Back to Journals » Clinical, Cosmetic and Investigational Dentistry » Volume 18
Oral Radiographic Changes in Severe Sickle Cell Anemia Patients: A Retrospective Comparative Study
Authors AlKhodier H ![]() , Bin Saleh F, AlRumi A, AlOjaym T
, Bin Saleh F, AlRumi A, AlOjaym T
Received 24 March 2026
Accepted for publication 14 May 2026
Published 22 May 2026 Volume 2026:18 611537
DOI https://doi.org/10.2147/CCIDE.S611537
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Renan Dal Fabbro
Haifa AlKhodier, Fahad Bin Saleh, Abdulmajeed AlRumi, Tariq AlOjaym
Department of Dentistry, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Correspondence: Haifa AlKhodier, Department of Dentistry, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia, Email [email protected]
Introduction: Sickle cell disease is caused by a point mutation in the β-globin gene, resulting in the production of abnormal hemoglobin. Various oral radiographic changes have been documented in the literature, though they are less frequently reported in patients with severe disease. This study aimed to evaluate specific radiographic features in pediatric patients with severe sickle cell disease.
Methods: This retrospective comparative study was conducted at the Dental Department of King Faisal Specialist Hospital and Research Centre. A total of 72 pediatric patients (aged 6– 14 years) with genetically confirmed severe sickle cell disease and 96 age-matched healthy controls were included. Dental radiographs were evaluated by four calibrated examiners for the presence of radiopaque areas, increased spacing of bony trabeculae, absence of mandibular canal corticalization, faint lamina dura, spiky roots, taurodontism, and haziness of the maxillary sinuses. Statistical analysis was performed using Chi-square test, with significance set at p < 0.05.
Results: The sickle cell group showed a significantly higher prevalence of increased trabecular spacing (58.3% vs. 19.8%), absence of mandibular canal corticalization (68.1% vs. 16.7%), faint lamina dura (19.4% vs. 2.1%), taurodontism (15.3% vs. 9.4%), and haziness of the maxillary sinuses (69.4% vs. 43.8%) (p < 0.05 for all comparisons).
Conclusion: Pediatric patients with severe sickle cell disease demonstrated a higher prevalence of specific radiographic changes, particularly increased trabecular spacing, loss of mandibular canal corticalization, and haziness of the maxillary sinuses. This study highlights the importance of recognizing these imaging features in the assessment of disease severity.
Keywords: sickle cell disease, bone marrow transplantation, pediatric dentistry
Introduction
Sickle cell disease (SCD) is an inherited hematologic disorder caused by a mutation in the β-globin gene.1,2 In affected individuals, glutamic acid is substituted by valine, which will produce a hemoglobin known as hemoglobin S.3 Red blood cells (RBCs) assume a sickle shape due to the presence of poorly soluble hemoglobin.1,2 The altered RBCs show a diminished capacity for oxygen transport, potentially leading to a vaso-occlusive crisis, which reduces the lifespan of RBCs and subsequently decreases hemoglobin levels in the body.4
Sickle Cell Disease is inherited as an autosomal recessive disorder.5 People who carry one copy of the affected HbS allele and one normal HbA allele are asymptomatic and are classified as having Sickle Cell Trait. Conversely, those who possess two copies of the affected allele (HbSS) are symptomatic and are diagnosed with the disease.6 The condition becomes apparent after the first six months of life, which is attributable to the protective effect provided by elevated levels of fetal hemoglobin (HbF) during early life stages.4
Approximately 300,000 infants are born annually worldwide with SCD. Among these populations, individuals from the Middle Eastern, Mediterranean, and Sub-Saharan regions constitute the largest affected groups. Additionally, males tend to exhibit a more aggressive disease presentation.7 Unfortunately, the number of affected individuals has significantly increased in recent years.8
Symptoms of Sickle Cell Disease vary among individuals.9 A classic symptom in all affected patients is episodic severe pain.10 Additionally, anemia, infections, and fatigue are prevalent among this population.11 Patients with SCD also face other significant complications, such as acute chest syndrome, avascular necrosis, renal failure, Moya-Moya disease, ischemic or hemorrhagic stroke, and silent cerebral infarction.12–17
For two decades, Hydroxyurea was the sole approved therapeutic agent for managing Sickle Cell Disease (SCD), followed by L-glutamine as a prophylactic measure against SCD-related complications.18 Notably, the curative treatment option for sickle cell disease is hematopoietic stem cell transplantation (HSCT) especially in severe forms. However, not all patients are suitable candidates for it.19,20 Hematopoietic stem cell transplantation (HSCT) is generally reserved for individuals suffering from severe complications such as stroke, acute chest syndrome, nephropathy, and osteonecrosis.20 Clinically, many patients with SCD present with various manifestations, including pallor of the mucosa, periodontitis, delayed tooth eruption, paresthesia of the mental nerve, necrotic pulps, and hypomineralization.21,22 Radiographically, multiple changes have been reported such as hypercementosis, stepladder appearance of the alveolar bone, and increased trabecular spacing.21–23
The aim of this study is to assess oral and maxillofacial radiographic changes in pediatric patients with severe sickle cell disease. Recognition of these radiographic features may facilitate early identification of disease-related skeletal involvement, support dental treatment planning, and contribute to the overall medical assessment and multidisciplinary management of affected patients, particularly in those with advanced disease.
