha-2-macroglobulin-like-1 is the p170 antigen recognized by paraneoplastic pemphigus autoantibodies in human. PLoS One. 2010;5:e12250. | ||
Maldonado F, Pittelkow MR, Ryu JH. Constrictive bronchiolitis associated with paraneoplastic autoimmune multiorgan syndrome. Respirology. 2009;14:129–133. | ||
Nikolskaia OV, Nousari CH, Anhalt GJ. Paraneoplastic pemphigus in association with Castleman’s disease. Br J Dermatol. 2003;149:1143. | ||
Fullerton SH, Woodley DT, Smoller BR, Anhalt GJ. Paraneoplastic pemphigus with autoantibody deposition in bronchial epithelium after autologous bone marrow transplantation. JAMA. 1992;267:1500–1502. | ||
Fang Y, Zhao L, Yan F, Cui X, Xia Y, Duren A, et al. A critical role of surgery in the treatment for paraneoplastic pemphigus caused by localized Castleman’s disease. Med Oncol. 2010;27:907. | ||
Sehgal VN, Srivastava G. Paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome. Int J Dermatol. 2009;48:162. | ||
Hertzberg MS, Schifter M, Sullivan J, Stapleton K. Paraneoplastic pemphigus in two patients with B-cell non-Hodgkin’s lymphoma: significant responses to cyclophosphamide and prednisolone. Am J Hematol. 2000;63:105. | ||
Heizmann M, Itin P, Wernli M, Borradori L, Bargetzi MJ. Successful treatment of paraneoplastic pemphigus in follicular NHL with rituximab: report of a case and review of treatment for paraneoplastic pemphigus in NHL and CLL. Am J Hematol. 2001;66:142–144. | ||
Anan T, Shimizu F, Hatano Y, Okamoto O, Katagiri K, Fujiwara S. Paraneoplastic pemphigus associated with corneal perforation and cutaneous alternariosis: a case report and review of cases treated with rituximab. J Dermatol. 2011;38:1084. | ||
Zhu X, Zhang B. Paraneoplastic pemphigus. J Dermatol. 2007;34:503. | ||
Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol. 2012;148:1165–1172. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.