Back to Journals » International Journal of Nanomedicine » Volume 13
Orlistat-loaded solid SNEDDS for the enhanced solubility, dissolution, and in vivo performance
Authors Kim DH, Kim JY, Kim RM, Maharjan P, Ji YG ![]() , Jang DJ
, Jang DJ ![]() , Min KA, Koo TS
, Min KA, Koo TS ![]() , Cho KH
, Cho KH
Received 23 July 2018
Accepted for publication 11 October 2018
Published 5 November 2018 Volume 2018:13 Pages 7095—7106
DOI https://doi.org/10.2147/IJN.S181175
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Dae Hun Kim,1,* Jae Yeol Kim,1,* Rae Man Kim,1 Pooja Maharjan,1 Yu-Geun Ji,2 Dong-Jin Jang,3 Kyoung Ah Min,1 Tae-Sung Koo,2 Kwan Hyung Cho1
1Department of Pharmacy, Inje Institute of Pharmaceutical Sciences and Research, Inje University, Gimhae, Republic of Korea; 2Graduate School of New Drug Discovery and Development, Chungnam National University, Daejeon, Republic of Korea; 3Department of Pharmaceutical Engineering, Inje University, Gimhae, Republic of Korea
*These authors contributed equally to this work
Background: The present study aimed to develop orlistat-loaded solid self-nanoemulsifying drug delivery system preconcentrate (SSP) with the minimum use of lipid excipients for the enhanced solubility, in vitro dissolution, lipase inhibition, and in vivo performance.
Materials and methods: In the screening of solubilizing vehicles, Solutol HS15 and Lauroglycol 90 were selected as the surfactant and oil phase, respectively. A pseudo-ternary phase diagram composed of Solutol HS15, Lauroglycol 90, and orlistat as an anti-obesity agent and lipid component was constructed, and the SSP regions were confirmed in terms of the particle size distribution in water, melting point by differential scanning calorimetry, and crystallinity by X-ray diffraction.
Results: Physicochemical interaction between Solutol HS15 and orlistat resulted in SSP with various melting points in the range of 26°C~33°C. The representative maximum orlistat-loaded SSP (orlistat/Solutol HS15/Lauroglycol 90=55/40/5, weight ratio) showed the melting point of 32.23°C and constructed uniform nanoemulsion with the particle size of 141.7±1.1 nm dispersed in water. In the dissolution test at pH 1.2 without any detergent, the SSP reached 98.12%±0.83% until 45 minutes, whereas raw orlistat showed no significant dissolution rate. The dissolution samples containing SSP showed a lipase inhibition of 90.42%±1.58% within 45 minutes. In terms of the reduction level of fat absorption in rats, the intake group of SSP gave a significantly higher fat excretion into stool than the one observed in the raw orlistat group (P<0.05).
Conclusion: In conclusion, the suggested novel SSP formulation would be an effective and promising candidate for the treatment of obesity.
Keywords: orlistat, solid self-nanoemulsifying drug delivery system, lipase inhibition, fat absorption
Introduction
Orlistat, a gastrointestinal lipase inhibitor that reduces dietary fat absorption by ~30%, has been widely used in treatment of extreme obesity through promotion of weight loss.1,2 Obesity induces multiple metabolic abnormalities that contribute to pathogenesis of diabetes mellitus, cardiovascular disease, cancer, and nonalcoholic fatty liver disease and is associated with increased morbidity and mortality.3,4 Orlistat has been clinically characterized by its good efficacy and safety based on the local action in gastrointestinal tract compared to other systemically acting anorectic agents, such as phentermine and lorcaserin, that have severe side effects.5
Orlistat, a fat-soluble drug having long lipophilic chains in the structure (Figure 1), is an off-white crystalline powder with the low melting point (between 40°C and 50°C), waxy- and fat-like nature; it is also difficult to handle in the formulation approach.6 The first approved commercial product containing orlistat by Food Drug Administration was Xenical® capsule (120 mg orlistat; Roche S.p.A, Milano, Italy). The standard recommended dose is 120 mg, to be taken three times a day, which is repetitive administration at a higher dose level.7 This product is in a pellet form that comes in a hard gelatin capsule shell; it is indicated to be administered during or after fatty meal for the solubilization of lipophilic orlistat.8 The solubility and dissolution rate of orlistat in the formulation have to be enhanced by fat intake for the lipase inhibitory action. Furthermore, sodium dodecyl sulfate (SDS) included in the commercial product is used for the easy mixing, dispersion, and partitioning of orlistat into the meal composition. SDS is widely used in cosmetics and oral and topical pharmaceutical formulations. However, it is a moderately toxic material with acute irritation to the mucous layer and the stomach.9 The exclusion of using SDS is desirable in the oral formulation and administration.
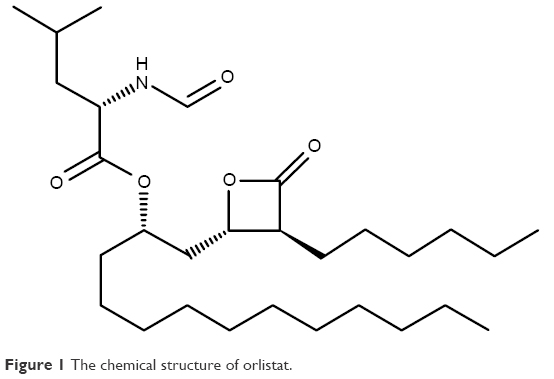 | Figure 1 The chemical structure of orlistat. |
Several pharmaceutical technologies for orlistat have been explored to enhance the solubility, dissolution, and/or lipase inhibition, including nano-sized particles prepared by melt emulsification and solid dosage forms (tablet or pellet) based on the self-nanoemulsifying drug delivery system (SNEDDS).6,10,11 In contrast to the sufficient dissolution rate in the aqueous medium without a detergent, these new solid dosage forms from the lipid-based formulation showed the enhanced dissolution rate only in the medium containing a detergent such as SDS. Therefore, in order to improve efficacy of orlistat, a new technology enabling sufficient solubility needs to be proposed.