Materials and Methods
This study was designed as a retrospective comparative study. It was conducted in the Dental Department at King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia. This study was conducted in accordance with the ethical standards of the institutional research committee and with the Declaration of Helsinki and its later amendments. Ethical approval was obtained from the Office of Research Affairs at KFSHRC. The requirement for informed consent was waived as the study involved a retrospective review of anonymized data without direct patient contact or intervention. Furthermore, no identifiable information was collected.
The study population recruited pediatric patients aged between 6 and 14 years. A total of 168 patients were included. The diagnosis of sickle cell disease was confirmed based on documented clinical and hematological criteria in the patients’ medical records, as established by specialist physicians.
Two groups were included in the study. Group A consisted of medically healthy children with no history of systemic illness.
Group B was made of patients with confirmed genetic diagnoses of severe SCD who had undergone hematopoietic stem cell transplantation. Severe SCD was defined as patients requiring hematopoietic stem cell transplantation due to recurrent vaso-occlusive crises, acute chest syndrome, stroke, or other life-threatening complications.
Inclusion criteria for group A consisted of being medically healthy, aged between six and fourteen years, and possessing diagnostic radiographs. Exclusion criteria for group A were: Having a confirmed diagnosis of any medical illness, falling outside the specified age range or not having diagnostic radiographs possibly due to unfavorable behavior of the child. 96 healthy individuals were recruited for group A.
Inclusion criteria for group B were an age range between six to fourteen years, a confirmed diagnosis of severe form Sickle Cell Disease (SCD) necessitating bone marrow transplantation, and the availability of diagnostic radiographs. Patients were excluded if they lacked confirmed diagnoses, fell outside the specified age range, did not undergo bone marrow transplantation or did not have diagnostic radiographs due to unfavorable behavior of the child.
A total of 93 medical records of SCD patients were reviewed. Of which, 72 met the inclusion criteria. The remaining 21 charts were excluded due to the unavailability of x-rays or the presence of non-diagnostic radiographs.
Radiographic changes were assessed according to predefined criteria adapted from previous literature on SCD-related maxillofacial changes, including evaluation of trabecular spacing, root morphology, taurodontism, mandibular canal corticalization, lamina dura, the presence of radiopaque areas and maxillary sinus appearance.
Radiographs were obtained using digital imaging units within the same institution. Patients with sickle cell disease were imaged in a dedicated clinical setting for medically compromised patients, while healthy controls were imaged in a separate clinic. Panoramic radiographs were acquired using Carestream CS 8200 3D and Carestream CS 9300 digital imaging systems (Carestream Dental, Atlanta, GA, USA) with standardized exposure parameters of approximately 70–75 kV, 8.0 mA, and an exposure time of 11.3 seconds. Periapical radiographs were obtained using a CS 2200 intraoral imaging unit (Carestream Dental, Atlanta, GA, USA) with exposure settings ranging from 60–70 kV, 7 mA, and approximately 0.119 seconds exposure time. Comparable digital imaging systems and standardized acquisition protocols were used across both groups to minimize inter-clinic variability.
Panoramic radiographs were the primary imaging modality used for analysis. When available, supplementary intraoral radiographs (bitewing and periapical images) were reviewed to provide a more detailed assessment of specific features, particularly the lamina dura.
All radiographs were evaluated for the following features: presence of radiopaque areas; increased spacing of bony trabeculae; absence of mandibular canal corticalization; spiky roots; faint lamina dura; taurodontism; and obliteration (haziness) of the maxillary sinuses. Four examiners performed the radiographic assessment; two were consultant dentists and the other were senior dental residents. Only images of acceptable diagnostic quality were incorporated into the final analysis. Radiographs containing artifacts, low resolution, or incomplete anatomical coverage were excluded.
Demographic and radiographic data were extracted, coded, and anonymized prior to performing statistical analysis. Data were analyzed using the Statistical Package for the Social Sciences (SPSS) version 26.0 (IBM Corp., Armonk, NY, USA). Categorical variables were expressed as frequencies and percentages, and comparisons between groups were conducted using the Chi-square test (χ2). A p-value of less than 0.05 was considered statistically significant.
Results
A total of 168 pediatric patients were included in the study: 96 in group A (healthy controls) and 72 in group B (patients with severe sickle cell disease).
