Back to Journals » Drug Design, Development and Therapy » Volume 9
Paclitaxel attenuates renal interstitial fibroblast activation and interstitial fibrosis by inhibiting STAT3 signaling
Authors Zhang L, Xu X, Yang R, Chen J, Wang S, Yang J, Xiang X, He Z, Zhao Y, Dong Z, Zhang D
Received 22 January 2015
Accepted for publication 27 February 2015
Published 15 April 2015 Volume 2015:9 Pages 2139—2148
DOI https://doi.org/10.2147/DDDT.S81390
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Lei Zhang,1,2,* Xuan Xu,1,* Ruhao Yang,1,* Jingwen Chen,1 Shixuan Wang,5 Junqin Yang,3 Xudong Xiang,1 Zhibiao He,1 Yu Zhao,4 Zheng Dong,2,5 Dongshan Zhang1
1Department of Emergency Medicine, 2Department of Nephrology, 3Department of Minimally Invasive Surgery, Second Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 4Department of Nephrology, Harbin First Hospital, Harbin, Heilongjiang, People’s Republic of China; 5Department of Cellular Biology and Anatomy, Medical College of Georgia at Georgia Regents University and Charlie Norwood VA Medical Center, Augusta, GA, USA
*Co-first authors in this study
Abstract: Recent studies have demonstrated that paclitaxel might inhibit renal fibrosis. However, the underlying molecular mechanism remains unclear. In this study, we hypothesized that low-dose paclitaxel may block the STAT3 (signal transducer and activator of transcription 3) signaling to attenuate fibrosis in a mouse model with unilateral ureteral obstruction. Both NRK-49F cells and mice with unilateral ureteral obstruction were treated with paclitaxel. The results showed that paclitaxel treatment resulted in a dose- and time-dependent decrease in tyrosine-phosphorylated STAT3, and inhibited the expression of fibronectin, alpha-smooth muscle actin (α-SMA), and collagen I in cultured NRK-49F cells. S3I-201, an STAT3 inhibitor, also suppressed the expression of fibronectin, α-SMA, and collagen I in cultured NRK-49F cells. Mechanistically, paclitaxel treatment blocked the STAT3 activity by disrupting the association of STAT3 with tubulin and inhibiting STAT3 nucleus translocation. Furthermore, paclitaxel also ameliorated renal fibrosis by down-regulating the expression of fibronectin, α-SMA, and collagen I, and suppressed the infiltration of macrophages and production of TNF-α, IL-1β, TGF-β, and ICAM-1 (intercellular adhesion molecule 1) by inhibition of STAT3 activity in obstructive nephropathy. These results suggest that paclitaxel may block the STAT3 activity by disrupting the association of STAT3 with tubulin and inhibiting STAT3 nucleus translocation, consequently leading to the suppression of renal interstitial fibroblast activation and the development of renal fibrosis, and inhibition of proinflammatory cytokine production.
Keywords: UUO, tubulointerstitial fibrosis, tubulin, paclitaxel, STAT3
Introduction
Paclitaxel, one of the most important anticancer drugs, has been used in the treatment of different types of cancers. Recently, it has been found that paclitaxel could be promising in treating noncancer diseases.1 For example, Zhang et al2 reported that paclitaxel significantly suppressed tubulointerstitial fibrosis by inhibiting TGF-β (transforming growth factor-beta)/Smad signaling in a rat model of unilateral ureteral obstruction (UUO). Karbalay-Doust et al3 also found that paclitaxel was more effective than taurine in suppressing renal fibrosis in the UUO model. Furthermore, paclitaxel showed an antifibrosis role by blocking TGF-β/Smad/miR-192 signaling.4 However, the underlying molecular mechanism is not fully understood.
Renal interstitial fibrosis is a progressive process. The key step is the transformation of the renal fibroblasts to alpha-smooth muscle actin (α-SMA)-positive myofibroblasts in the evolution of chronic kidney disease.5,6 Signal transducer and activator of transcription 3 (STAT3) is an important member of the STAT family (STAT14, STAT5a/5b, and STAT6) and mediates cell survival and proliferation.7–9 Multiple growth factors and cytokines may activate STAT3 tyrosine phosphorylation that promotes STAT3 to form dimers and translocate to the cell nucleus to regulate the transcription of target genes.7–9 It has been reported that STAT3 activation mediates the activation of renal interstitial fibroblasts and the progression of renal fibrosis in UUO models.10,11 Interestingly, Walker et al12 reported that paclitaxel might inhibit STAT3 signaling in several tumor cell lines. In view of these findings, this study was initiated to assess whether paclitaxel can, by blocking STAT3 signaling, attenuate the activation of renal interstitial fibroblasts and the progression of renal fibrosis.
Materials and methods
Reagents and antibodies
S3I-201 was purchased from Calbiochem (La Jolla, CA, USA). Antibodies were obtained from different sources: anti-GAPDH, anti-α-SMA, anticollagen I, and antifibronectin from Santa Cruz Biotechnology (Santa Cruz, CA, USA), antimacrophage and Gr-1 from Abcam (Cambridge, MA, USA), anti-α-tubulin from Sigma-Aldrich (St Louis, MO, USA), anti-STAT3 and anti-p-STAT3 from Cell Signaling Technology (Danvers, MA, USA). The kit for protein isolation of cytoplasm and nucleus was purchased from NobleRyde (Beijing, People’s Republic of China). All secondary antibodies were from Thermo Fisher Scientific (Waltham, MA, USA).
Cell culture and treatments
NRK-49F cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum, 0.5% penicillin, and streptomycin in an atmosphere of 5% CO2 and 95% air at 37°C.
Care and use of laboratory animals
Animal experiments were performed in accordance with the regulations set by the Institutional Committee for the Care and Use of Laboratory Animals of Second Xiangya Hospital, People’s Republic of China, and approved by local authorities. C57BL/6 mice were housed on a 12-hour light/dark cycle and were allowed free access to food and water.