Among lipid-based formulations, in particular, SNEDDS has attracted much attention for the formulation of poorly water-soluble and lipophilic drug.12 SNEDDS is generally an isotropic mixture composed of oil, surfactant, and/or co-surfactant, and water, designed to form spontaneously oil-in-water nanoemulsion when dispersed in the aqueous medium. In the composing of SNEDDS, drug molecules can be incorporated into dispersed oil droplets and solubilized in the carrier system. The drug loaded in SNEDDS consistently takes action on the target without any food effects and rapidly releases the drug onto the specific target site.13 However, SNEDDS requires a much greater amount of lipidic excipients to solubilize drug depending on its solubility. Furthermore, SNEDDS is thermodynamically unstable and tends to cause instability issues, such as particle size increase or precipitation of water-insoluble drug during storage. Therefore, the use of SNEDDS is limited by its drug-loading capacity and the used excipients amount. Surfactants and co-surfactants would be irritable to the gastrointestinal tract at high doses and may have limitation in their daily uptake levels.14 Formulation for maximum drug-loading capacity while using the minimum possible amounts of solubilizing excipients is thus needed. Recently, in order to enhance oral bioavailability of drug and physical stability of the dispersion system, solid SNEDDS, prepared by incorporating the conventional liquid SNEDDS preconcentrate into solid carrier, has been introduced.15 Solid SNEDDS combines the advantages of liquid SNEDDS (enhancing oral bioavailability) with those of solid dosage forms (stability and easy handling).16 However, solid SNEDDS is generally complicated and time-consuming in terms of the preparation processes and requires a large amount of excipients for emulsification and subsequent solidification.17,18
In the present study, a novel solid SNEDDS preconcentrate (SSP) was prepared according to a pseudo-ternary phase diagram containing orlistat as a solid lipid component with the minimum use of lipid excipients for the enhanced in vitro dissolution and lipase inhibition, as well as for in vivo performance. Various SSPs were prepared and investigated in terms of the physical state, melting temperature, crystallinity, and dispersed particle size. The optimized formulation was then evaluated in the dissolution without any addition of detergent and in vitro lipase inhibition, and in vivo performance in rat model.
Materials and methods
Materials
Orlistat (purity >99.0%) was purchased by Chongqing Zein Pharmaceutical CO., Ltd. (Chongqing, China). Solutol HS15 (polyoxyl 15 hydroxystearate), Kolliphor P 407 (poloxamer 407), Kolliphor PS 80 (polysorbate 80), and Myritol 318 PH (medium-chain triglycerides) were kindly given as a gift by CTC Bio Inc. (Hwaseong, South Korea). Lauroglycol 90 (propylene glycol monolaurate, type II), Labrafac PG (propylene glycol dicaprylate/dicaprate), Labrafil M 1944 CS (oleoyl polyoxyl-6 glycerides), Labrafil M 2125 CS (linoleoyl polyoxyl-6 glycerides), Labrasol (caprylocaproyl polyoxyl-8 glycerides), and Capmul MCM C8 EP (mono/diglycerides of caprylic acid) were kindly provided as a gift by MASUNG & CO., Ltd. (Seoul, South Korea). 4-nitrophenyl palmitate (p-NPP), lipase powder from porcine pancreas (L3126, Type II, 358 units/mg protein using olive oil or 50 units/mg protein using triacetin), sodium deoxycholate, and sodium taurodeoxycholate were all purchased from Sigma Aldrich Korea (Youngin, South Korea). All other chemicals were of reagent grade and used without further purification.
HPLC condition
The HPLC analysis of orlistat in the samples was conducted using a Waters 2695 HPLC system (Waters, Milford, MA, USA) equipped with a UV-Vis detector (Waters 2487, Waters, Milford, MA, USA). Orlistat was analyzed using the reverse column with C18, 5 μm, 4.5 mm ×25 cm (Shiseido, Tokyo, Japan). Mobile phase consisted of acetonitrile, water, and phosphoric acid (860:140:0.05, v/v/v). The HPLC analysis was performed with the flow rate of 1.5 mL/min. The injected volume of the sample was 10 μL, and UV detection was monitored at 195 nm. Data acquisition and processing were carried out using the Waters LC Solution software.
Solubility studies
The solubility of orlistat in various oils and 1% (w/v) surfactants aqueous solution was determined. Lauroglycol 90, Labrafac PG, Labrafil M 1944 CS, Labrafil M 2125 CS, or Myritol 318 PH were tested as oil components, while Solutol HS15, Kolliphor P 407, Kolliphor PS 80, Labrasol, and Capmul MCM C8 EP were tested as surfactants. An excess of orlistat powder was added to 2 mL of each oil type and 1% surfactant aqueous solution. The mixture was stirred for 24 hours at ambient temperature. In the next step, 1 mL of supernatant was obtained after centrifuge at 15,000 rpm for 5 minutes. As mentioned above, the orlistat amount was measured using the HPLC method.