The four examiners re-established intra-examiner reliability after one month. Following that, inter- and intra-examiner reliability were evaluated using Cohen’s kappa statistics, yielding a kappa value of 0.712, indicating substantial agreement.
In relation to sex, 57% of patients in group B were females, while 51% of group A were males. The mean age was 10.38 ± 2.77 years in group B and 10.23 ± 2.17 years in group A. Overall, no significant differences in age or sex were observed between the two groups.
Collectively, analysis of radiographs demonstrated that several radiographic changes were significantly more prevalent in patients with SCD compared to controls. (Table 1).
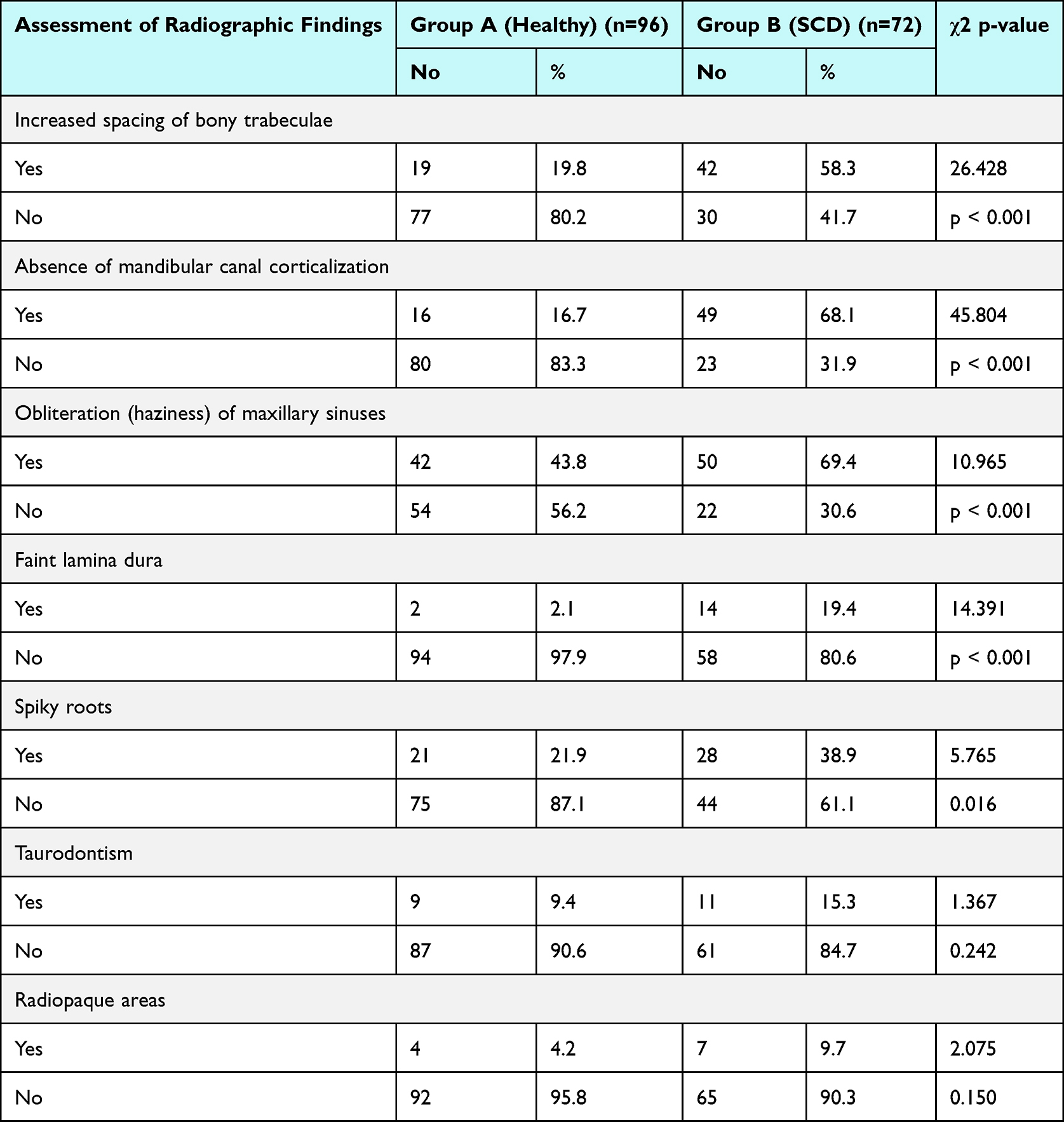 |
Table 1 Distribution of Radiographic Findings in Group A and Group B |
Increased spacing of bony trabeculae was observed in 58.3% of group B compared to 19.8% of group A (χ2 = 26.428, p < 0.001). Similarly, absence of mandibular canal corticalization was present in 68.1% of patients in group B compared to 16.7% of patients in group A (χ2 = 45.804, p < 0.001). Additionally, obliteration (haziness) of the maxillary sinuses was also significantly more frequent in group B (69.4%) than in group A (43.8%) (χ2 = 10.965, p < 0.001). Representative radiographic changes are illustrated in Figure 1.