Animal model
The UUO model was established in male C57 black mice that weighed 20–25 g (Shanghai animal center, Shanghai, People’s Republic of China), as previously described.2 Four groups of mice comprising eight animals each (total 32) were divided as follows: 1) Sham group, 2) Sham with paclitaxel (Taxol; Sigma-Aldrich) group, 3) UUO group, and 4) UUO with paclitaxel group. Paclitaxel was injected intraperitoneally at a dose of 0.3 mg/kg twice a week. The control group was administered with saline. Mice were sacrificed on day 7 after UUO or sham operation. The kidneys were harvested for various biochemical and morphological studies.
Immunoprecipitation
The immunoprecipitation was performed as previously described.12,13 Briefly, approximately 500 μg of cellular protein was immunoprecipitated with 2 μg of antibodies to STAT3 or α-tubulin for 1 hour at 4°C, followed by the addition of 15 μL of protein A/G-agarose beads (Santa Cruz Biotechnology). The precipitates were washed four times with lysis buffer and subjected to immunoblotting for p-STAT3 and STAT3.
Histology and immunohistochemistry
For the histological analysis, kidney tissues fixed with 4% buffered paraformaldehyde were embedded in paraffin and 3 μm thick sections were prepared. The sections were then stained with hematoxylin–eosin and Masson’s trichrome methods.2 The interstitial fibrosis and interstitial gross area were separately analyzed by an image analysis software of Image-Pro Plus 6 (Media Cybernetics, Inc., Bethesda, MD, USA); then we calculated the percent positive area of renal interstitial fibrosis versus renal interstitial gross area. Immunohistochemical analyses were performed using antiphospho-STAT3, anti-α-SMA, anticollagen I, antifibronectin, and antimacrophage according to the previous protocol.2,14 The total number of positive cells for p-STAT3 (as identified by nuclear staining) and macrophage in tubulointerstitium was quantified by counting the number of stained cells per field, for p-STAT3. We collected 25–30 images of a kidney from each animal at 20× magnification. Areas of positive staining for α-SMA collagen type I and fibronectin in the entire cortical tubulointerstitium (a cross section of the kidney) were examined using quantitative software of Image-Pro Plus 6 (Media Cybernetics, Inc). Briefly, the detected area of tubulointerstitium was labeled, the positive-staining parts were discriminated, and the percent positive area in the detected tubulointerstitium was then measured.2,14
Immunoblot analysis
Immunoblot was carried out as previously described.14 Briefly, cells or kidney tissues were treated with a lysis buffer (Sigma-Aldrich) containing phosphatase inhibitors (Calbiochem). Equal amounts of proteins were loaded in each well for electrophoresis using SDS–PAGE gel, followed by transferring to polyvinylidene fluoride membranes. The blots were incubated with primary antibodies overnight at 4°C and probed by the horseradish peroxidase-conjugated secondary antibodies. Bands of target and internal control protein were separately outlined, and then gray level was analyzed by an image J software (NIH, Bethesda, MD, USA); then we calculated gray ratio of target protein versus internal control protein.
Measurement of TNF-α, IL-1β, and TGF-β
The levels of TNF-α (tumor necrosis factor-α), IL-1β (interleukin-1β), and TGF-β were determined using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems Inc, Minneapolis, MN, USA) as described previously.11,13 Renal cytokine levels were normalized by the protein concentration.
Real-time polymerase chain reaction
Real-time quantitative reverse transcriptase PCR amplifications were performed in 20 μL reactions as described previously.4 The primer sets used for various genes were as follows: ICAM-1 (intercellular adhesion molecule-1): forward 5′-CTTCCAGCTACCATCCCAAA-3′, reverse 5′-CTTCAGAGGCAGGAAACAGG-3′; GADPH: forward 5′-TGCTGAGTATGTCGTGGAGTCTA-3′, reverse 5′-AGTGGGAGTTGCTGTTG AAATC-3′.
Statistical analysis
Data were calculated as mean ± SEM (standard error of the mean). One-way ANOVA, followed by the Tukey’s post hoc test, was used to compare multiple treatment groups. Two-way ANOVA was used to assess the statistical significance of the differences between multiple treatment groups at different time points. Statistical significance was set at P<0.05 or <0.01.
Results
Paclitaxel suppresses the expression of fibronectin, α-SMA, and collagen I by inhibiting STAT3 activation in cultured renal interstitial fibroblasts
Previous results have demonstrated that STAT3 mediated the activation of renal interstitial fibroblasts, characterized by the expression of fibronectin, α-SMA, and collagen I.11,15 To further investigate whether low-dose paclitaxel suppresses the activation of renal interstitial fibroblasts by inhibiting STAT3 activation, we examined the effect of paclitaxel on STAT3 activation and expression of fibronectin, α-SMA, and collagen I in NRK-49F cells. As shown in Figure 1A–D, treatment with paclitaxel resulted in a dose- and time-dependent decrease in tyrosine-phosphorylated STAT3 in NRK-49F cells. Furthermore, the expression level of fibronectin, α-SMA, and collagen I was higher in NRK-49F cells (Figure 1E and F), indicating that the cultured renal interstitial fibroblasts had the phenotypic features of myofibroblasts. Treatment with paclitaxel also significantly decreased the expression of fibronectin, α-SMA, and collagen I in a time-dependent manner. This result suggests that paclitaxel suppresses the activation of renal interstitial fibroblasts by inhibiting STAT3 activation.
