Back to Archived Journals » Oncolytic Virotherapy » Volume 7
Oral poliovirus vaccine-induced programmed cell death involves both intrinsic and extrinsic pathways in human colorectal cancer cells
Authors Zhand S, Hosseini SM, Tabarraei A ![]() , Saeidi M, Saghaeian Jazi M, Kalani MR
, Saeidi M, Saghaeian Jazi M, Kalani MR ![]() , Moradi A
, Moradi A
Received 14 June 2018
Accepted for publication 30 August 2018
Published 30 October 2018 Volume 2018:7 Pages 95—105
DOI https://doi.org/10.2147/OV.S177260
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Athanasios Kyritsis
Sareh Zhand,1 Seyed Masoud Hosseini,1 Alijan Tabarraei,2 Mohsen Saeidi,2 Marie Saghaeian Jazi,3 Mohamad Reza Kalani,4 Abdolvahab Moradi2
1Department of Microbiology, Faculty of Biological Sciences and Technology, Shahid Beheshti University, Tehran, Iran; 2Department of Microbiology, School of Medicine, Golestan University of Medical Sciences, Gorgan, Iran; 3Metabolic Disorders Research Center, Golestan University of Medical Sciences, Gorgan, Iran; 4Department of Biochemistry, School of Molecular and Cell Biology, University of Illinois at Urbana Champaign, Urbana, IL, USA
Purpose: Colorectal cancer (CRC) is one of the most common causes of cancer death throughout the world. Replication-competent viruses, which are naturally able to infect and lyse tumor cells, seem to be promising in this field. The aim of this study was to evaluate the potential of oral poliovirus vaccine (OPV) on human CRC cells and elucidate the mechanism of apoptosis induction.
Materials and methods: Protein and gene expression of poliovirus (PV) receptor (CD155) on four human CRC cell lines including HCT116, SW480, HT-29, and Caco-2 and normal fetal human colon (FHC) cell line as a control were examined by flow cytometry and SYBR Green Real-Time PCR, respectively. Cytotoxicity of OPV on indicated cell lines was tested using MTT assay. The ability of OPV on apoptosis induction for both intrinsic and extrinsic pathways was examined using caspase-8 and caspase-9 colorimetric assay kits. The PV propagation in mentioned cell lines was investigated, and the quantity of viral yields (cells associated and extracellular) was determined using TaqMan PCR.
Results: CD155 mRNA and protein were expressed significantly higher in studied CRC cell lines rather than the normal cell line (P=0). OPV induced cell death in a time- and dose-dependent manner in human CRC cells. Apoptosis through both extrinsic and intrinsic pathways was detected in CRC cells with the minimum level found in FHC. PV viral load was significantly correlated with apoptosis via extrinsic (R=0.945, P=0.0001) and intrinsic (R=0.756, P=0.001) pathways.
Conclusion: This study suggests that OPV has potential for clinical treatment of CRC. However further studies in animal models (tumor xenografts) are needed to be certain that it is qualified enough for treatment of CRC.
Keywords: oncolytic virotherapy, oral poliovirus vaccine, colorectal cancer cells, apoptosis, CD155
Introduction
Colorectal cancer (CRC) is one of the most common causes of cancer death throughout the world with equal mortality in both genders. It occurs as a result of multistep processes caused by the accumulation of genetic/epigenetic changes.1 In Iran, CRC is regarded as the fourth leading cause of death2 and the third most commonly diagnosed cancer.3 Regular CRC screening is one of the most powerful weapons against CRC. Screening can often find CRC early, when it is small, has not spread, and might be easier to treat. Regular screening can even prevent CRC. When CRC is found at an early stage before it has spread, the 5-year relative survival rate is ~90%. However, only approximately four out of 10 CRCs are found at this early stage. When cancer has spread outside the colon or rectum, survival rates are lower.4 Radiotherapy and chemotherapy, which are commonly used for treating cancers, act in an unspecific manner and cause damage to normal cells and even surrounding noncancerous tissues.1 Despite huge advances made in diagnosis, surgery, and systemic therapy, the disease still remains one of the most common causes of death, highlighting the necessity to invent new strategy to combat the disease.5,6 The most common site of metastases for CRC is the liver;7,8 therefore, liver resection is a common choice for treating the disease.9 Unfortunately, two-thirds of patients with successful liver resection may experience the disease recurrence, possibly due to microscopic residual disease.10 Moreover, only one-third of patients with unresectable liver metastases respond to palliative chemotherapy.11 These drawbacks in treatment have stimulated the quest for novel therapies that are applicable. Replication-competent viruses, which are naturally able to infect and lyse tumor cells but not normal cells, seem to be promising in this field.12
Viral oncolysis seems to be a new alternative for cancer treatments, which can combat cancer through different mechanisms and can lead to tumor cell lysis through viral replication or expression of viral cytotoxic proteins.13 The use of viruses for treatment of human cancers has been investigated for almost 50 years.14–17 Virotherapy can overcome potential resistance mechanism developed against standard therapies. Oncolytic virus (OV) not only possesses unique mechanisms of action but also its self-perpetuating nature provides an ideal platform for therapeutic transgenic insertion.18 Majority of tumor cells are resistant to antiproliferative effects of interferons (IFNs) due to various defects in the IFN signal-transduction pathway19 that makes these cells more sensitive to IFNs with a variety of viruses.20–24 Therefore, viruses have engineered to have the ability to selectively replicate in tumor cells25,26 or encode a cytotoxic protein inducing “suicide gene” expression.27 Besides engineered DNA viruses (such as adenovirus, herpes simplex virus, vaccinia virus, and parvovirus) that replicate specifically in tumor cells, RNA viruses with inherent tumor specificity have been developed as well. These OVs include reovirus,28 Newcastle disease virus,29 measles virus,30 vesicular stomatitis virus,6 poliovirus (PV),31 mutant HSV (herpes simplex virus),41 mutant VZV varicella zoster virus),42 and nonpathogenic enterovirus B.43 OVs such as the PV can independently destroy tumor cells without waiting for the host genes to be expressed. The exact mechanism of PV-mediated cytolysis still remains unclear. Combination of shutoff of cellular protein synthesis, inhibition cellular glycoprotein transportation, and the proteolytic digestion of transcription factors have been reported to completely destroy primary cell lines.31
PV, the causative agent of paralytic poliomyelitis, is a non-enveloped positive-stranded RNA virus belonging to the Picornaviridae family. Oral poliovirus vaccines (OPV) are predominantly used in the fight to eradicate polio. There are different types of OPV, which may contain one, a combination of two, or all three different serotypes of attenuated vaccine. PV allows selective targeting of motor neurons via the distribution of its cellular receptors CD155 (also known as poliovirus receptor [PVR]) and nectin-like molecule-5.32–34 CD155 has a critical role in cell adhesion, migration, polarization, and proliferation.35 CD155 expression has been reported to significantly increase several human cancers with various origins, including epidermoid carcinoma, breast carcinoma, osteocarcinoma, colorectal carcinoma, neuroblastoma, and glioblastoma.36–38 CD155 can be used as a unique tumor marker for future PV virotherapy, because it is dominantly expressed in tumor cells.36,37
Results of our previous study indicated that CD155 expression is significantly elevated in CRC cell lines.39 The aim of this study was to evaluate the potential of OPV in cell death induction by activating apoptosis pathway in CRC cell lines rather than normal colon cell lines in order to use as a potential therapeutic agent.