Construction of pseudo-ternary phase diagram
A pseudo-ternary phase diagram composed of orlistat, Solutol HS15, and Lauroglycol 90 was constructed in the following three steps. In the first step, various mixtures (the compositions shown in Figure 2A) of orlistat, Solutol HS15, and Lauroglycol 90 were weighed in a vial (a total of 200 mg), followed by standing it in a 60°C chamber (HB-501M, HANBAEK, Bucheon, South Korea) for 30 minutes to produce a melted liquid of a homogeneous mixture. The vial was then transferred to a refrigerator and stored at 4°C overnight. The vial was withdrawn and left at the temperature of 25°C or 37°C in a chamber for 5 hours to equilibrate the mixture with the temperature. By the appearance at the temperature (25°C or 37°C), the mixture was classified into either the solid or the liquid state.
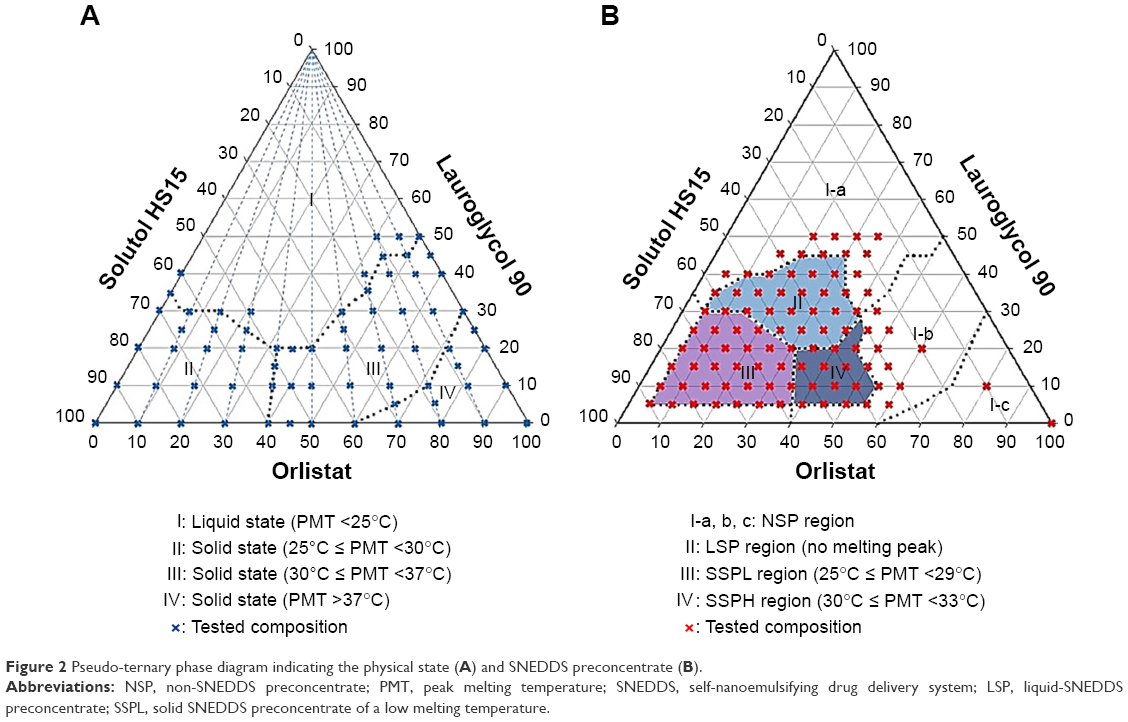 | Figure 2 Pseudo-ternary phase diagram indicating the physical state (A) and SNEDDS preconcentrate (B). |
For the second step, the mixture (the compositions shown in Figure 2B) of the liquid state (a total of 200 mg) was dispersed into 3 mL of water with a mild agitation for 1 minute. The particle size of dispersions was measured and classified as a SNEDDS preconcentrate or a non-SNEDDS preconcentrate (NSP) region. In the third step, the melting points of the mixtures positioned in SNEDDS preconcentrate region were measured using the differential scanning calorimetry (DSC) method. Depending on the peak melting temperature (PMT) in the DSC curve, the SNEDDS preconcentrate region was divided into four regions involving NSP, liquid-SNEDDS preconcentrate (LSP) showing no melting peak, solid SNEDDS preconcentrate of a low melting temperature (SSPL) showing 25°C ≤MPT <29°C, and solid SNEDDS preconcentrate of a high melting temperature (SSPH) showing 30°C ≤MPT <33°C.
Measurement of particle size
Particle size was measured using a particle size analyzer (NanoBrook 90Plus, Brookhaven instruments Corporation, Holtsville, NY, USA). Each sample was prepared as dispersed in distilled water. The measurement was repeated thrice.
DSC measurement
The measurement of DSC was performed using a DSC Q20 (TA Instruments, New Castle, DE, USA). The device was calibrated using indium, and the samples were scanned under nitrogen gas purging (20 mL/min). For the measurements, various 10 mg samples were placed in an aluminum pan (Tzero Pan) and covered with an aluminum lid (Tzero lid). The standby temperature was set at 25°C, while the ramping rate was set at 10°C/min. The analysis was assessed in the range of 20°C~100°C.
X-ray diffraction (XRD) patterns measurement
The XRD patterns were measured by using an X-ray diffractometer (Bruker AXS D8 discover, Bruker AXS Inc., Pittsburgh, WI, USA) equipped with a LynxEye 1D detector. Each sample was added to the grid, and the diffraction pattern of sample was measured using a Cu Kα radiation source (40 kV and 40 mA) with the acquisition time of 0.5 seconds per step.