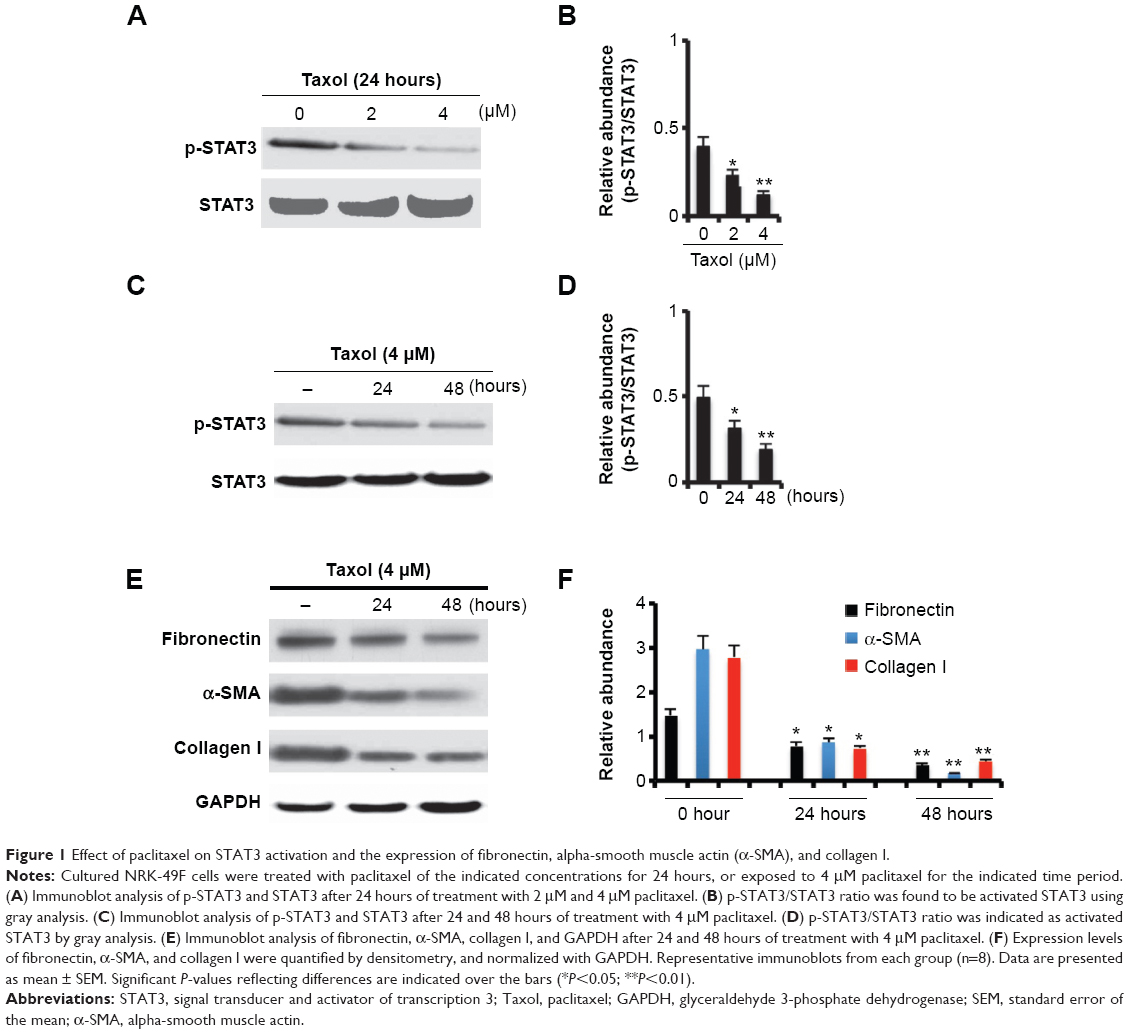 | Figure 1 Effect of paclitaxel on STAT3 activation and the expression of fibronectin, alpha-smooth muscle actin (α-SMA), and collagen I. |
Paclitaxel blocks tyrosine-phosphorylated STAT3 (Tyr705) by inhibiting the association of STAT3 with tubulin and STAT3 nucleus translocation
On the basis of the findings that STAT3 directly interacted with tubulin and that paclitaxel was a microtubule stabilization agent, we presume that paclitaxel inhibits STAT3 activation by interfering with its association with tubulin. NRK-49F cells were treated with vehicle, paclitaxel, or S3I-201, which is a novel selective inhibitor of STAT3.16 First, we confirmed that S3I-201 suppressed the expression of fibronectin, α-SMA, and collagen I by blocking STAT3 activation (Figure 2A and B). However, S3I-201 also increased the expression of p-STAT1 but did not affect that of p-STAT5 at the concentration of 50 μM (data not shown). Briefly, the cell lysate was immunoprecipitated with an anti-STAT3 antibody, followed by the immunoblot detection of STAT3 tyrosine phosphorylation (Tyr705). Both paclitaxel and S3I-201 inhibited STAT3 tyrosine phosphorylation (Tyr705) (Figure 2C and D). We further examined the effect of paclitaxel on STAT3/tubulin association by immunoprecipitation assay. The cell lysate was immunoprecipitated with an antitubulin antibody, followed by the immunoblot detection of STAT3. IgG was used as the input control. The association of STAT3 with tubulin was suppressed by paclitaxel treatment (Figure 2E and F). However, S3I-201 treatment did not affect the association between STAT3 and tubulin. Thus, paclitaxel disrupted the association of STAT3 with tubulin, independently of the effect on STAT3 phosphorylation. Because paclitaxel might inhibit the STAT3 binding to tubulin, we presume that paclitaxel might suppress STAT3 translocation. Proteins of cytoplasm and nucleus from kidneys were separately extracted using a commercial kit, followed by immunoblot. The results demonstrated that paclitaxel blocked STAT3 translocation from the cytoplasm to the nucleus (Figure 2G and H), which further supported that paclitaxel blocked STAT3 activation by inhibiting the association of STAT3 with tubulin.