Materials and methods
Cell lines
Four human colon adenocarcinoma cell lines including HCT116 (C570), SW480 (C506), HT-29 (C466) and Caco-2 (C139) were obtained from National Cell Bank of Iran (NCBI). Fetal human colon (FHC) (CRL-1831) cell line was obtained from American Type Cell Culture (ATCC).
Cell culture
HCT116 and Caco-2 cell lines were cultured in DMEM (F12). SW480 and HT-29 cell lines were adapted with RPMI 1640 containing sodium bicarbonate and l-glutamine (Thermo Fisher Scientific, Waltham, MA, USA). FHC (CRL-1831) cell line medium and DMEM: F12 had an extra 10 mM HEPES (for a final concentration of 24 mM), 10 ng/mL cholera toxins, 0.005 mg/mL transferrin, 0.005 mg/mL insulin, and 100 ng/mL hydrocortisone. Cells were grown at 37°C and humidified 5% CO2 atmosphere. CRC cell lines were passaged twice a week. However, the normal colon cell line was passaged once per 14 days.
Total RNA extraction and real-time PCR analysis
Total RNA was extracted from 3×106 of each cell lines according to the TRI Reagent (Sigma-Aldrich Co., St Louis, MO, USA) protocol, and reverse transcriptase was done with Revert Aid First Strand cDNA Synthesis Kit from Thermo Fisher Scientific (Lithuania) using 4 µg of RNA as template and according to the manufacturer’s instructions. SYBR Green Real-Time quantitative PCR analysis was carried out using an ABI Prism 7300 sequence detector system (Applied Biosystems, USA), and β-actin gene was used as an endogenous housekeeping gene for normalization. All primer sets are listed in Table 140 and were purchased from Bioneer (Daejeon, South Korea). The primers used to quantify expression of CD155 gene were located in a region corresponding to the extracellular domain of the protein, which is conserved in all spliced variants. Real-time quantitative PCR cycling conditions were as follows: an initial step at 95°C for 15 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 30 seconds. All assays were performed in triplicate and repeated three times.
 | Table 1 Primer sequences and their nucleotide binding site for expression of CD155 using real-time PCR40 |
Flow cytometry
All cell lines were trypsinized and counted as 5–10×105 cells/tube to be suitable for flow cytometry analysis. All cell lines stained with phycoerythrin (PE) antihuman CD155 (PVR) antibody (BioLegend, San Diego, CA, USA) and with PE Mouse IgG2a, κ Isotype Control (BioLegend) were then analyzed by a flow cytometer (Accuri™ C6; BD Biosciences, San Jose, CA, USA).
Endpoint titration assay (50% tissue culture infectious dose [TCID50])
An OPV containing the Sabin 1 and 3 strains (Razi Vaccine and Serum Research Institute, Karaj, Iran) was used as an OV. The virus titer was determined by measuring the 50% TCID50 in all cell lines. Briefly, normal and all CRC cell lines suspended in a suitable culture medium (as described in the “Materials and methods” section) were seeded in a 96-well micro titer plate at a density of approximately 3.6×104 cells/well and allowed to grow for 24 hours at 37°C in a humidified 5% CO2 environment. The cellar monolayers were thoroughly washed with PBS (pH=7.4) and then incubated with virus (100 µL/well, different dilutions of -1:10 to 1:106). After 1 hour virus attachment, the viruses containing media were depleted and the wells filled with fresh mediums. The virus-infected cell cultures were then maintained at 37°C in a humidified 5% CO2 environment in FBS-free medium (100 µL/well) until complete cytopathic effect (CPE) was visible (up to 7 days). When complete CPE happened, the cells were photographed using microscopy, and those with observable CPE (≥30% of well surface) were marked as “positive CPE” and otherwise were marked as “negative CPE”. Further calculations were conducted using the Reed–Muench formula.
MTT assay
Initially, a 1:2 dilution of the OPV stock with determined TCID50 was prepared followed by a further 10 log2 dilutions and added to cells in quadruplet. Cells were seeded on 96-well plates (1.0×104 cells per well) and treated with OPV at multiplicity of infection (MOI) of 8, 4, 2, 1, 0.5, 0.25, 0.125, 0.0625, 0.031, 0.015, and 0.007 TCID50/cell. The plates were incubated for 1 hour at room temperature for virus attachment. The virus-containing media were depleted, and fresh serum-free equivalents were added to the wells. After 24 and 48 hours incubation at 37°C in a humidified atmosphere containing 5% CO2, the viability was assayed using the MTT test. In all, 20 µL of the 100 µL of MTT solution (at a final concentration of 0.45 mg/mL, prepared in serum-free RPMI; Sigma-Aldrich Co.) was added to each well and incubated at 37°C. After 4 hours, the solution was decanted and the wells were filled with 100% dimethyl sulfoxide (DMSO; Sigma–Aldrich Co.). The plate was shacked slightly to solubilize formazone crystals. The color change was measured using a microplate reader (BioTek, Winooski, VT, USA) at wavelengths of 570–630 nm.