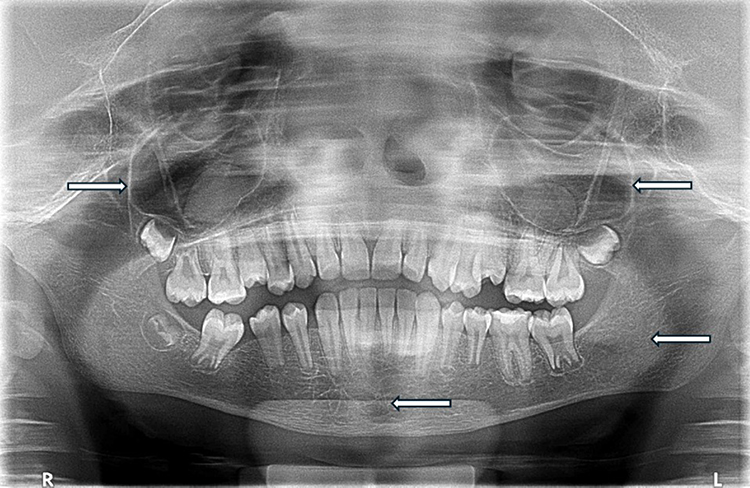 |
Figure 1 Panoramic radiograph of a pediatric patient with severe sickle cell disease demonstrating haziness of the maxillary sinuses, increased spacing of bony trabeculae, and absence of mandibular canal corticalization. White arrows indicate representative areas of interest. |
In addition, faint lamina dura was observed in periapical and bitewing x-rays and found to be more frequent among patients in group B (19.4%) in comparison to group A (2.1%) with a statistical significance (χ2 = 14.391, p < 0.001). A representing periapical radiograph demonstrating faint lamina dura is presented in Figure 2.
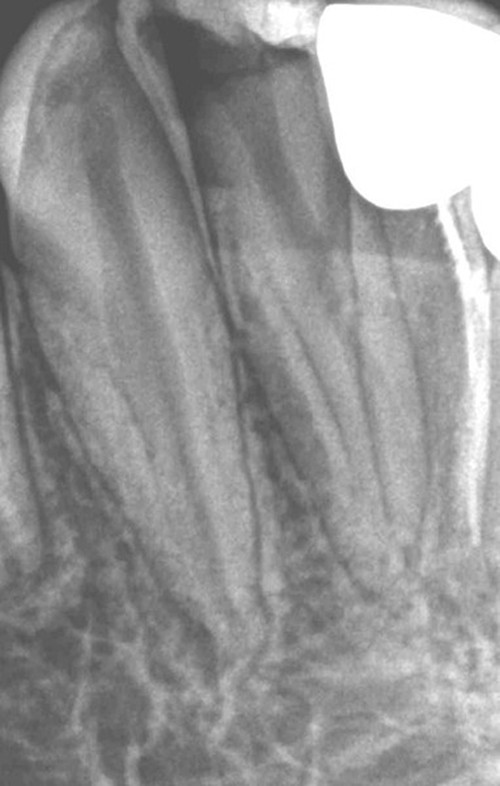 |
Figure 2 Periapical radiograph of a pediatric patient with severe sickle cell disease demonstrating faint lamina dura. |
Moreover, spiky roots were more prevalent in group B (38.9%) when compared to group A (21.9%) with this difference reaching statistical significance (χ2 = 5.765, p = 0.016). In contrast, certain findings showed no substantial differences between the two groups. In this regard, despite the fact that taurodontism was more frequent in group B (15.3%) when compared to group A (9.4%), the difference was statistically insignificant (χ2 = 1.367, p = 0.242). Similarly, radiopaque areas were assessed using panoramic x-rays and were observed in 9.7% of group B and 4.2% of group A without a significant difference (χ2 = 2.075, p = 0.150). Additional representative radiographic changes are presented in Figure 3. Finally, it is worth mentioning that no other changes considered to be related to this disease were observed.