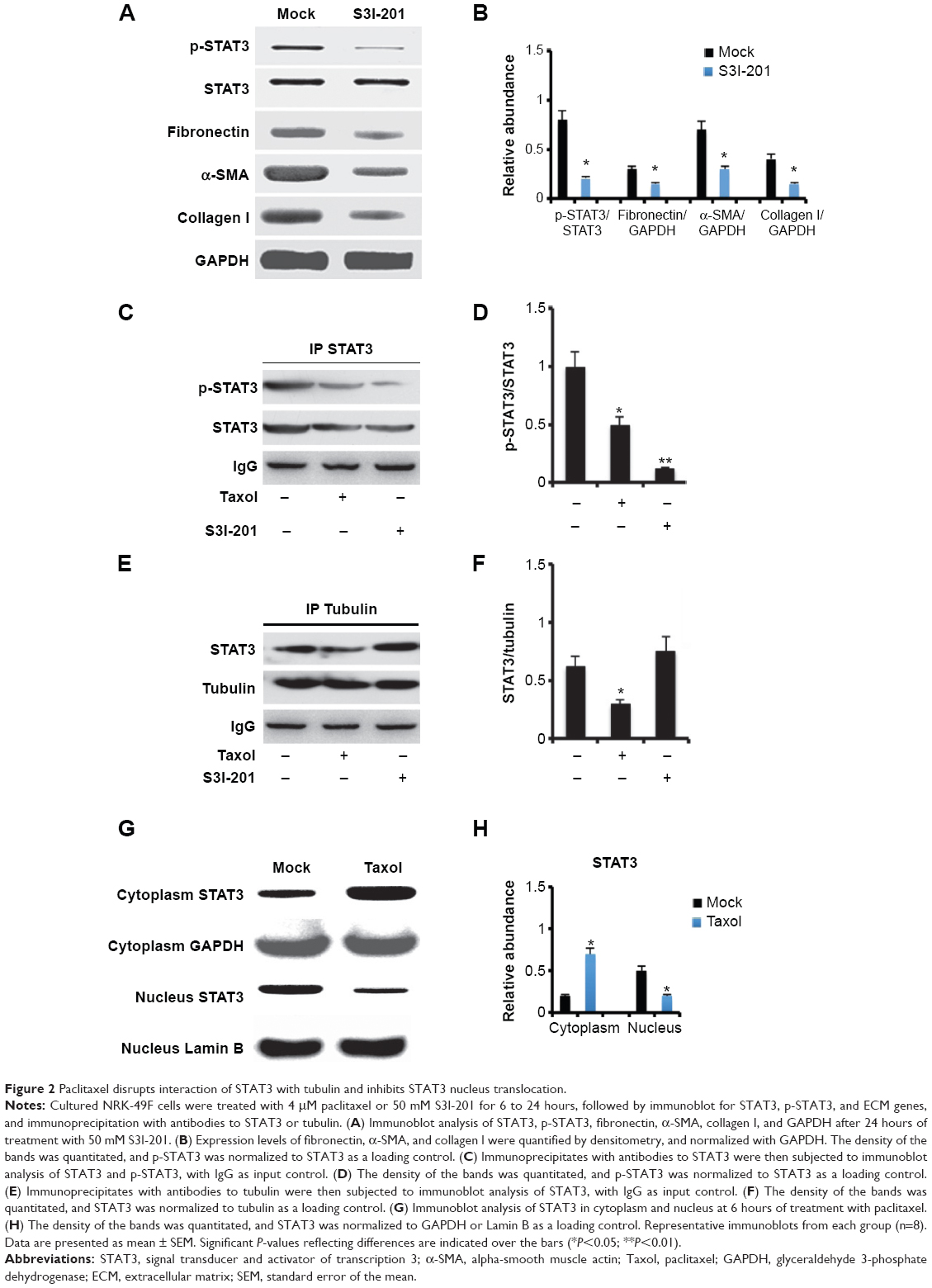 | Figure 2 Paclitaxel disrupts interaction of STAT3 with tubulin and inhibits STAT3 nucleus translocation. |
Paclitaxel attenuates tubulointerstitial fibrosis in mice obstructive nephropathy
Previous results demonstrated that paclitaxel attenuated fibrosis in a rat UUO model. In this experiment, we confirmed that paclitaxel also ameliorated tubulointerstitial fibrosis and tubular injury in a mouse UUO model. Briefly, hematoxylin-eosin staining showed that UUO animals exhibited significant tubular atrophy on day 7, which was notably reduced with paclitaxel treatment (Figure 3A). Masson’s trichrome staining demonstrated that UUO animals exhibited marked tubulointerstitial fibrosis on day 7, which was significantly reduced by paclitaxel treatment (Figure 3B and C).
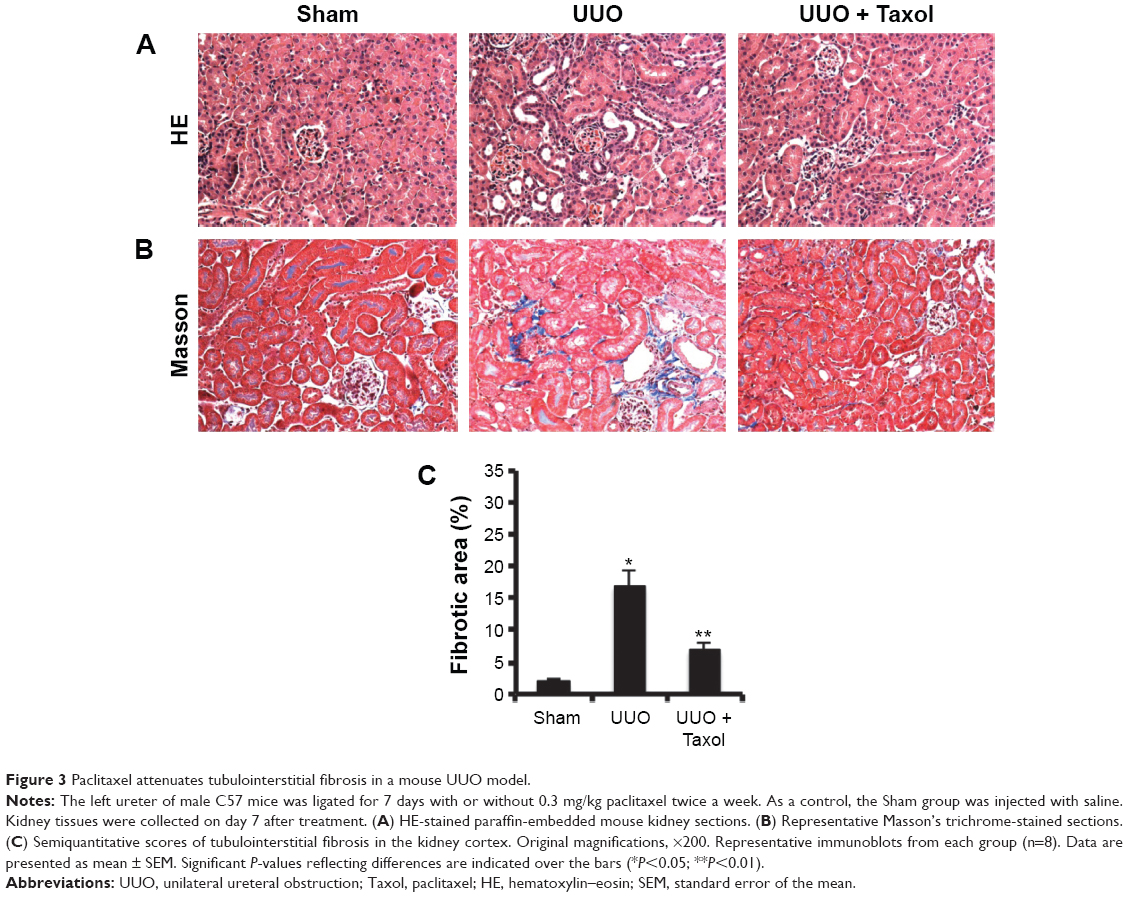 | Figure 3 Paclitaxel attenuates tubulointerstitial fibrosis in a mouse UUO model. |
Blockade of STAT3 activation is a key mechanism by which paclitaxel prevents renal fibrosis in mouse obstructive nephropathy
We have demonstrated that paclitaxel suppressed STAT3 activation in vitro. To further investigate whether paclitaxel suppresses STAT3 activation and expression of collagen I, fibronectin, and α-SMA in mouse obstructive nephropathy, we examined the effect of paclitaxel. By immunohistochemistry, we found that the expression of p-STAT3, collagen