Apoptosis detection by measuring caspase activity
To demonstrate whether cell death in CRC cells following treatment with PV was due to apoptosis, the occurrence of apoptotic cell death was examined using caspase colorimetric assay. The activities of caspases-8/-9 were measured to signify what mechanism (extrinsic or intrinsic pathway) is involved in cell death. For this purpose, four human CRC cell lines (SW480, HCT116, HT-29, and Caco-2) and a normal colon cell line (FHC) were incubated in the presence of OPV at an MOI from 8 to 0.003 TCID50/cell and their viability was measured by the MTT assay. Apoptotic cells were detected using caspase-8 and caspase-9 colorimetric assays. Summarily, the cells were seeded in six-well plates (1.0×106 cells/well) and incubated overnight at 37°C in a humidified atmosphere containing 5% CO2. The cells were incubated with OPV (at an MOI of 1, 0.5, and 0.25 TCID50/cell) for 1 hour at room temperature. The virus-containing media were depleted, and the wells filled with fresh serum-free equivalents. The plates were incubated at 37°C in a humidified atmosphere containing 5% CO2 for 24 and 48 hours, during which the activity of caspase-8 and caspase-9 was assayed by FLICE/caspase-8 colorimetric assay kit (BioVision, Milpitas, CA, USA) and caspase-9 colorimetric assay kit (BioVision), respectively.
Determination of PV viral load
Since OPV was able to induce apoptosis in all studied colon cancer cells, we decided to investigate PV propagation in these cells. To this mean, the cells were exposed to OPV for 24 hours (at the same titer used for apoptosis assay), and the quantity of viral yields (cells associated and extracellular) were determined using TaqMan PCR. Briefly, monolayer of ~106 cells was infected with OPV with appropriate MOIs. After slowly stirring the dish for 60 minutes at room temperature, the cells were thoroughly washed with the medium to remove unbound virus and then incubated in the serum-free medium at 37°C for 24 hours. After virus inoculation, the extracellular virus and the corresponding cell-associated virus were recovered after three consecutive freeze–thaw cycles. The suspensions were clarified by centrifugation 10,000 × g for 5 minutes at 4°C, and 200 µL of each supernatant was used for RNA extraction using PureLink™ Viral RNA/DNA Mini Kit, according to the manufacture’s instruction. Oligonucleotide primers and probe were designed as listed in Table 2. Synthetic oligodeoxynucleotides were prepared, purified, and analyzed. The degenerate primers were used for amplifying PV sequences.
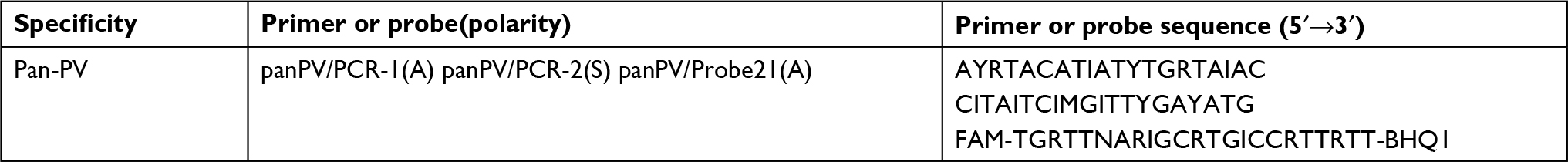 | Table 2 Primers and probe used for determination of PV viral load Note: M, A, and C; R=A and T; I, degenerated base analog. Abbreviation: PV, poliovirus. |
Amplification mixture containing 1X Super ScriptIII Platinum One-Step Quantitative RT-PCR System (Invitrogen), 100 ng of extracted RNA, 1.14 μΜ of forward primer and 1.88 μΜ reverse primer, 0.28 μΜ pan PV probe, and 0.7 mM MgSO4 was added. ROX (6-carboxy-X-rhodamine) reference dye was used, and 0.35 mM of DTT (dithiothreito) was added in a total volume of 35 µL of distilled water. The reverse transcriptase and profile started with an initial reverse transcriptase at 42°C for 45 minutes, followed by reverse transcriptase inactivation for 3 minutes at 95°C. Real-Time PCR program was started at 95°C for 24 seconds, followed by 40 cycles of amplification including denaturation for 30 seconds at 44°C, ramp between annealing and extension for 30 seconds at 52°C, and primer annealing for 1 minute at 60°C.
Preparation of reference standards
For preparation of reference standards, RNA extracted from 200 µL of the OPV with determined TCID50 was used (Invitrogen) following manufacturer’s instructions and the extracted RNA was serial diluted. A seven dilution series of the reference standard was prepared with copy numbers as follows: 0.4×101, 0.4×102, 0.4×10,3 0.4×104, 0.4×105, and 0.4×106 PV viral RNA copies/mL. The results of measurements of each standard viral load were used for the design of a general standard curve for mean CT values in order to calculate copy numbers in samples of different cell lines when mean values had to be compared.
Statistical analysis
CD155 expression level in cell lines was compared using two-way ANOVA (post hoc) test. Statistical significance was determined to be P<0.05. All statistical analyses were performed using SPSS 22. Treatments for real-time PCR, MTT assay, and TCID50 were applied in triplicate, triplicate, and quadruplicate, respectively.
Results
End point titration (TCID50) for OPV
Results of our previous work39 demonstrated that CD155 gene expression is increased significantly (P<0.001) in all studied human colon cancer cell lines compared to the normal cell line. Quantification of obtained signal quantification indicated that CD155 mRNA expression increases up to 491.14-, 461.44-, 24.25-, and 12.04-fold in SW480, HCT116, HT-29, and Caco-2 cell lines, respectively. CD155 protein expression across four different human colon adenocarcinoma cell lines and a normal human colon cell line as a control was examined by flow cytometry analysis. CD155 was detected by PE antihuman PVR CD155 monoclonal antibody using a flow cytometer (BD Accuri C6), and an extensive level of expression was observed in adenocarcinoma cell lines. CD155 protein was expressed on 98.1%, 96.7%, 87.7%, and 57.9% of SW480, HCT116, HT-29, and Caco-2 cell lines, respectively. Collectively, these data indicate a frequently elevated CD155 expression in CRC cell lines.39 To examine whether OPV induces cell death in colon cancer cell lines, the morphological changes in all cell lines 24 and 48 hours after exposure were investigated.