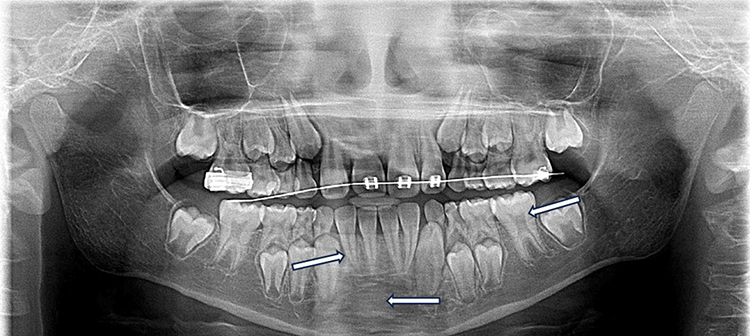 |
Figure 3 Panoramic radiograph of a pediatric patient with severe sickle cell disease demonstrating taurodontism, spiky roots and radiopaque areas. White arrows indicate representative areas of interest. |
Discussion
Sickle cell disease (SCD) encompasses a group of inherited disorders caused by mutations in the HBB gene, which encodes the β-globin subunit of hemoglobin.24 Every year, an estimated 300,000 infants are born worldwide with SCD. While approximately 100,000 individuals with SCD reside in the United States, the majority are in sub-Saharan Africa, India, the Mediterranean region, and the Middle East.2 Although the incidence of SCD is not associated with sex differences, morbidity and mortality rates may differ between males and females.7
A previous retrospective study examined the clinical records of 39 children diagnosed with SCD to investigate sex disparities across various clinical features, including acute manifestations and long-term complications. They reported higher morbidity rates among male patients.7 Similarly, another study with comparable findings was undertaken among patients aged 14 years and older has revealed that males were more frequently admitted to stabilization units for pain management. Notably, older males appeared to experience less severe symptoms.25 However, contrasting results have been reported in a study at a Yemeni hospital. The study recruited 102 children under the age of 16, of whom 56 were males. Interestingly, the prevalence of severe symptoms was higher in females and increased with older ages.26
In our study, all patients exhibited a severe clinical course that ultimately necessitated bone marrow transplantation. Among them, females represented a slightly higher proportion (57%). Additionally, two-thirds of the participants fell within the 10–14-year age group. For the radiographic findings, sex and age, no significant difference was found between age in years and the findings of interest in groups A and B. Similarly, no significant difference was found between sex and radiographic findings in both groups.
This study demonstrated a greater prevalence of widened bony trabecular spacing in 58.3% of the affected group (group B), compared to only 19.8% within the control group (group A). Similarly, a prior study reported that 70% of patients diagnosed with sickle cell anemia displayed a “stepladder” trabecular pattern, characterized by widened marrow spaces.27 Furthermore, another study involving 71 patients with sickle cell disease found a significantly higher incidence of increased spacing of bony trabeculae in the affected group compared to the control group.23 This observation may be attributed to erythroblastic hyperplasia and medullary hypertrophy, which result in the loss of trabeculae and the expansion of marrow spaces.27,28
Furthermore, our study found that 68.1% of subjects within the affected group (group B) exhibited the absence of mandibular canal corticalization, in contrast to 16.7% within the control group (group A).
Similarly, a study conducted by Neves and coworkers evaluated panoramic radiographs of patients with sickle cell disease, reported a significantly higher prevalence of absent mandibular canal corticalization in the affected group.23 Further analysis by Neves and colleagues suggested a correlation between the severity of sickle cell disease and the absence of corticalization of the mandibular canal.29 This observation was consistent with our study as all the subjects presented with severe forms of sickle cell disease, which ultimately required bone marrow transplantation. It is noteworthy that absent corticalization of the mandibular canal may reflect marrow hyperplasia and cortical bone remodeling associated with chronic anemia.30
Of particular relevance in numerous studies was the presence of radiopaque areas.31 In a study conducted by Neves and coworkers, an increased incidence of radiopaque areas within the jaws of SCD patients was reported.23
This finding was of particular importance because the presence of radiopaque regions has been attributed to bony infarctions due to less blood supply.32 However, in our study most of the affected individuals in group B did not exhibit this finding (90.3%). Furthermore, there was no statistically significant difference between the two groups.
This lack of statistical significance may be explained by variability in disease expression and the possibility that such radiographic changes occur intermittently or at different stages of the disease, which may limit their detection in retrospective radiographic assessments.
In a related vein, changes in tooth formation have also been documented in patients suffering from SCD.33 Enamel, dentin, and cementum formation generally follow analogous processes. Any disruptions at any stage of odontogenesis may result in permanent tooth defects, whether it was systemic or localized.34 Notably, reports have documented occurrences of hypomineralization and hypomaturation in both enamel and dentin among patients with SCD.35 In our study, spiky roots were slightly more prevalent in group B (38.9%) than in group A (21.9%). In this context, another study compared dental and bony changes in the jaws of patients with sickle cell anemia and those with sickle cell trait. The study found that the most common dental changes in the sickle cell anemia group included pulp calcification, partial or complete loss of lamina dura, and external root resorption. In contrast, the sickle cell trait group exhibited more frequent changes in root size, shape, and periapical areas, including spiky roots. However, both homozygous and heterozygous patients showed alterations in their jawbones.36
As a further point, our study documented a 19.4% incidence of lamina dura loss in group B in comparison to 2% in the group A. Significantly, this could be attributed to local factors associated with periodontal disease in patients with inherited blood disorder.37 It was found that poor oral hygiene, malocclusion, and drying of the gingival tissues due to lip incompetence may contribute to the higher prevalence of periodontal disease in individuals with blood disorders. Additionally, chronic anemia has been shown to further complicate the condition.37
From a related perspective, taurodontism was observed more commonly in patients with SCD when compared to control groups.38 It is generally believed that taurodontism arises from the failure of Hertwig’s epithelial root sheath to invaginate at the appropriate horizontal level.39 In this regard, individuals presenting with more severe forms of taurodontism have been found to exhibit chromosomal changes more frequently.40 However, in our study the incidence of taurodontism was 15.3% in group B and 9.4% in group A which was not statistically significant.