I, fibronectin, and α-SMA were markedly increased in UUO than in the Sham group on day 7, which was significantly reduced by paclitaxel treatment (Figure 4A–E). Western blot analysis indicated similar results (Figure 4F and G). These findings suggest that paclitaxel suppresses the activation of renal fibroblasts to attenuate renal fibrosis by inhibiting STAT3 activation in a mouse UUO model, being consistent with in vitro findings.
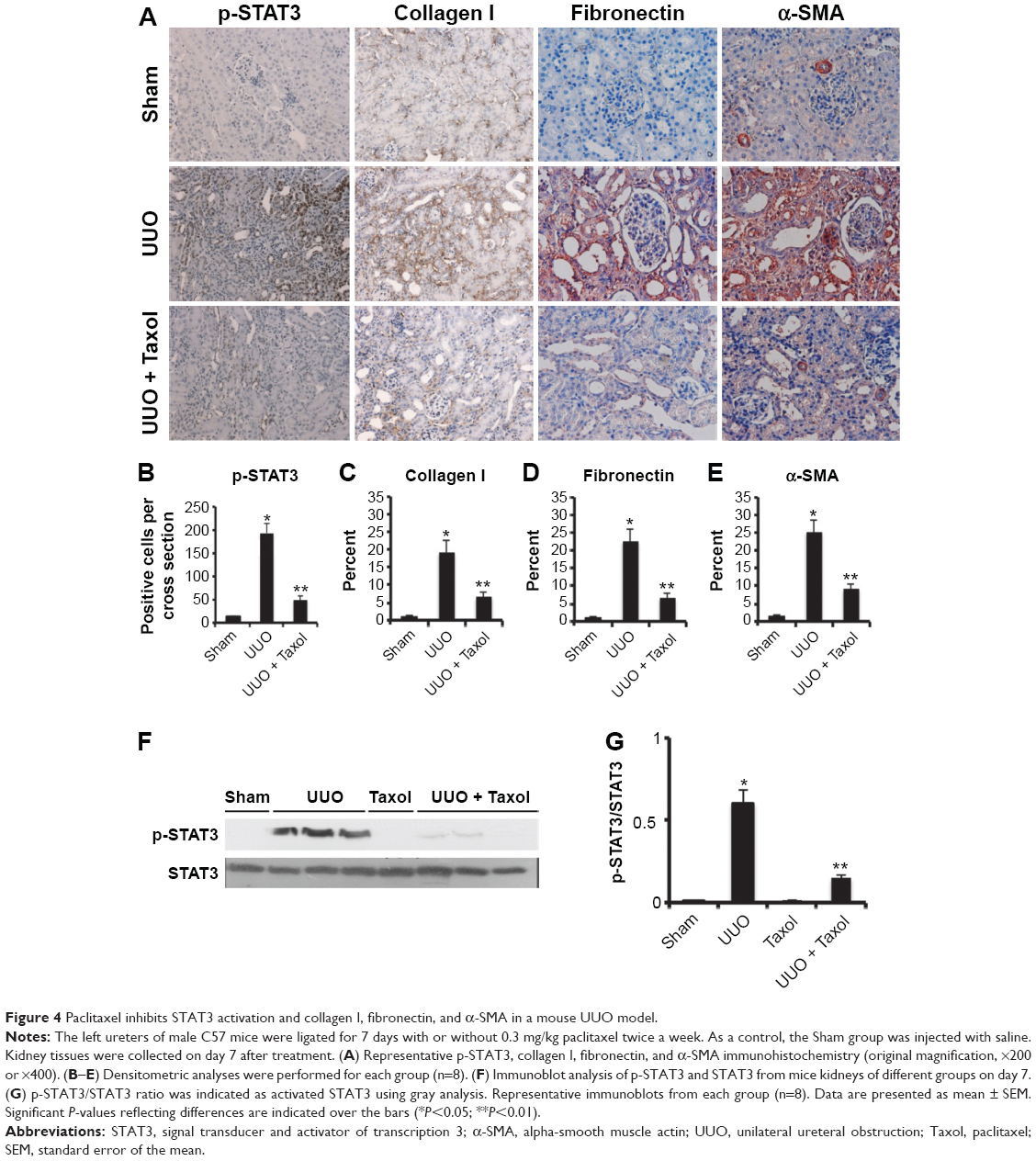 | Figure 4 Paclitaxel inhibits STAT3 activation and collagen I, fibronectin, and α-SMA in a mouse UUO model. |
Effect of paclitaxel on macrophages and neutrophils infiltration and expression of TNF-α, IL-1β, TGF-β, and ICAM-1 in mouse obstructive nephropathy
We examined the effect of paclitaxel on macrophage and neutrophil infiltration and the expression of TNF-α, IL-1β, TGF-β, and ICAM-1. Analysis of kidney sections by immunochemistry showed a prominent interstitial infiltration of macrophages and neutrophils after obstructive injury, which was significantly reduced by paclitaxel (Figure 5A–C). The expression levels of TNF-α, IL-1β, TGF-β, and ICAM-1 were increased after obstructive injury, which were also markedly reduced by paclitaxel (Figure 6A–D). These data suggest that paclitaxel suppresses macrophage and neutrophil infiltration and proinflammatory cytokines by inhibiting STAT3 activity in UUO-induced renal fibrosis.