Measurement of scanning electron microscope (SEM)
Morphology of raw orlistat, NSP, SSPL, and SSPH was measured using the field emission scanning electron microscope (FE-SEM; Mira3 LM, TESCAN Korea INC., Seoul, Korea). NSP, SSPL, and SSPH were diluted with distilled water for the measurement. Each sample was placed on the carbon tape and vacuum-coated with platinum with a coater (108Auto, Cressington scientific instruments Ltd., Watford, England). All measurements were conducted at ambient temperature.
Dissolution studies
Dissolution test was performed using the USP (35) dissolution apparatus II with 0.1 N HCl solution (pH 1.2) with the media volume of 900 mL at 37.5°C±0.5°C. The rotational speed was adjusted to 50 rpm. The selected LSP, SSPL, and SSPH filled in a gelatin capsule (equivalent to an orlistat amount of 120 mg) were prepared and placed into a dissolution tester with a sinker. At each predetermined interval, the aliquot (5 mL) of the medium was collected and filtered through a membrane filter (pore size: 0.45 μm). The concentration of orlistat in the filtrate was determined using the HPLC method.
Lipase inhibition test
A lipase inhibition test was performed with the related reference6 and for the dissolution samples previously obtained at each predetermined time point for raw orlistat, LSP, SSPL, or SSPH. For the enzymatic reaction test, solutions A, B, and C were prepared. Solution A was prepared by mixing 41 mM Tris buffer (pH 8.4), 1.8 mM sodium deoxycholate, and 7.2 mM of sodium tauro deoxycholate. Solution B was prepared by mixing 0.1 mM calcium chloride and 1.6 mM of tartrate buffer (pH 4.0). Solution C was prepared by mixing Solution A and Solution B at the ratio of 8.5:1.5. The stock solution of substrate was prepared by dissolving p-NPP in acetonitrile with the concentration of 10 mM. This stock solution was diluted to 0.25 mM concentration with Solution C as a working solution of substrate and stored at −20°C. All reagents were brought to room temperature for the experiments. Lipase suspension was prepared by suspending lipase powder (5 mg/mL) into a Tris buffer with a pH of 8.4. The suspension was then centrifuged and a supernatant was used as the lipase suspension for the enzymatic reaction. The basic principle of lipase inhibition assay is to analyze the yellow colored p-nitrophenol product produced by the enzymatic hydrolysis of the chromogenic ester.
The lipase inhibition analysis was performed using the following procedure. On a 96-well microplate, 78 μL of Solution C, 20 μL of lipase suspension, and 2 μL of dissolution sample were added. This was followed by a 20-minute incubation at 35°C with 400 rpm in a thermoshaker (MB100-2A, Thermo Shaker, Hangzhou Zhejia, China). A total of 100 μL of substrate solution was then added to each well. After the dissolution test, a dissolution sample at pH 1.2 medium was diluted with a pH 1.2 buffer for the lipase inhibition test. The absorbance was measured at 405 nm using a Multiskan™ Go UV/Vis microplate reader (Thermo scientific, Waltham, MA, USA), at 37°C with the kinetic loop mode of 1 minute. Each sample was repeatedly measured at 1-minute intervals for 20 minutes. For negative control, a mixture consisting of 80 μL of solution C, 20 μL of lipase suspension, and 100 μL of substrate solution was used. The lipase inhibition (%) was calculated as follows (Eq. (1)):
|
|
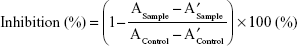
where A is an endothermic spectrum value at 20 minutes, A′ is that at 5 minutes, and control denotes the values of negative control. The test was conducted three times for the dissolution samples.
Animals
All animal care and procedures were performed following the Guiding Principles in the Use of Animals in Toxicology, as adopted in 1989, revised in 1999, and amended in 2008 by the Society of Toxicology. The protocols for the animal studies were also approved by the Institute of Laboratory Animal Resources of Chungnam National University (approval IRB number CNU-00972).
In vivo inhibition of fat absorption in rats
Seven-week-old male Sprague Dawley rats (280–300 g) and high-fat diet for rodents (D12492) were purchased from Orient Bio (Seongnam, Korea) and Central Lab. Animal Inc. (Seoul, Korea), respectively. All animals were kept in an animal room with the controlled illumination (12/12 hours light/dark cycle), temperature (20°C~25°C), and humidity (over 40%).
The rats were divided into four groups, with eight animals in each group. Depending on the group, vehicle (5% gum Arabic solution), orlistat (10 mg/kg in 5% gum Arabic solution), SSPL9, or SSPH6 via oral gavage was administered. All animals were fed with high-fat diet (20 g/day/animal) for 3 days before the experiment and kept separately in animal cages. On the experiment day, the lights were turned off after feeding the diet. After 1 hour, the feces were removed from cages, and the samples were orally administered to the groups. The feces for 24 hours after administration were weighted and stored at −70°C. After feeding the diet, the above procedure was repeated and the feces up to the next 24 hours were collected.
Analysis of fat in stool
The Folch method was used to measure the amount of fat in the feces.19 For the extraction of fat from the feces, first, homogenization was done and then 700 mg of homogenized feces and 14 mL of chloroform/methanol (2/1, v/v) were placed in the tube. The mixture was vortex-mixed for 20 minutes and centrifuged at 1,900 g for 10 minutes. Ten milliliters of the liquid phase was collected and washed with 2 mL of 0.9% NaCl solution. After vortex-mixing for 10 seconds and centrifuging at 1,900 g for 10 minutes, the upper phase was removed by suctioning, and 5 mL of the lower chloroform phase containing fat were transferred to the weighted vial. Then, the solution in vials was evaporated at 1,700 rpm for 120 minutes at 60°C, and the weight was measured.