This finding may be attributed to the multifactorial nature of taurodontism, which is not specific to sickle cell disease and may also occur in the general population. Additionally, the relatively limited sample size and variability in its expression may have reduced the ability to detect a statistically significant difference.
Of particular relevance is another observation of maxillary sinus obliteration which could be attributed to bone marrow hyperplasia.41 It was found that greater sino-nasal morbidity and a broader spectrum of dysfunction have been associated with hemoglobinopathies.42 With respect to these findings, affected patients are generally more susceptible to infections such as chronic sinusitis.43 Moreover, an increased incidence of sinus infections has been observed following bone marrow transplantation due to treatment-induced immunosuppression.44 Consistent with previous information, our study has reported 69.4% maxillary sinus obliteration in the affected group.
In conclusion, the oral radiographic changes associated with SCD are often underappreciated. Furthermore, there is limited evidence in the literature linking oral radiographic characteristics to the severity of SCD. Recognizing such a potential connection could improve prognostic assessment and clinical management of both oral and systemic complications in SCD patients.
This study is a well-characterized pediatric cohort which has assessed a range of variables providing valuable insights into these changes. All the findings were observed in patients with severe forms of SCD who ultimately required bone marrow transplantation. Additionally, the comparative nature of the present study, as opposed to descriptive ones, adds strength to the results.
Collectively, the radiographic alterations observed in this study appear to reflect the cumulative skeletal effects of severe sickle cell disease. Chronic anemia, marrow hyperplasia, and recurrent vaso-occlusive events may contribute to trabecular remodeling, cortical alterations, and changes in bone density within the craniofacial region. These underlying processes likely account for the spectrum of radiographic findings identified in the affected group.
Notably, the findings of the present study have important implications for oral prognosis and clinical management in pediatric patients with SCD. The observed radiographic features, including increased trabeculation and spiky root morphology, likely reflect underlying bone marrow hyperplasia and may indicate altered bone quality, which could influence dental procedures such as extractions and healing capacity. Taurodontism may pose challenges during endodontic treatment, while maxillary sinus haziness may suggest underlying inflammatory changes requiring careful assessment. Furthermore, loss of mandibular canal corticalization may increase the risk of neurovascular complications during surgical interventions. The presence of radiopaque areas may indicate regions of sclerosis or previous bone infarction, reflecting chronic or severe disease involvement, whereas their absence may suggest less advanced osseous changes. Collectively, these findings highlight the importance of careful radiographic evaluation and emphasize the need for a cautious, individualized approach to dental treatment and follow-up in this patient population. Early recognition of these features may contribute to improved treatment planning and better long-term oral outcomes. As a limitation, detailed clinical dental parameters, such as tooth loss, restorative status, and occlusal characteristics, were not included in the analysis, as the primary objective of this study was to assess radiographic features on imaging.
Conclusion
This study demonstrated a significantly higher prevalence of specific oral and maxillofacial radiographic changes in pediatric patients with severe sickle cell disease compared to healthy controls. These findings included increased trabecular spacing, absence of mandibular canal corticalization, faint lamina dura, and haziness of the maxillary sinuses, with taurodontism and radiopaque areas observed to a lesser extent.
These radiographic alterations likely reflect underlying hematologic changes and marrow hyperplasia associated with severe disease. The findings highlight the importance of dental radiographic evaluation in the early identification and clinical management of oral manifestations in pediatric patients with severe sickle cell disease. Further studies are recommended to evaluate these findings across different disease severities and broader populations.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are not publicly available due to institutional and patient confidentiality regulations but can be provided by the corresponding author on reasonable request and with permission from the institution.
Ethics Statement
This study was conducted following approval from the Research Ethics Committee of King Faisal Specialist Hospital & Research Centre (RAC #2231150). As a retrospective study, it involves the review and analysis of existing patient records and radiographic data, without any direct interaction or intervention with patients. A waiver of informed consent was granted by the Research Ethics Committee due to the retrospective nature of the study and the use of previously collected clinical data. No attempt was made to contact patients, and no additional procedures were performed.
All data were handled with strict confidentiality. Personally identifiable information was not collected unless necessary, and any identifiers were removed or coded to ensure patient anonymity. Access to the data was restricted to authorized research team members only, and all data were stored securely in compliance with institutional and national data protection regulations.