All cell lines were titrated based on the traditional TCID50 method. Visible cell shrinkage and rounding using a phase-contrast microscope were considered symptoms of CPE (Figure 1). Visible cell shrinkage and rounding were considered symptoms of CPE by which further calculations were conducted (using Reed–Muench formula). The highest titer of PV belonged to HCT 116 (4.62×106 TCID50). SW480, HT-29, Caco-2, and FHC had TCID50l of 1.52×106, 5.3×105, 4.95×105, and 2.5×105 per 0.1 mL of OPV, respectively. Interestingly, the TCID50 is directly related to the CD155 expression level at both RNA (R=0.77, P=0.009) and protein (R=0.55, P=0.097) levels, indicating that PV uptake depends on the PVR of cancer cells, as it was expected. Thus, the most killing effect of OPV was observed in the SW480 cell line, which has the highest expression level of CD155.
 | Figure 1 Morphological changes observed by phase-contrast microscope for Caco-2, HT-29, HCT116, FHC, and SW480 cell lines before and after incubation with OPV at highest MOI (MOI=8 TCID50/cell). Notes: Magnification of cell=10×20. (A), 0 hour post infection; (B), 24 hours post infection; (C), 48 hours post infection. Abbreviations: FHC, fetal human colon; MOI, multiplicity of infection; OPV, oral poliovirus vaccine; TCID50, 50% tissue culture infectious dose. |
OPV decreases viability of CRC cell lines at different MOIs
OPV was found to strongly induce cell death in a time- and dose-dependent manner in all studied human colon cancer cell lines (Figure 2A and B). Statistical analysis showed that at MOIs from 8 to 1, the viability of normal FHC cell line in both studied timetable was significantly higher than that of other studied cancer cell lines (detailed statistical analysis is shown in Figure 2A and B), which makes OPV as a suitable candidate for virotherapy. FHC exhibited a lower degree of morphological changes. It was considered that the FHC cell line was not susceptible to morphological change for OPV. In this study, the MOIs lower than one-fourth of TCID50/cell was not considered for further studies because of the lower virus particle. The viability of the FHC cells exposed to the PV was significantly higher than that of the cancerous cells at 48 hours or more after infection (data not shown). The present study showed that OPV can kill various types of CRC cell lines (5.43%–99.6% toxicity), with less toxicity (0.89%–61%) on normal FHC colon cell, and consequently, there is more viral propagation in infected cancer cell. According to statistical analysis, this difference is significant (P=0.005). This finding illustrates the specificity of OPV in CRC cell growth inhibition and promises its potential for in vivo viral therapeutics.
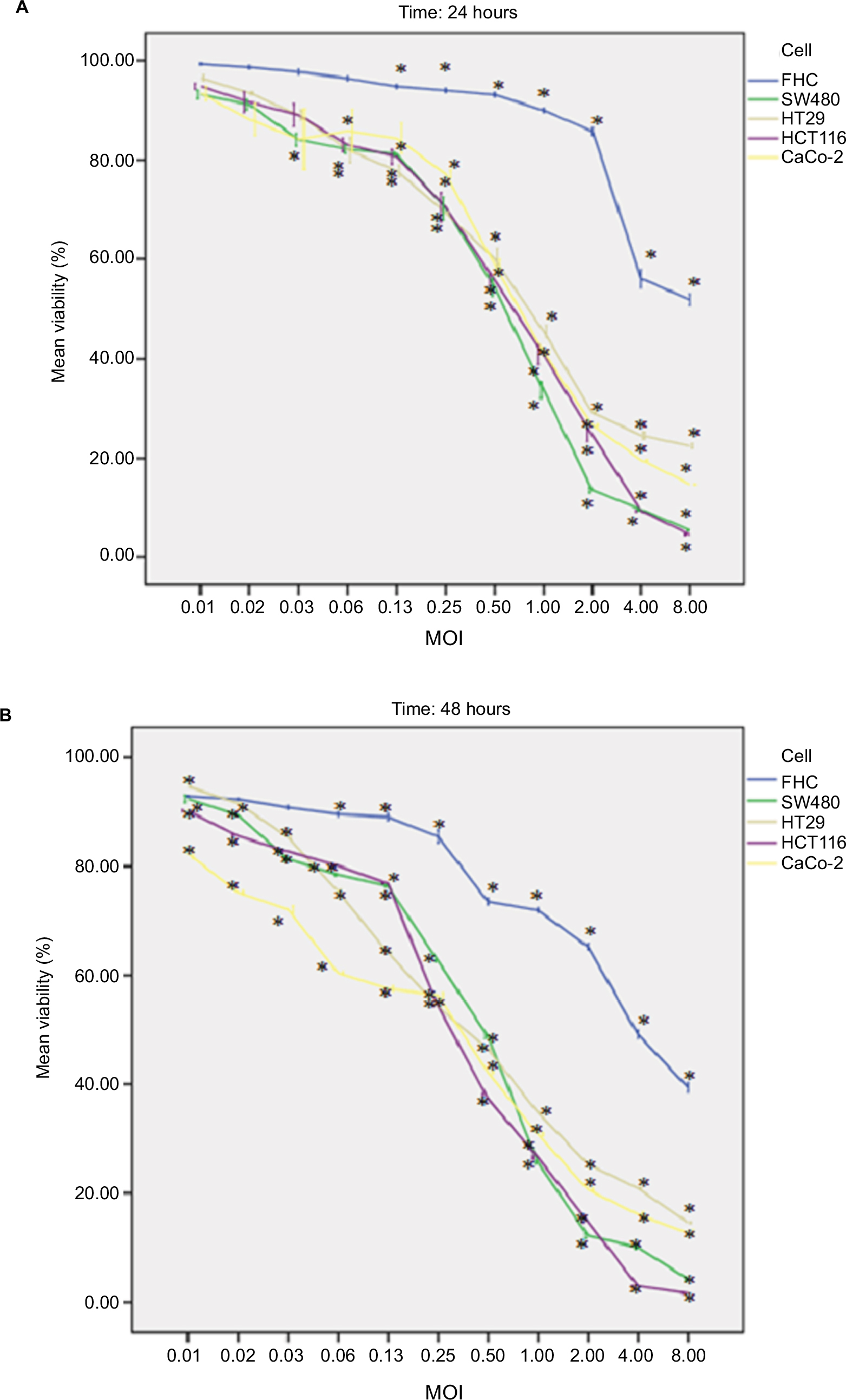 | Figure 2 Effect of OPV on human colon cancer and normal cell lines’ viability. Notes: Cell lines were incubated with OPV for 24 and 48 hours at the indicated MOI. (A) At 24 hours and (B) 48 hours interval, the cell viability was assessed by the MTT assay. OPV induced cell death in a time- and dose-dependent manner in CRC cell lines. Bars represent the mean±SEM. *P<0.05; post-Hoc test applied. Error bars ±1SE. Abbreviations: CRC, colorectal cancer; FHC, fetal human colon; MOI, multiplicity of infection; OPV, oral poliovirus vaccine; SE, standard error; SEM, standard error of the mean. |
Because of the lowest expression of CD155 on FHC cells, the PV could rarely infect FHC cells. The mean viral production of OPV in HCT116 was 4.5×106 TCID50l, while the mean production of FHC was 2.5×105 TCID50 per 0.1 mL of OPV.