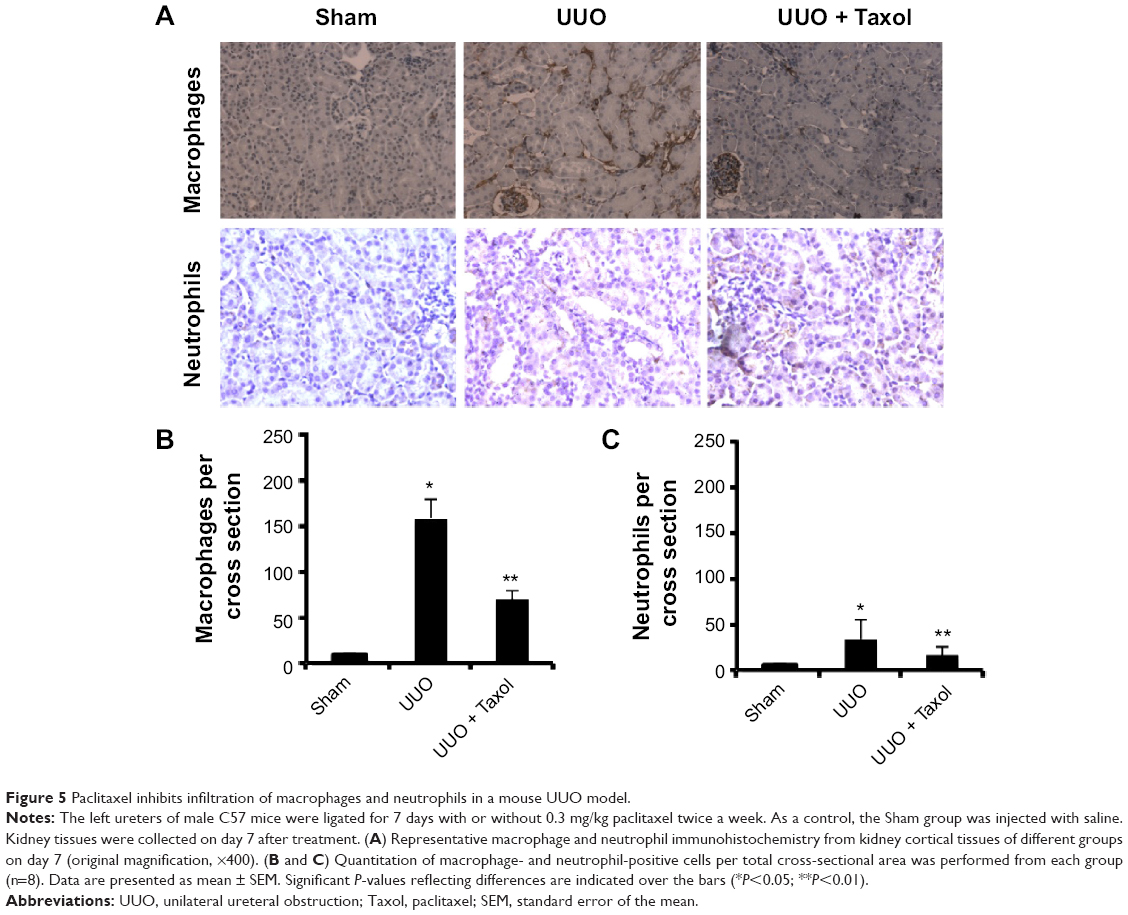 | Figure 5 Paclitaxel inhibits infiltration of macrophages and neutrophils in a mouse UUO model. |
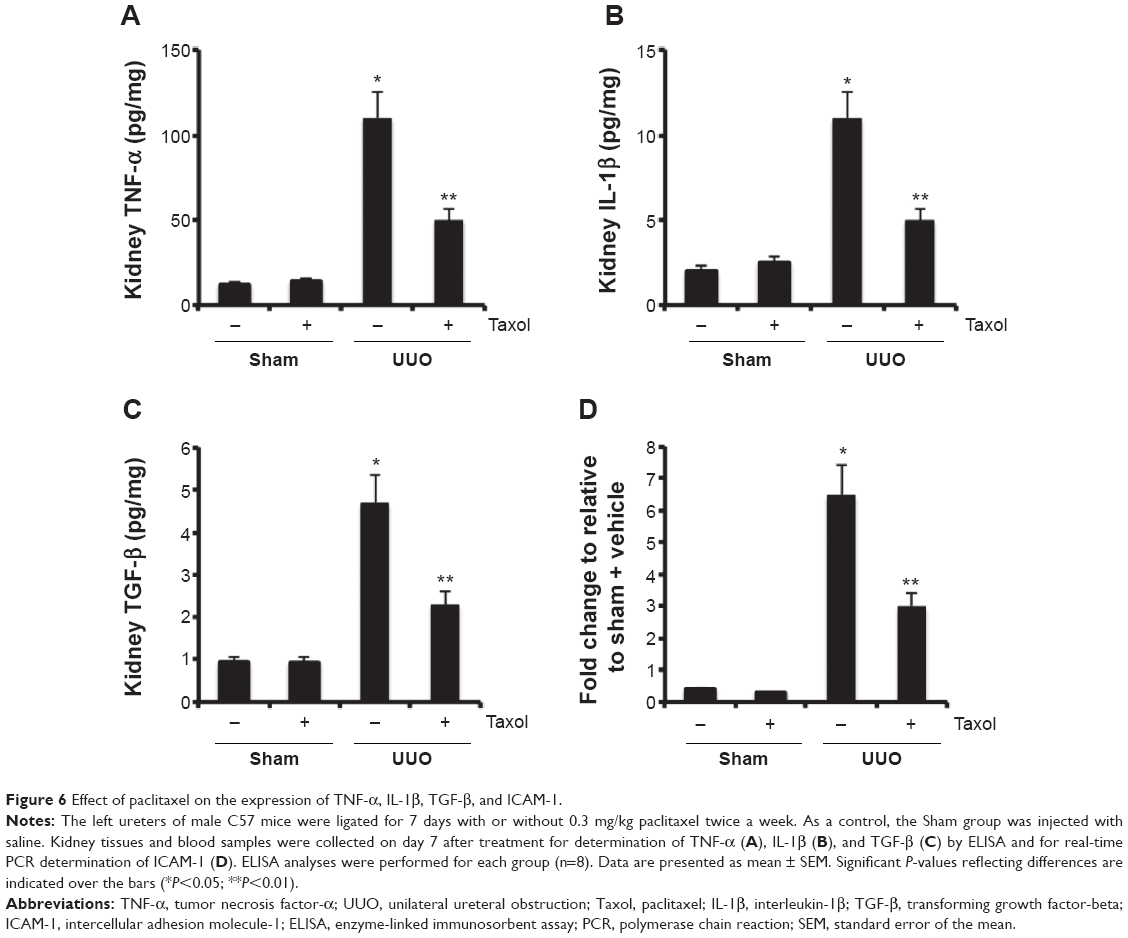 | Figure 6 Effect of paclitaxel on the expression of TNF-α, IL-1β, TGF-β, and ICAM-1. |
Discussion and conclusion
Previous results demonstrated that paclitaxel might ameliorate renal fibrosis in a rat UUO model.2 However, the mechanism remains unclear. In this study, the results show that paclitaxel treatment inhibits the expression of α-SMA and extracellular matrix proteins in renal interstitial fibroblasts, ameliorates renal fibrosis, and suppresses the infiltration of macrophages and proinflammatory cytokines in obstructive nephropathy.
Mechanistically, we observed that paclitaxel treatment blocked the STAT3 activity in cultured renal interstitial fibroblasts and the fibrotic mouse kidney. TGF-β1, platelet-derived growth factor (PDGF), and IL-6 can induce STAT3 activation,17–20 which was detected in several human diseases associated with the development of fibrosis, including liver fibrosis, scar formation after spinal cord injury, glomerulonephritis, and diabetic nephropathy.9,21–25 Furthermore, Pang et al11 reported that STAT3 played an important role in mediating the activation of renal interstitial fibroblasts and the development of renal fibrosis following UUO injury. In this study, we demonstrated that paclitaxel ameliorated fibrosis by inhibiting STAT3 activity in cultured renal interstitial fibroblasts and mouse UUO models. We wanted to know how the microtubule-targeting agents inhibit STAT3 activation. Mechanistically, the interaction between STAT3 and microtubules was prevented by paclitaxel. Paclitaxel may also have an effect on nonphosphorylated forms of STAT3, thereby preventing their translocation to the nucleus. Walker et al12 demonstrated that phosphorylated STAT3 was significantly inhibited by paclitaxel at a concentration of 7 μM in MDA-MB-468, MDA-MB-231, and MCF-7 cell lines. Interestingly, paclitaxel at