Inhibition of fat absorption and statistical analysis
To evaluate the differences in fat weight in feces among the four treatment groups, one-way ANOVA with Prism software (ver. 7.0; GraphPad Software, La Jolla, CA, USA) was used. In the event of differences, Dunnett’s test was used to determine if these differences were statistically significant.
Results and discussion
Solubility of orlistat in surfactants and oils
To compose SNEDDS containing orlistat, the saturated solubility was tested in various 1% surfactant solutions and pure oils. Orlistat powder is extremely water-insoluble (0.49±0.12 μg/mL), because it has a very lipophilic and hydrophobic group of the long hydro-carbon chain (C14) in the structure (Figure 1). While the types of surfactants were screened, the mucous-irritable surfactants, such as SDS and cremophore, were excluded from the tested candidates, and orally safe surfactants were included (Table 1). Physical states of surfactants varied as solid (Kolliphor P 407), semi-solid (Capmul MCM C8), and liquid forms (Labrasol and Kolliphor PS 80). Therefore, the solubilizing effect of surfactant was evaluated in the 1% aqueous solution, rather than in the surfactant itself. Among the tested surfactants, Solutol HS15 demonstrated the highest solubilizing effect (47.08±0.24 μg/mL) on orlistat. Its solubility capacity was about 96-fold higher than that in water. In the oils, Lauroglycol 90 dissolved orlistat at the highest level (239.66±9.70 mg/mL) as compared to the other oils showing <200 mg/mL. The degree of solubility in oils was high due to the lipid-like structure and properties of orlistat. As a result, Solutol HS15 and Lauroglycol 90 that showed good affinity with orlistat and enhanced the solubility were selected as a surfactant and oil, respectively, for further development of SNEDDS.
 | Table 1 Solubility of orlistat in surfactants and oils |
Pseudo-ternary phase diagram
Pseudo-ternary phase diagram composed of orlistat, Solutol HS15, and Lauroglycol 90 (Figure 3) was constructed to find out the optimum SSPH displaying appropriate a solid state in room temperature being efficiently dispersed in water with a small particle size and having a high orlistat weight % with the minimum excipient required. Generally, pseudo-ternary phase diagram for confirmation of SNEDDS preconcentrate region was composed of oil, surfactant, and co-surfactant. Then, the drug material was dissolved in optimized formulations containing those components. However, in the present study, we introduced orlistat as a drug substance and a lipid component to develop SNEDDS preconcentrate, resulting in a high drug-loading efficiency (%). Binary mixture of orlistat and Solutol HS15 at various weight ratios (10:0, 9:1, 8:2, 7:3, 6:5, 5:5, 4:6, 3:7, 2:8, 1:9, and 0:10) were first prepared, and Lauroglycol 90 was added to the mixture to determine a physical state (solid or liquid) at the setting temperature (25°C or 30°C). There were four different regions including PMT <25°C (region I), 25°C~30°C (region II), 30°C~37°C (region III), and >37°C (region IV) (Figure 2A). The combined use of Solutol HS15 and orlistat with some addition of Lauroglycol 90 resulted in the solid and liquid states of various melting points.
 | Figure 3 3D plots of particle size (nm) vs. weight ratio of each ingredient (%); Orlistat and Lauroglycol 90 (A), Orlistat and Solutol HS15 (B), Lauroglycol 90 and Solutol HS15 (C). |
To determine SNEDDS preconcentrate regions, 91 mixtures of different compositions (Figure 2B) were prepared and evaluated using visual observation and then, after being dispersed in 37°C water, particle sizes were measured. Furthermore, PMTs were determined by DSC for selective 35 mixtures out of 91 preparations. In this performance, four regions indicating for NSP, LSP, SSPL, and SSPH were separated. SNEDDS preconcentrate of LSP, SSPL, and SSPH except for NSP formed the mean particle size below 200 nm with a mild agitation. Due to the low free energy to be dispersed in water,20 the formation of this nanoemulsion was thermodynamically spontaneous. In the present study, the standard particle size for the designation as the SNEDDS preconcentrate was set at 200 nm, which is generally known to be an acceptable value.21 As a dispersion medium, water (37°C) was used based on the melting points of SSPL and SSPH and their applications to the gastrointestinal tract at body temperature. The maximum weight ratios for Lauroglycol 90, Solutol HS15, and orlistat were 45%/65%/40% in LSP, 30%/90%/38% in SSPL, and 28%/56%/55% in SSPH, respectively. As compared to the other LSP and SSPH, the SSPH regions gave the highest orlistat weight ratio of 55% and a higher PMT >30°C, which might be related to the combination use of orlistat (solid) and Solutol HS15 (semisolid).