OPV causes CPE faster in HCT116 than in FHC, and 6CPE was found after 48 hours and only at the highest MOI in this cell line, whereas CPE was found in all tested MOIs after 24 hours in all indicated CRC cell lines. This indicates that the same viral serotype may have a different impact when infecting cancer cells derived from the same original cancer and also may support the clinical safety of OPV for in vivo studies
OPV induced apoptosis in studied CRC cell lines via extrinsic and intrinsic pathways
Results showed that PV induced the activation of caspase-8 and caspase-9 in a time- and dose-dependent manner (Figure 3). In the studied MOIs, cancer cell lines rather than normal FHC cell lines show the highest caspase activity in both extrinsic and intrinsic pathways. Statistical analysis indicated that there was a significant relation between induction of caspase-8 activity in MOI=0.5 and MOI=0.25 in both timetables among all studied CRC cell lines rather than the normal cell line (P=0.008 and P=0.006, respectively). Analysis of individual cell lines at indicated MOIs also showed that OPV significantly induces caspase-8 activity in both timetables in HCT116 (P=0.014 and P=0.013, respectively) and SW480 (P=0.006 and P=0.012, respectively) against normal FHC cell line. Data showed that there was a significant relation between induction of caspase-9 activity in all studied MOIs (1, 0.5, and 0.25) in both timetables among all studied CRC cell lines rather than the normal cell line (P=0.005, P=0.036, and P=0.041, respectively). Analysis of individual cell lines at indicated MOIs also showed that OPV significantly induces caspase-9 activity in both timetables in SW480 against normal FHC cell line (P=0.003, P=0.020, and P=0.029, respectively). Hence, we concluded that OPV induced apoptosis in studied CRC cell lines via extrinsic and intrinsic pathways. The most caspase activity was observed in the SW480 cell line; of note, this cell line harbored the highest CD155 cellular level and consequently low viability of infected cell. On the other hand, the normal colon cell line, FHC, showed no CPE in low viral doses with the minimum caspase activity.
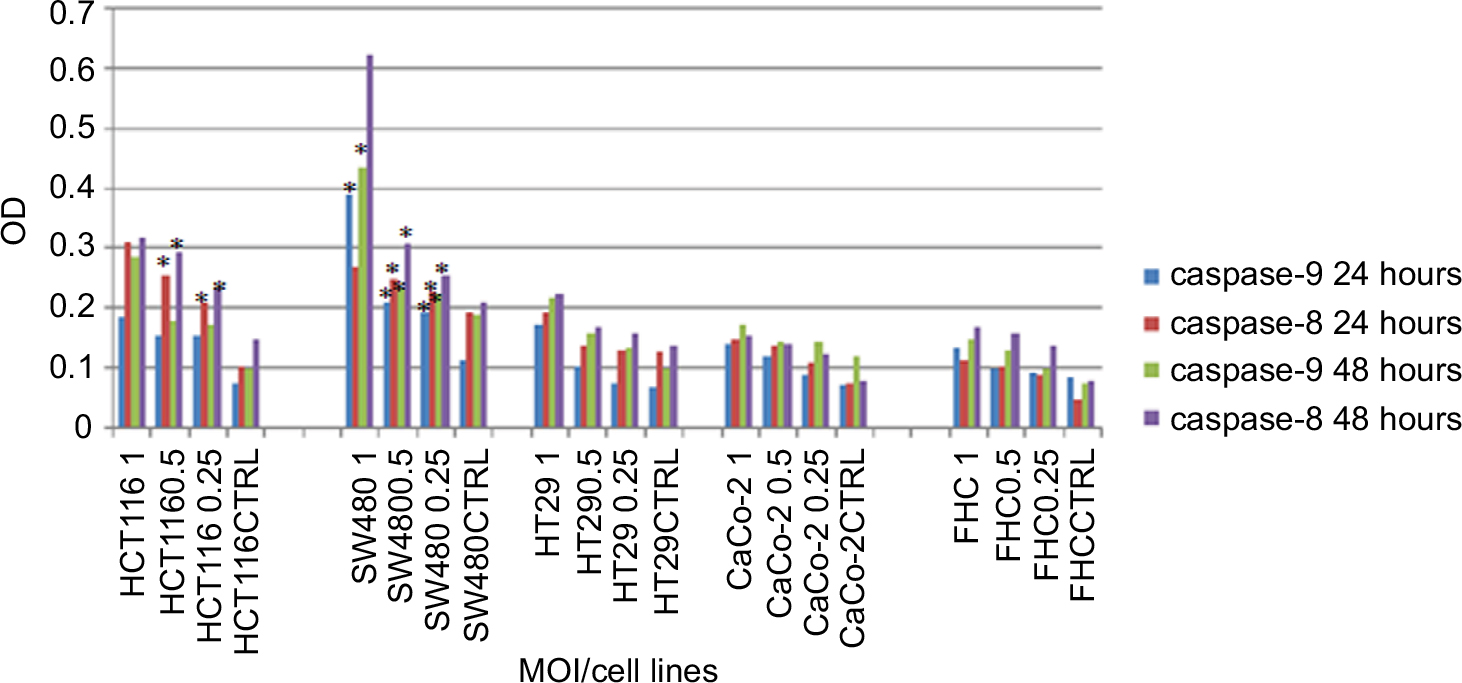 | Figure 3 Caspase-8 and caspase-9 colorimetric assays after 24 and 48 hours exposure to OPV in all colon cancer and normal colon cell lines. Note: 1, MOI 1; 0.5, MOI 0.5; 0.25, MOI 0.25. Abbreviations: CTRL, control; FHC, fetal human colon; MOI, multiplicity of infection; OPV, oral poliovirus vaccine. |
Propagation of OPV in colon cancer and normal cell lines
Viral load of PV in both normal and cancerous cell lines after 24 hours exposure is listed in Table 3. The results showed that PV viral load was significantly correlated with apoptosis via extrinsic (R=0.945, P=0.0001) and intrinsic (R=0.756, P=0.001) pathways. There was not any significant difference between CRC and normal colon cell lines with respect to apoptosis via intrinsic and extrinsic pathways (P=0.1).