concentrations 2 μM or 4 μM also blocked STAT3 activation in renal interstitial fibroblasts, which indicated that different cell lines had different levels of sensitivity for paclitaxel treatment. However, paclitaxel at concentration 4 μM was a relatively high dose in most cell lines according to value of the 50% inhibitory concentration (IC50). Furthermore, paclitaxel as a chemotherapeutic agent is usually used at a dosage range from 10 to 15 mg/kg in nude mice.26,27 Consequently, paclitaxel at 0.3 mg/kg in the UUO model is lower than the dosages used in nude mice, but we found that paclitaxel at this dose also markedly inhibited p-STAT3 activation.
Inflammatory responses could accelerate the progression of renal fibrosis, in which multiple inflammatory mediators play a pivotal role.28,29 Pang et al11 reported that inhibition of STAT3 activity by S3I-201 could reduce the infiltration of neutrophils and monocytes in the obstructed kidney. In this study, we observed that the administration of paclitaxel also inhibited macrophage infiltration and the proinflammatory cytokines TNF-α, IL-1β, TGF-β, and ICAM-1. Zhang et al30 demonstrated that STAT3 activity in monocytes/macrophages was a critical mediator of macrophage migration, which supported our results. As we know, TGF-β is the major cytokine in a variety of fibrogenic processes, which initiates the transition of renal tubular epithelial cells to myofibroblasts, the cellular source for extracellular matrix synthesis.31,32 Previous results have shown that low-dose paclitaxel can significantly suppress the exacerbated TGF-β/Smad signaling in the kidney and lessen the interstitial fibrosis in the rat UUO model.2 STAT3 and Smad3 are independent signaling pathways for the regulation of fibrosis development. Ogata et al9 reported that STAT3 might directly regulate TGF-β gene expression. In our study, TGF-β expression is also reduced by paclitaxel. These results suggest that paclitaxel inhibition on inflammatory responses was another mechanism of anti-renal fibrosis.