Characterization of LSP, SSPL, and SSPH
The particle size in SNEDDS is a critical attribute in SNEDDS formulation. The smaller particle size ensures a better stability of the dispersion system, rapid drug release, and a more effective action on the target enzyme. The mean particle size range for LSP, SSPL, and SSPH was 25.1~143.2 nm, 17.9~40.6 nm, and 85.1~173.3 nm, respectively (Table 2). In SSPLs, the used amount (%) of Solutol HS15 (55%~80%) was higher than that in LSP (25%~60%) and SSPH (35%~50%), and this might be related to the small particle size of SSPLs. Solutol HS15 as a surfactant decreased interfacial tension in dispersion, which enabled to form a stable nanoemulsion of small particle size. However, orlistat weight % for SSPLs was quite low (10%~30%), and, as the melting occurred at <30°C, the preparation could not exist as a solid at room temperature. Whereas, in the case of SSPHs, the mean particle size is larger than that of SSPL due to the higher orlistat weight %, and all mean size values below 200 nm are acceptable. The achievement of particle size of <100 nm for nanoemulsion is critical in terms of the thermodynamic stability. In the present study, we prepared optimized SSPHs as the final formulation that should be dispersed and easily form nanoemulsions in the stomach by the contact with dispersion medium. A long-term stability issue of dispersion system during storage did not arise, and the reasonable particle size of <200 nm was sufficiently small to achieve the enhanced solubilization and enzyme inhibition efficiency of orlistat. In LSPs, formulations containing not more than 40% of orlistat showed the mean particle size of 143.2 nm, though all formulations were in the liquid state at the room temperature, because the weight percentage of Lauroglycol 90 was higher as compared to those of the other SSPLs and SSPHs. The effect of three components on the particle size is represented on a three-dimensional plot (Figure 3). The higher weight percentage of orlistat increased the mean particle size in the reconstituted nanoemulsion, but the inclusion of Solutol HS15 showed the opposite result (Figure 3A–C). The used weight % of Lauroglycol 90 as an oil was in the narrow range of below 40%, and the effect on mean particle size was restricted, showing no significant tendency on the mean particle size change.
 | Table 2 Particle size and peak melting temperature of orlistat formulations |
In the DSC analysis, raw orlistat and treated orlistat (recrystallization after melting and cooling) showed a similar curve with the PMT of 45°C (Figure 4G and H). Treated orlistat recovered its crystallinity after melting followed by cooling down. Solutol HS15 that was in the solid state at room temperature showed no significant melting with its amorphous form (Figure 4F). In the case of binary mixture of orlistat and Solutol HS15, two endothermic melting peaks appeared at 29.71°C and 36.10°C, respectively (Figure 4E). This thermodynamic behavior showing multiple melting peaks may imply coexistence of two different crystallinities in the binary mixture of orlistat and Solutol HS15. This nonuniformity in the crystal form may affect dispersity in medium and cause a high variation in the particle size distribution.22 SSPH6, SSPL9, and NSP6 showed a single melting peak at 32.23°C, 28.63°C, and 36.31°C, respectively (Figure 4A, B and D). The use of Lauroglycol 90 might have contributed to changing the double melting peaks in the binary mixture into a single melting peak in the ternary mixture and could have resulted in the single thermodynamic behavior.23,24 LSP11 had no significant endothermic peaks, which was consistent with its liquid state observed at 25°C (Figure 4C).
 | Figure 4 Differential scanning calorimetry curve of the mixtures of orlistat, Solutol HS15, and Lauroglycol 90; SSPH6 (A), SSPL6 (B), LSP11 (C), NSP6 (D), binary mixture of orlistat and Solutol HS15 (50:50, w/w) (E), Solutol HS15 (F), treated orlistat (G), or raw orlistat (H). |
Raw orlistat and treated orlistat showed different XRD patterns, which might be due to the slight changes in the crystal structure (Figure 5F and G). The binary mixture of orlistat and Solutol HS15 yielded a new XRD pattern including similar peaks in the orlistat-treated group (Figure 5D). The interaction of orlistat and Solutol HS15 produced new crystal forms having intrinsic peaks at 17.16°, 17.93°, 19.59°, and 22.66°. These new XRD patterns clearly appeared commonly in SSPH6 and SSPL9, with noticeably weakened or excluded peaks as compared to those in the group treated with orlistat (Figure 5A and B). Compared to SSPL9, SSPH6 exhibited a higher ratio of the new crystal forms with the higher peak intensity. This result explains the higher melting temperature of SSPH6 than that of SSPL9, as well as is consistent with the DSC analysis results (Figure 4A and B). The use of Lauroglycol 90 contributed to weakening peaks from treated orlistat and produced highly pure new crystal forms. No significant peak for LSP11 and Solutol HS15 was found because they have no crystalline property (Figure 5C and E).
 | Figure 5 The X-ray diffraction curve of the mixtures of orlistat, Solutol HS15, and Lauroglycol 90; SSPH6 (A), SSPL9 (B), LSP11 (C), binary mixture of orlistat and Solutol HS15 (50:50, w/w) (D), Solutol HS15 (E), treated orlistat (F), or raw orlistat (G). |
The SEM images of raw orlistat and dispersed particles from NSP7, SSPH6, and SSPL9 were obtained (Figure 6). The agglomeration of micrometer-size particles with a rough surface was observed in raw orlistat and NSP7 (Figure 6A and B). SSPH6 and SSPL9 showed little aggregation of spherical particles with a smooth surface (Figure 6C and D).
 | Figure 6 A scanning electron microscopy image of raw orlistat (A), NSP7 (B), SSPL9 (C), or SSPH6 (D). |
In summary, our results suggest that SSPH6 is an optimized formulation with the high melting point (>30°C), particle size of <200 nm, and the maximum orlistat weight ratio of 55%. This was used for further in vitro drug dissolution and lipase inhibition and in vivo study.