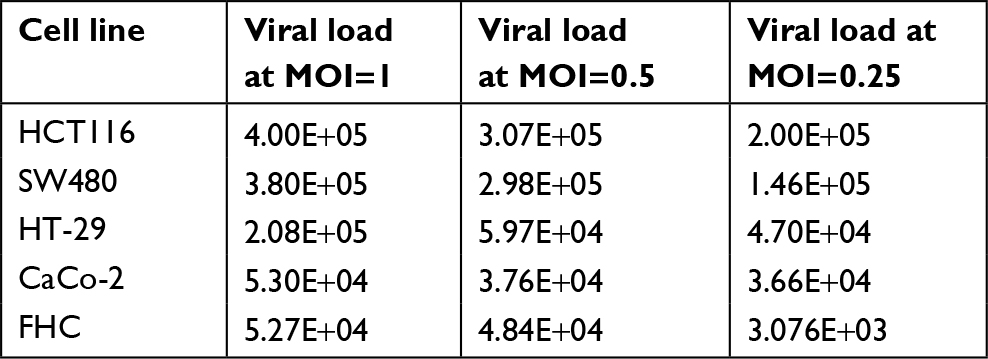 | Table 3 Viral load of PV in all normal and cancerous cell lines after 24 hours exposure Abbreviations: FHC, fetal human colon; MOI, multiplicity of infection; PV, poliovirus. |
Our findings revealed that the extrinsic apoptotic pathway (caspase-8) and the intrinsic programmed cell death (caspase-9) is the main responsible mechanism of CRC cell death. The cross talk between both intrinsic and extrinsic apoptosis pathways may explain this process.
Previous reports on the apoptotic pathways induced by PV infection in Caco-2 CRC cell have demonstrated that mitochondria play a key role in PV-induced apoptosis,41 but other tested colorectal cell lines are not included. However, we found both extrinsic and intrinsic apoptosis responses were effective in all tested colon cancer cells.
In this study, for the first time, we tested the viral load of virus after virotherapy to be confident that the observed apoptosis is directly for virus replication and we found out that viral load is also directly correlated with the infected cell caspase activity level, supporting the fact that productive viral infection can be associated with the cell death. Interestingly, the polio viral load is significantly related to apoptosis via extrinsic (R=0.945, P=0.0001) and intrinsic (R=0.756, P=0.001) pathways, indicating that all studied cell lines’ death via apoptosis depends on the PV replication, as it was expected.
Discussion
Despite huge advances in the field of CRC therapy, the disease still remains a major cause of death.42 Viral oncolysis seems to be a new alternative for cancer treatments, which can combat cancer through different mechanisms and it can lead to tumor cell lysis through viral replication or expression of viral cytotoxic proteins.13 Virotherapy can overcome potential resistance mechanism developed against standard therapies. OV not only possesses unique mechanisms of action but also its self-perpetuating nature provides an ideal platform for therapeutic transgenic insertion.18
Virus strains used as human vaccines need to be engineered genetically in order to increase their tumor targeting ability prior to being applied as OVs. Because of the attenuated nature, the need for removing pathogenic factors is not a priority for OVs. The polio and measles vaccines are prominent examples of viral vaccines developed for virotherapy.43 OVs such as the PV can independently destroy tumor cells without waiting for the host genes to be expressed. The exact mechanism of PV-mediated cytolysis still remains unclear. Combination of shutoff of cellular protein synthesis, inhibition of cellular glycoprotein transportation, and the proteolytic digestion of transcription factors have been reported to completely destroy primary cell lines.31
OVs such as mutant HSV, mutant VZV, and non-pathogenic enterovirus B species have been reported to be efficient in killing CRC cells. The feasibility of using G207, a multi-mutated, replication-competent type-1 herpes simplex virus in the treatment of human CRC, was examined on five human CRC cell lines to determine efficacy of infection and tumor cell kill. The results of measuring response to G207 therapy in vitro was 1%–93%.44 The priority of our study is that we test the OPV in both normal and cancer cell lines to predict the possible outcome in in vivo experiments in order to prevent undesired issues. Moreover, we specified our study according to specific receptor of PV CD155 in CRC cell lines too.
Oncolytic activity of recombinant wild-type and M protein mutant vesicular stomatitis viruses in RKO, Hct116, and LoVo CRC cells, as well as in human fibroblast and hepatocyte primary cultures, was tested. RKO and Hct116 cells were sensitive to both viruses, whereas LoVo cells were resistant.45 Viability of RKO cells reduced to approximately 10%–20% of mock-infected controls by 48 hours post infection, and Hct116 cells were intermediate in their sensitivity. The results of the present study showed that the sensitivity of Hct116 to OPV was much higher than that to VZV and the toxicity of OPV was 99.6% after 24 hours.