The collected data were used solely for the purposes of this research and were not disclosed to any third parties. No secondary use or sharing of identifiable data occurred. Any amendments to the study protocol, or new information affecting the risk-benefit balance, will be reported to the appropriate regulatory authorities in accordance with institutional policies. This study adhered to the ethical principles outlined in relevant international guidelines and the regulations of the Kingdom of Saudi Arabia.
Informed Consent
Informed consent was waived as the study involved retrospective review of anonymized data with no direct patient contact or intervention, and no identifiable information was collected.
Acknowledgments
The authors would like to express their gratitude to the designated Converis system employees at the Office of Research Affairs for their valuable support.
Funding
There is no funding to report.
Disclosure
The authors declare they have no conflict of interest, financial or non-financial benefit in the subject matter discussed in this study.
References
1. Piccin A, Murphy C, Eakins E, et al. Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. Eur J Haematol. 2019;102(4):319–10. doi:10.1111/ejh.13212
2. Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: a review. JAMA. 2022;328(1):57–68. doi:10.1001/jama.2022.10233
3. Steinberg MH. Genetic etiologies for phenotypic diversity in sickle cell anemia. Sci World J. 2009;9:46–67. doi:10.1100/tsw.2009.10
4. Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev. 2003;17(3):167–178. doi:10.1016/S0268-960X(03)00003-1
5. Orelaru F, Bolanle G, Tolulope I, Ishmael J. Assessing knowledge of sickle cell trait/disease inheritance in metropolitan detroit. J Natl Med Assoc. 2019;111(6):656–664. doi:10.1016/j.jnma.2019.09.003
6. Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. Am J Med. 2009;122(6):507–512. doi:10.1016/j.amjmed.2008.12.020
7. Ceglie G, Di Mauro M, Tarissi De Jacobis I, et al. Gender-related differences in sickle cell disease in a pediatric cohort: a single-center retrospective study. Front Mol Biosci. 2019;6:140. doi:10.3389/fmolb.2019.00140
8. Thomson AM, McHugh TA, Oron AP, Collaborators GBDSCD. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: a systematic analysis from the global burden of disease study 2021. Lancet Haematol. 2023;10(8):e585–e99. doi:10.1016/S2352-3026(23)00118-7
9. Elendu C, Amaechi DC, Alakwe-Ojimba CE, et al. Understanding sickle cell disease: causes, symptoms, and treatment options. Medicine. 2023;102(38):e35237. doi:10.1097/MD.0000000000035237
10. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37–47. doi:10.1016/j.blre.2006.07.001
11. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease -- life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. doi:10.1056/NEJM199406093302303
12. Adesina O, Brunson A, Keegan THM, Wun T. Osteonecrosis of the femoral head in sickle cell disease: prevalence, comorbidities, and surgical outcomes in California. Blood Adv. 2017;1(16):1287–1295. doi:10.1182/bloodadvances.2017005256
13. Adeyoju AB, Olujohungbe AB, Morris J, et al. Priapism in sickle-cell disease; incidence, risk factors and complications – an international multicentre study. BJU Int. 2002;90(9):898–902. doi:10.1046/j.1464-410X.2002.03022.x
14. Ataga KI, Orringer EP. Renal abnormalities in sickle cell disease. Am J Hematol. 2000;63(4):205–211. doi:10.1002/(SICI)1096-8652(200004)63:4<205::AID-AJH8>3.0.CO;2-8
15. Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The cooperative study of sickle cell disease. Blood. 1994;84(2):643–649. doi:10.1182/blood.V84.2.643.643
16. DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119(20):4587–4596. doi:10.1182/blood-2011-02-272682
17. Farooq S, Testai FD. Neurologic complications of sickle cell disease. Curr Neurol Neurosci Rep. 2019;19(4):17. doi:10.1007/s11910-019-0932-0
18. Neumayr LD, Hoppe CC, Brown C. Sickle cell disease: current treatment and emerging therapies. Am J Manag Care. 2019;25(18 Suppl):S335–S43.
19. Kanter J, Liem RI, Bernaudin F, et al. American society of hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv. 2021;5(18):3668–3689. doi:10.1182/bloodadvances.2021004394C
20. Krishnamurti L, Neuberg DS, Sullivan KM, et al. Bone marrow transplantation for adolescents and young adults with sickle cell disease: results of a prospective multicenter pilot study. Am J Hematol. 2019;94(4):446–454. doi:10.1002/ajh.25401
21. da Fonseca M, Oueis HS, Casamassimo PS. Sickle cell anemia: a review for the pediatric dentist. Pediatr Dent. 2007;29(2):159–169.