In vitro dissolution and lipase inhibition
An in vitro dissolution test is useful to evaluate the performance of the product, especially the one that contains water-insoluble and lipophilic drugs. All the formulations in oral administration route expose to an acidic condition due to the secretion of HCl in stomach. Thus, simulated gastric fluid of pH 1.2 was used as the priority dissolution medium for dissolution tests of the candidate formulations based on the characterization results mentioned above. As shown in Figure 7, LSP11, SSPL9, and SSPH6 showed significantly faster dissolution curves and reached the mean values of 94.34%, 96.70%, and 98.12%, respectively, at pH 1.2 within 45 minutes. No significant differences in dissolution behaviors among the three formulations were observed, whereas they showed varied physicochemical characteristics in terms of the physical state, melting points, crystallinity, or particle sizes. SSPH6, an optimized formulation, showed a comparably good dissolution profiles as LSP11 and SSPL9. This rapid dissolution at the simulated gastric fluid can contribute for the enhanced solubilization and lipase inhibition activity without any help of fat intake and gastric motility.12 However, due to the absence of detergent like SLS in the medium, no significant detection was possible for raw orlistat during the test time of 90 minutes. In several previous studies, the similar dissolution test setup was used, but SLS has been included as a dissolution enhancer for its poor water solubility of orlistat.6,12,13 In the present study, the suggested formulations such as SSPH6 provided sufficient solubilization and rapid dissolution in an aqueous medium without the support of SLS.
 | Figure 7 Dissolution profiles of raw orlistat, LSP11, SSPL9, or SSPH6 at pH 1.2 (n=4). |
To predict the lipase inhibition efficiency by orlistat in various formulations, lipase inhibition test was conducted with the obtained dissolution samples as in Figure 7. The lipase inhibition % of LSP11, SSPL9, and SSPH6 increased rapidly, reaching the mean values of 90.16%, 89.34%, and 90.42% at 45 minutes, respectively (Figure 8). This inhibition curve showed a similar pattern to the dissolution test with the time. The theoretical concentration of orlistat at 100% dissolution was calculated as 133.33 μg/mL, achieving the almost completed lipase inhibition. Overall, SSPH6 showed a relatively good performance in dissolution and lipase inhibition.
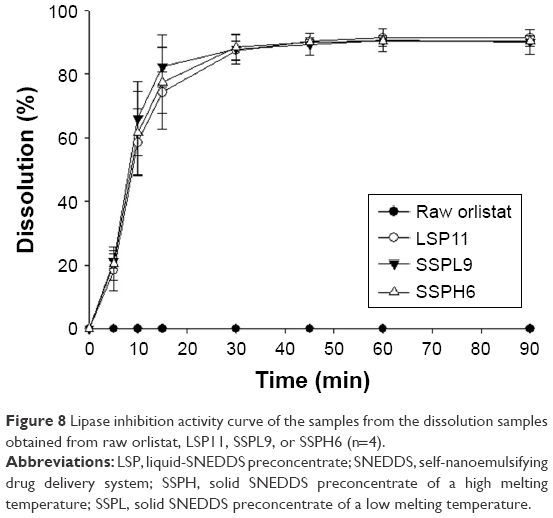 | Figure 8 Lipase inhibition activity curve of the samples from the dissolution samples obtained from raw orlistat, LSP11, SSPL9, or SSPH6 (n=4). |
In vivo inhibition of fat absorption in rats
Lipid amount in the feces in male SD rats after high-fat diet and orlistat preparation is shown in Figure 9. After a vehicle oral dosing, the lipid amount of feces at 0~24, 24~48, and 0~48 hours were 16.44±2.16, 16.35±1.75, and 32.79±3.15 g, respectively. After an orlistat oral dosing, the lipid amount of feces at 0~24, 24~48, and 0~48 hours amounted to 58.79±18.15, 68.84±9.59, and 127.63±19.70 g, respectively. After SSPH6 oral dosing, the lipid amount of feces at 0~24, 24~48, and 0~48 hours was 74.99±19.81, 90.73±22.23, and 165.71±33.61 g, respectively. At 24 hours, the lipid amount of feces in orlistat group was significantly higher than that in the vehicle group, but no differences were found with SSPH6 group. The reason for the lack of significant differences in the lipid amount of feces between raw orlistat and SSPH6 groups seems to be that feces at 0~24 hours are a mixture of stools affected or not affected by orlistat because of drug dissolution and absorption process in the gastrointestinal tract. For the next 24 hours, the lipid amount of orlistat group was also significantly higher than in the vehicle group and significantly lower than SSPH6. In both results, on the first and the next 24 hours and the combined results, all orlistat-treated groups effectively blocked the absorption of fat. SSPH6 was found to be more effective than in the control group, and significant differences were observed in the SSPH6-treated group, suggesting that it has the best effect. We did not observe changes in body weight over a long period of time using obese animal models. However, as lipid absorption is significantly impeded by the novel orlistat formulation via lipase inhibition activity, some level of weight loss effect is expected over a long period in an obesity model. More research is needed to obtain reliable weight loss results.
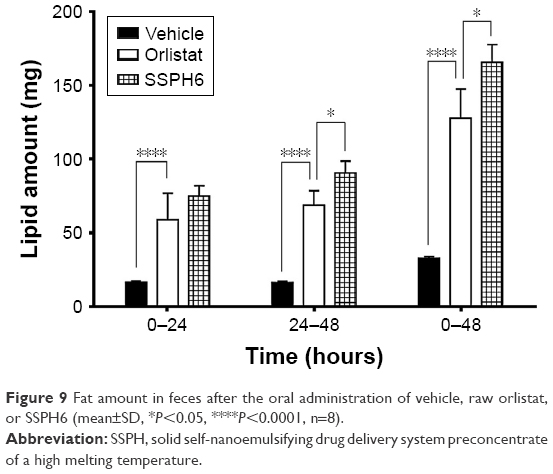 | Figure 9 Fat amount in feces after the oral administration of vehicle, raw orlistat, or SSPH6 (mean±SD, *P<0.05, ****P<0.0001, n=8). |
Conclusion
Obesity has been known to be a major health problem worldwide, inducing many metabolic abnormalities, which lead to high morbidity and mortality rates. Up to date, orlistat-containing products failed to show efficient inhibition activity on lipase, which might be related to the poor aqueous solubility or limited formation of dispersions in gastrointestinal tract. Therefore, a strategy that would improve effects of the orlistat is necessary to increase treatment rates of the obesity. Based on our results, the efficacy of orlistat could be increased by adopting the novel SNEDDS formulation technology. The enhancement in the drug solubility and dissolution rate strongly supported the physical changes of orlistat in the formulation. Furthermore, the lipase inhibition activity of this novel formulation (SSPH6) was confirmed by lipase inhibition assay results both in the in vitro and in the in vivo rat model. Therefore, it can be concluded that the novel SSP formulation of orlistat is an effective way for obesity treatment.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2015R1C1A1A01051698 and NRF-2018R1A2B6002686).