The oncolytic potential of several low-pathogenic human enterovirus B species echovirus 12, 15, 17, 26, and 29 with rare clinical occurrence on six colon cancer cell lines (Caco-2, HT-29, LoVo, SW480, SW620, and T84) and CPEs as well as viral replication efficacy has been investigated. Echoviruses 12, 17, 26, and 29 replicated efficiently in almost all cell lines and were considered as highly cytolytic.46
The limitation of their study against the result of the present study was that E12 replicated in HT-29, SW480, and Caco-2 cell lines, but no CPE was observed after 7 days, whereas in our study, the indicated cell lines showed complete CPE after 2 days infection with OPV, and also, no infection was indicated with E15 in SW480, whereas in our study, this cell line showed the highest CD155/PVR expression level, had higher TCID50/mL, and showed CPE after 2 days post infection.
The small virion size (~30 nm in diameter) of picornaviruses could be an oncolytic advantage. Picornaviruses have a relatively fast replication cycle, ranging from 5 to 10 hours. This could also be an advantage for viral oncolysis, since a faster replicating virus may reduce a tumor in size more effectively than a slower replicating virus.46
However, when using oncolytics in vivo, targeting the cancer cell is very important to prevent toxic side effects in normal colon cells. For CRC viral therapy, using PVs may seem to be more specific since it has been reported that CRC cells express more PVR (CD155) than normal colon cells.40 Our previous experiments also support CD155 overexpression at both mRNA and protein levels in different CRC cell lines (SW480, HCT116, HT-29, and Caco-2) in comparison with normal colon cell line.39 CD155 differential overexpression can be useful when using viral therapeutics strategies, since it is sufficient for all binding and entry functions, leading to uncoating of the viral genome and virus uptake.47,48
The result of present study in contrast with results of Eswaran et al49 showed the higher TCID50 for OPV, and this difference of TCID50 for the studied cell lines may relate directly to the CD155 expression level.
In this study, we showed that OPV possesses an inherent capacity to induce apoptosis in CRC cell lines in vitro and has ability to use in in vivo studies.
It has been reported that PV infection can trigger apoptosis in neuroblastoma and bone and soft tissue sarcoma cells expressing CD155, as shown by DNA fragmentation, activation of effector caspase, mitochondrial dysfunction, and TUNNEL assay.50–52 Our findings similarly showed that the PV infection leads to programmed cell death in CRC cell lines in a time- and dose-dependent manner.
It has been shown that cells respond to PV infection by switching on the apoptotic program, implementation of which is usually suppressed by viral antiapoptotic functions. PV infection activates the apoptotic pathway, involving mitochondrial damage, cytochrome c efflux, and consecutive activation of caspase-9 and caspase-3.53 Apart from the involvement of activation of caspases, the mechanism of apoptosis triggered by PV infection remains unknown. An important point concerns relationships between the induction of the antiapoptotic state and commitment to CPE.53
There are also some limitations of this study, which include the inability of virus to home to some tumor types and the therapeutic window. In the in vivo study, we try to administrate the OPV intratumor to decrease the effect of anti-polio antibodies, present following pediatric vaccination, and also try to use some protocols to boost the ability of virus to home to tumor cells.
Conclusion
Our results show that OPV can induce apoptosis in CRC cell lines either by intrinsic (mitochondrial pathway) or extrinsic pathways. In addition, CPE observed at all indicated MOIs and after short period of time in studied CRC cell lines. The results of this study suggest that OPV has potential for the clinical treatment of CRC. However, further studies in animal models (tumor xenografts) need to be certain that it is qualified enough for treatment of CRC. The safety of normal cells also needs to be assured before proceeding with clinical trials.
Acknowledgments
We thank Dr Farzaneh and Dr Nasimian from Iranian Biological Resource Center for their compassionate effort in cell culture and also Golestan University of Medical Sciences for the continuous encouragement during this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Davis D, Lahiri S. Application of Oncolytic Viruses for Cure of Colorectal Cancer. Cancer Res J. 2015;3(4):76. | ||
Malekzadeh R, Bishehsari F, Mahdavinia M, Ansari R. Epidemiology and molecular genetics of colorectal cancer in Iran: a review. Arch Iran Med. 2009;12(2):161–169. | ||
Kolahdoozan S, Sadjadi A, Radmard AR, Khademi H, Five common cancers in Iran. Arch Iran Med. 2010;13(2):143. | ||
Society AC, editor. Colorectal Cancer Early Detection, Diagnosis, and Staging. American Cancer Society Atlanta: American Cancer Society; 2017. | ||
Balachandran S, Barber GN. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life. 2000;50(2):135–138. | ||
Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6(7):821–825. | ||
Weiss L, Grundmann E, Torhorst J, et al. Haematogenous metastatic patterns in colonic carcinoma: an analysis of 1541 necropsies. J Pathol. 1986;150(3):195–203. | ||
Willett CG, Tepper JE, Cohen AM, Orlow E, Welch CE. Failure patterns following curative resection of colonic carcinoma. Ann Surg. 1984;200(6):685–690. | ||
Fong Y, Cohen AM, Fortner JG, et al. Liver resection for colorectal metastases. J Clin Oncol. 1997;15(3):938–946. | ||
Panis Y, Ribeiro J, Chrétien Y, Nordlinger B. Dormant liver metastases: an experimental study. Br J Surg. 1992;79(3):221–223. | ||
Fong Y, Kemeny N, Paty P, Blumgart LH, Cohen AM, editors. Treatment of Colorectal Cancer: Hepatic Metastasis. Seminars in Surgical Oncology. New York: Wiley Online Library; 1996. | ||
Kenney S, Pagano JS. Viruses as oncolytic agents: a new age for “therapeutic” viruses? J Natl Cancer Inst. 1994;86(16):1185–1186. | ||
Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4(2):101–117. | ||
Asada T. Treatment of human cancer with mumps virus. Cancer. 1974;34(6):1907–1928. | ||