22. Rouse LE, Hays GL. Dental considerations in sickle cell anemia. General Dentistry. 1979;27(6):18–19.
23. Neves FS, de Almeida DA, Oliveira-Santos C, et al. Radiographic changes of the jaws in HbSS and HbSC genotypes of sickle cell disease. Spec Care Dentist. 2011;31(4):129–133. doi:10.1111/j.1754-4505.2011.00195.x
24. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi:10.1038/nrdp.2018.10
25. Udezue E, Girshab AM. Differences between males and females in adult sickle cell pain crisis in eastern Saudi Arabia. Ann Saudi Med. 2004;24(3):179–182. doi:10.5144/0256-4947.2004.179
26. Al-Saqladi A-WM, Delpisheh A, Bin-Gadeem HA, Brabin BJ. Severity of sickle cell disease in Yemeni children. J Trop Pediatr. 2009;55(3):208–209. doi:10.1093/tropej/fmn109
27. Taylor LB, Nowak AJ, Giller RH, Casamassimo PS. Sickle cell anemia: a review of the dental concerns and a retrospective study of dental and bony changes. Spec Care Dentist. 1995;15(1):38–42. doi:10.1111/j.1754-4505.1995.tb00469.x
28. Sanger RG, McTigue DJ. Sickle cell anemia--its pathology and management. J Dentistry Handicapped. 1978;3(2):9–21.
29. Neves FS, Passos CP, Oliveira-Santos C, et al. Correlation between maxillofacial radiographic features and systemic severity as sickle cell disease severity predictor. Clin Oral Investig. 2012;16(3):827–833. doi:10.1007/s00784-011-0577-0
30. Erdoğan Ö, Kisa HI, editors.. Sickle-Cell Disease: A Review of Oral Manifestation and Presentation of a Case with an Uncommon Complication of the Disease. 2013.
31. Cox GM, Soni NN. Pathological effects of sickle cell anemia on the pulp. ASDC J Dent Child. 1984;51(2):128–132.
32. Kavadia-Tsatala S, Kolokytha O, Kaklamanos EG, Antoniades K, Chasapopoulou E. Mandibular lesions of vasoocclusive origin in sickle cell hemoglobinopathy. Odontology. 2004;92(1):68–72. doi:10.1007/s10266-004-0036-3
33. Mello SMF, Paulo CAR, Alves C. Oral considerations in the management of sickle cell disease: a case report. Oral Health Dent Manag. 2012;11(3):125–128.
34. Tran HQ, Jenssen L. Classification of Severe Tooth Discolorations and Treatment Options: Universitetet I Tromsø. 2011.
35. Pithon MM. Orthodontic treatment in a patient with sickle cell anemia. Am J Orthod Dentofacial Orthop. 2011;140(5):713–719. doi:10.1016/j.ajodo.2010.02.039
36. Souza S, de Carvalho H, Costa C, Thomaz E. Association of sickle cell haemoglobinopathies with dental and jaw bone abnormalities. Oral Dis. 2018;24(3):393–403. doi:10.1111/odi.12742
37. Hattab FN. Periodontal condition and orofacial changes in patients with thalassemia major: a clinical and radiographic overview. J Clin Pediatr Dent. 2012;36(3):301–308. doi:10.17796/jcpd.36.3.45763534u3n44k7w
38. de Carvalho H, Rolim JYS, Thomaz ÉBAF, Souza SDFC. Are dental and jaw changes more prevalent in a Brazilian population with sickle cell anemia? Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;124(1):76–84. doi:10.1016/j.oooo.2017.02.016
39. Bharti R, Chandra A, Tikku AP, Wadhwani KK. “Taurodontism” an endodontic challenge: a case report. J Oral Sci. 2009;51(3):471–474. doi:10.2334/josnusd.51.471
40. Yeh S-C, Hsu T-Y. Endodontic treatment in taurodontism with Klinefelter’s syndrome: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;88(5):612–615. doi:10.1016/S1079-2104(99)70094-6
41. Clark CA, Worden CP, Thorp BD, et al. Extramedullary hematopoiesis in the sinonasal cavity: a case report and review of the literature. Allergy Rhinol. 2020;11:2152656720918874. doi:10.1177/2152656720918874
42. Ragab A, Ragab SM, Shawki M. Impact of beta thalassemia on maxillary sinuses and sino-nasal passages: a case control study. Int J Pediatr Otorhinolaryngol. 2015;79(12):2253–2259. doi:10.1016/j.ijporl.2015.10.016
43. Ochocinski D, Dalal M, Black LV, et al. Life-threatening infectious complications in sickle cell disease: a concise narrative review. Front Pediatr. 2020;8:38. doi:10.3389/fped.2020.00038
44. Martino F, Di Mauro R, Paciaroni K, et al. Pathogenesis of chronic rhinosinusitis in patients affected by β-thalassemia major and sickle cell anaemia post allogenic bone marrow transplant. Int J Pediatr Otorhinolaryngol. 2018;106:35–40. doi:10.1016/j.ijporl.2018.01.002
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.