Disclosure
The authors report no conflicts of interest in this work.
References
Heck AM, Yanovski JA, Calis KA. Orlistat, a new lipase inhibitor for the management of obesity. Pharmacotherapy. 2000;20(3):270–279. | ||
Morales P, Fujio S, Navarrete P, et al. Impact of dietary lipids on colonic function and microbiota: an experimental approach involving orlistat-induced fat malabsorption in human volunteers. Clin Transl Gastroenterol. 2016;7(4):e161. | ||
Pozza C, Isidori AM. What’s Behind the Obesity Epidemic. Imaging in Bariatric Surgery. New York: Springer International Publishing AG. 2018:1–8. | ||
Aung K, Lorenzo C, Hinojosa MA, Haffner SM. Risk of developing diabetes and cardiovascular disease in metabolically unhealthy normal-weight and metabolically healthy obese individuals. J Clin Endocrinol Metab. 2014;99(2):462–468. | ||
Kushner RF. Weight loss strategies for treatment of obesity. Prog Cardiovasc Dis. 2014;56(4):465–472. | ||
Dolenc A, Govedarica B, Dreu R, Kocbek P, Srcic S, Kristl J. Nanosized particles of orlistat with enhanced in vitro dissolution rate and lipase inhibition. Int J Pharm. 2010;396(1–2):149–155. | ||
Menendez JA, Vellon L, Lupu R. Antitumoral actions of the anti-obesity drug orlistat (XenicalTM) in breast cancer cells: blockade of cell cycle progression, promotion of apoptotic cell death and PEA3-mediated transcriptional repression of Her2/neu (erbB-2) oncogene. Ann Oncol. 2005;16(8):1253–1267. | ||
Pandey HC, Radhakrishnan RB, Mohammed SF, Vaghela D, Narayana RGL. INVENTIS DDS PVT Ltd. Pharmaceutical composition of orlistat. U.S. Patent. 2010; Application No 12/738 145. | ||
Lindenmüller IH, Lambrecht JT. Oral care. Topical Applications and the Mucosa. Basel: Karger Publishers. 2011;40:107–115. | ||
Gade MM, Hurkadale PJ. Formulation and evaluation of self-emulsifying orlistat tablet to enhance drug release and in vivo performance: factorial design approach. Drug Deliv Transl Res. 2016;6(3):276–288. | ||
Sangwai M, Sardar S, Vavia P. Nanoemulsified orlistat-embedded multi-unit pellet system (MUPS) with improved dissolution and pancreatic lipase inhibition. Pharm Dev Technol. 2014;19(1):31–41. | ||
Madhav KV, Kishan V. Self microemulsifying particles of loratadine for improved oral bioavailability: preparation, characterization and in vivo evaluation. J Pharm Investig. 2018;48(4):497–508. | ||
Kumar A, Sharma S, Kamble R. Self emulsifying drug delivery system (SEDDS): future aspects. Int J Pharm Pharm Sci. 2010;2(4):7–13. | ||
Narang AS, Delmarre D, Gao D. Stable drug encapsulation in micelles and microemulsions. Int J Pharm. 2007;345(1–2):9–25. | ||
Nazzal S, Khan MA. Controlled release of a self-emulsifying formulation from a tablet dosage form: stability assessment and optimization of some processing parameters. Int J Pharm. 2006;315(1–2):110–121. | ||
Ahsan MN, Verma PRP. Enhancement of in vitro dissolution and pharmacodynamic potential of olanzapine using solid SNEDDS. J Pharm Investig. 2018;48(3):269–278. | ||
Seo YG, Kim DH, Ramasamy T, et al. Development of docetaxel-loaded solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced chemotherapeutic effect. Int J Pharm. 2013;452(1–2):412–420. | ||
Balakrishnan P, Lee BJ. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm. 2009;72(3):539–545. | ||
Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226(1):597–509. | ||
Feng J, Zhang Q, Liu Q, Zhu Z. Application of nanoemulsions in formulation of pesticides. In: McClements DJ, Jafari SM, editors. Nanoemulsions. Cambridge: Academic Press; 2018:379–413. | ||
Inugala S, Eedara BB, Sunkavalli S, et al. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: in vitro and in vivo evaluation. Eur J Pharm Sci. 2015;74:1–10. | ||
Zhuang CY, Li N, Wang M, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm. 2010;394(1–2):179–185. | ||
Mohsin K, Shahba AA, Alanazi FK. Lipid based self emulsifying formulations for poorly water soluble drugs-an excellent opportunity. Ind J Pharm Edu Res. 2012;46(2):88–96. | ||
Madhav KV, Kishan V. Self microemulsifying particles of loratadine for improved oral bioavailability: preparation, characterization and in vivo evaluation. J Pharm Investig. 2018;48(4):497–508. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.