Moore AE. Viruses with oncolytic properties and their adaptation to tumors. Ann N Y Acad Sci. 1952;54(6):945–952. | ||
Southam CM. Division of microbiology: present status of oncolytic virus studies. Trans N Y Acad Sci. 1960;22(8 Series II):657–673. | ||
Taylor MW, Cordell B, Souhrada M, Prather S. Viruses as an aid to cancer therapy: regression of solid and ascites tumors in rodents after treatment with bovine enterovirus. Proc Natl Acad Sci U S A. 1971;68(4):836–840. | ||
Motalleb G. Virotherapy in cancer. Iran J Cancer Prev. 2013;6(2):101–107. | ||
Wong LH, Krauer KG, Hatzinisiriou I, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J Biol Chem. 1997;272(45):28779–28785. | ||
Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330(1):34–49. | ||
Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4(12):e353. | ||
Haralambieva I, Iankov I, Hasegawa K, Harvey M, Russell SJ, Peng KW. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol Ther. 2007;15(3):588–597. | ||
Elankumaran S, Chavan V, Qiao D, et al. Type I interferon-sensitive recombinant newcastle disease virus for oncolytic virotherapy. J Virol. 2010;84(8):3835–3844. | ||
Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9(9):1163–1176. | ||
Khuri FR, Nemunaitis J, Ganly I, et al. a controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6(8):879–885. | ||
Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. Embo J. 1998;17(12):3351–3362. | ||
Klatzmann D, Philippon J, Valery CA, Bensimon G, Salzmann J-L. Gene Therapy for Glioblastoma in Adult Patients: Safety and Efficacy Evaluation of an In Situ Injection of Recombinant Retroviruses Producing Cells Carrying the Thymidine Kinase Gene of the Herpes Simplex Type 1 Virus, to be Followed with the Administration of Ganciclovir. Laboratoire Immunologie B, Hôpital Pitié-Salpêtrière, Paris Cedex, France. Hum Gene Ther. 1996;7(1):109–126. | ||
Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282(5392):1332–1334. | ||
Sinkovics JG, Horvath JC. Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol. 2000;16(1):1–15. | ||
Springfield C, Fielding A, Peng K, Galanis E, Russell SJ, Cattaneo R. Measles virus: improving natural oncolytic properties by genetic engineering. In: Horvarth J, Sinkovics J, editors. Viruses Therapy of Human Cancers. New York: Marcel Dekker Inc.; 2004. | ||
Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci U S A. 2000;97(12):6803–6808. | ||
Ren RB, Costantini F, Gorgacz EJ, Lee JJ, Racaniello VR. Transgenic mice expressing a human poliovirus receptor: a new model for poliomyelitis. Cell. 1990;63(2):353–362. | ||
Koike S, Taya C, Kurata T, et al. Transgenic mice susceptible to poliovirus. Proc Natl Acad Sci U S A. 1991;88(3):951–955. | ||
Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A. 1996;93(6):2370–2375. | ||
Takai Y, Miyoshi J, Ikeda W, Ogita H. Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol. 2008;9(8):603–615. | ||
Solecki D, Wimmer E, Lipp M, Bernhardt G. Identification and characterization of the cis-acting elements of the human CD155 gene core promoter. J Biol Chem. 1999;274(3):1791–1800. | ||
Solecki D, Bernhardt G, Lipp M, Wimmer E. Identification of a nuclear respiratory factor-1 binding site within the core promoter of the human polio virus receptor/CD155 gene. J Biol Chem. 2000;275(17):12453–12462. | ||
Gromeier M, Solecki D, Patel DD, Wimmer E. Expression of the human poliovirus receptor/CD155 gene during development of the central nervous system: implications for the pathogenesis of poliomyelitis. Virology. 2000;273(2):248–257. | ||
Zhand S, Hosseini SM, Tabarraei A, Moradi A, Saeidi M. Analysis of poliovirus receptor, CD155 expression in different human colorectal cancer cell lines: Implications for poliovirus virotherapy. J Can Res Ther. Epub 2017 Jan. | ||
Masson D, Jarry A, Baury B, et al. Overexpression of the CD155 gene in human colorectal carcinoma. Gut. 2001;49(2):236–240. | ||
Blondel B, Autret A, Brisac C, et al. Apoptotic signaling cascades operating in poliovirus-infected cells. Front Biosci. 2009;14:2181–2192. | ||
Mishra J, Drummond J, Quazi SH, et al. Prospective of colon cancer treatments and scope for combinatorial approach to enhanced cancer cell apoptosis. Crit Rev Oncol Hematol. 2013;86(3):232–250. | ||
Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. 2012;18(1):69. | ||
Kooby DA, Carew JF, Halterman MW, et al. Oncolytic viral therapy for human colorectal cancer and liver metastases using a multi-mutated herpes simplex virus type-1 (G207). FASEB J. 1999;13(11):1325–1334. | ||
Stewart JH, Ahmed M, Northrup SA, Willingham M, Lyles DS. Vesicular stomatitis virus as a treatment for colorectal cancer. Cancer Gene Ther. 2011;18(12):837–849. | ||
Israelsson S, Jonsson N, Gullberg M, Lindberg AM. Cytolytic replication of echoviruses in colon cancer cell lines. Virol J. 2011;8:473. | ||
Toyoda H, Franco D, Fujita K, Paul AV, Wimmer E. Replication of poliovirus requires binding of the poly(rC) binding protein to the cloverleaf as well as to the adjacent C-rich spacer sequence between the cloverleaf and the internal ribosomal entry site. J Virol. 2007;81(18):10017–10028. | ||
Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56(5):855–865. | ||
Eswaran SP, Praharaj AK, Chander Y, Nagendra A. Potency titration of oral polio vaccine by estimation of live virus content using tissue culture technique. Med J Armed Forces India. 2003;59(2):105–107. | ||
Toyoda H, Ido M, Hayashi T, et al. Experimental treatment of human neuroblastoma using live-attenuated poliovirus. Int J Oncol. 2004;24(1):49–58. | ||
Gosselin AS, Simonin Y, Guivel-Benhassine F, et al. Poliovirus-induced apoptosis is reduced in cells expressing a mutant CD155 selected during persistent poliovirus infection in neuroblastoma cells. J Virol. 2003;77(1):790–798. | ||
Atsumi S, Matsumine A, Toyoda H, et al. Oncolytic virotherapy for human bone and soft tissue sarcomas using live attenuated poliovirus. Int J Oncol. 2012;41(3):893–902. | ||
Belov GA, Romanova LI, Tolskaya EA, Kolesnikova MS, Lazebnik YA, Agol VI. The major apoptotic pathway activated and suppressed by poliovirus. J Virol. 2003;77(1):45–56. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.