In conclusion, our study demonstrates that paclitaxel can suppress the activation of renal fibroblasts, progression of renal fibrosis, and proinflammatory response in UUO kidney injury, suggesting a potential use of the chemotherapy drug in the treatment of renal fibrotic diseases. Mechanistically, paclitaxel treatment blocks the STAT3 activity by disrupting the association of STAT3 with tubulin and STAT3 nucleus translocation.
Acknowledgment
The study was supported in part by grants from the National Natural Science Foundation of China (81100507/H0509) and the Central South University Master Freedom Investigation program (2014zzts354).
Author contributions
DZ and ZD conceived and designed the experiments; LZ, RY, JY, and XX carried out the experiments; JC, YZ, and ZH analyzed the data; XX, LZ, and RY contributed reagents/materials/analysis tools; DZ wrote the paper. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang D, Yang, R, Wang S, Dong Z. Paclitaxel: new uses for an old drug. Drug Des Devel Ther. 2014;20(8):279–284. | ||
Zhang D, Sun L, Xian W, et al. Low-dose paclitaxel ameliorates renal fibrosis in rat UUO model by inhibition of TGF-β/Smad activity. Lab Invest. 2010;90(3):436–447. | ||
Karbalay-Doust S, Noorafshan A, Pourshahid SM. Taxol and taurine protect the renal tissue of rats after unilateral ureteral obstruction: a stereological survey. Korean J Urol. 2012;53(5):360–367. | ||
Sun L, Zhang D, Liu F, et al. Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J Pathol. 2011;225(3):364–377. | ||
Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. | ||
Neilson EG. Mechanisms of disease: fibroblasts–a new look at an old problem. Nat Clin Pract Nephrol. 2006;2(2):101–108. | ||
Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci. 2000;25(10):496–502. | ||
Fuller GM, Zhang Z. Transcriptional control mechanism of fibrinogen gene expression. Ann N Y Acad Sci. 2001;936:469–479. | ||
Ogata H, Chinen T, Yoshida T, et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene. 2006;25(17):2520–2530. | ||
Kuratsune M, Masaki T, Hirai T, et al. Signal transducer and activator of transcription 3 involvement in the development of renal interstitial fibrosis after unilateral ureteral obstruction. Nephrology (Carlton). 2007;12(6):565–571. | ||
Pang M, Ma L, Gong R, et al. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 2010;78(3):257–268. | ||
Walker SR, Chaudhury M, Nelson EA, Frank DA. Microtubule-targeted chemotherapeutic agents inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol Pharmacol. 2010;78(5):903–908. | ||
Zhang D, Li Y, Liu Y, et al. Paclitaxel ameliorates lipopolysaccharide-induced kidney injury by binding myeloid differentiation protein-2 to block toll-like receptor 4-mediated nuclear factor-κB activation and cytokine production. J Pharmacol Exp Ther. 2013;345(1):69–75. | ||
Zhang D, Liu Y, Wei Q, et al. Tubular p53 regulates multiple genes to mediate AKI. J Am Soc Nephrol. 2014;25(10):2278–2289. | ||
Grande MT, Lopez-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol. 2009;5(6):319–328. | ||
Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104(18):7391–7396. | ||
Yamamoto T, Matsuda T, Muraguchi A, et al. Cross-talk between IL-6 and TGF-beta signaling in hepatoma cells. FEBS Lett. 2001;492(3):247–253. | ||
Vij N, Sharma A, Thakkar M, et al. PDGF-driven proliferation, migration, and IL8 chemokine secretion in human corneal fibroblasts involve JAK2 STAT3 signaling pathway. Mol Vis. 2008;14(23):1020–1027. | ||
Kretzschmar AK, Dinger MC, Henze C, et al. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J. 2004;377:289–297. | ||
Lim CP, Phan TT, Lim IJ, et al. Cytokine profiling and Stat3 phosphorylation in epithelial–mesenchymal interactions between keloid keratinocytes and fibroblasts. J Invest Dermatol. 2009;129(4):851–861. | ||
Lane A, Johnson DW, Pat B, et al. Interacting roles of myofibroblasts, apoptosis and fibrogenic growth factors in the pathogenesis of renal tubulointerstitial fibrosis. Growth Factors. 2002;20 (3):109–119. | ||
Vesey DA, Cheung CW, Cuttle L, et al. Interleukin-1beta induces human proximal tubule cell injury, alpha-smooth muscle actin expression and fibronectin production. Kidney Int. 2002;62(1):31–40. | ||
Arakawa T, Masaki T, Hirai T, et al. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol Dial Transplant. 2008;23(11):3418–3426. | ||
Berthier CC, Zhang H, Schin M, et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes. 2009;58(2):469–477. | ||
Herrmann JE, Imura T, Song B, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28(28):7231–7243. | ||
Milanović D, Braun F, Weber W, et al. The influence of the combined treatment with Vadimezan (ASA404) and taxol on the growth of U251 glioblastoma xenografts. BMC Cancer. 2012;12:242. | ||
Abubaker K, Luwor RB, Escalona R, et al. Targeted disruption of the JAK2/STAT3 pathway in combination with systemic administration of paclitaxel inhibits the priming of ovarian cancer stem cells leading to a reduced tumor burden. Front Oncol. 2014;4:75. | ||
Stenvinkel P, Heimbürger O, Paultre F, et al. Strong association between malnutrition, inflammation, and atherosclerosis in chronic renal failure. Kidney Int. 1999;55(5):1899–1911. | ||
Vidt DG. Inflammation in renal disease. Am J Cardiol. 2006;97(2A):20A–27A. | ||
Zhang C, Li Y, Wu Y, et al. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem. 2013;288(7):1489–1499. | ||
Zeisberg M, Maeshima Y, Mosterman B, et al. Renal fibrosis. Extracellular matrix microenvironment regulates migratory behavior of activated tubular epithelial cells. Am J Pathol. 2002;160(6):2001–2008. | ||
Lan HY. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens. 2003;12(1):25–29. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.