Back to Archived Journals » Open Access Animal Physiology » Volume 6
Optogenetics: illuminating the neural bases of rodent behavior
Authors Francis TC, Chaudhury D, Lobo MK
Received 17 June 2014
Accepted for publication 3 September 2014
Published 8 December 2014 Volume 2014:6 Pages 33—51
DOI https://doi.org/10.2147/OAAP.S42339
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Peter Koulen
T Chase Francis,1 Dipesh Chaudhury,2 Mary Kay Lobo1
1Department of Anatomy and Neurobiology, University of Maryland School of Medicine, Baltimore, MD, USA; 2Department of Biology, New York University Abu Dhabi, Abu Dhabi, United Arab Emirates
Abstract: In vivo optogenetics has provided researchers with the ability to delve deeper into the neural basis of behavior by driving cell-type specific circuit connections within and between brain regions. The diverse toolbox available for circuit- and cell-specific manipulations is ever growing. Using these tools in conjunction with established and novel genetic and behavioral methods, neuroscience research has experienced an explosion in the understanding of the roles of specific cell subtypes in behavior. This review aims to outline recent advances in in vivo optogenetic tools for manipulation of behavior related to movement, pain and sensation, motivation, reward, emotion, learning, sleep, and epilepsy.
Keywords: in vivo optogenetics, behavior, neural circuits, transgenics, viral constructs
Introduction
Optogenetics, the integration of optics and genetics to manipulate cellular functions and activity, has been in use for nearly 10 years and its utility in neuroscience research is ever increasing. Neuroscientists initially used naturally occurring microbial opsins and later mutated versions of these opsins to alter the flux of ions across membranes to drive cell activity.1–3 Microbial opsins originally from archaebacteria2,3 or algae,1,3,4 contain a ligand binding site that is sensitive to distinct wavelengths of light and insensitive to chemical ligands. Through a series of selective and non-selective mutations, a large library of effective, non-toxic, temporal, and wavelength specific optogenetic constructs have been generated.3,5 The versatility of the available optogenetic constructs allows use in a widevariety of applications including in vivo behavioral manipulation.
The most commonly used membrane-bound optogenetic constructs fall under two major classes: 1) stimulatory/depolarizing opsins, often light-driven cation channels (channelrhodopsin; ChR2), and 2) inhibitory/hyperpolarizing opsins, light-driven chloride (halorhodopsin or Natronomonas pharaonis; NpHR) or proton (bacteriorhodopsin) pumps. However, other opsin constructs have emerged from the wealth of information about opsin mechanisms including light-driven constructs to regulate G-protein mediated signaling,6–8 intracellular signaling molecules,6,9–13 and gene expression.14,15 Microbial opsins have been incorporated in combination with numerous mouse transgenic technologies and viral constructs. These tools and constructs are utilized in vivo and ex vivo to drive specific cell subtypes in a region and temporal specific manner. Optogenetic manipulation is currently used in neuroscience to understand temporal and activity specific alterations in neuronal circuits, circuit connectivity, signaling mechanisms, and gene regulation that underlie behavior. With new and emerging technologies, the utility of optogenetic methods is ever growing. In this review, we highlight recent advances in optogenetic tools and constructs with a particular focus on its uses in behavioral systems.
Techniques and technological advances
A decade of optogenetic research has assembled simple, reliable, and effective means for opsin expression and light activated manipulation of neuronal activity in vivo. Opsin constructs are expressed in the central nervous system (CNS) with the use of viral vectors in wild-type or transgenic rodent models (Table 1). Specific control over particular circuits and neuronal populations within a brain region is achieved by implanting optic fibers, which are attached to a light source.16–18 Additionally, researchers have developed minimally invasive and wireless technologies for in vivo optogenetics.19,20 Further, optogenetics coupled with in vivo recording or calcium imaging has the potential to create the ability to analyze temporally specific single-cell and circuit level function within awake and behaving animals.
 | Table 1 Transgenic mouse lines used for optogenetic manipulation of behavior |
Viral and transgenic technological advances have paved the way for innovative targeting of opsin expression. Two major strategies are used to mediate opsin expression in the CNS: 1) direct opsin transgene expression, driven by a specific promoter in rodents, or 2) indirect expression of the opsin with a secondary promoter-driven element, such as Cre recombinase. These strategies have been used with both viral constructs and transgenic3 and knock-in rodent lines.21 Some of the first optogenetic studies utilized transgenic animals to express ChR2 or NpHR using neuron-specific promoters.22,23 Use of rodent transgenics and knock-in technology to express opsins in neurons has the selective advantage of spatial homogeneity and abundant opsin expression and this technology was successfully used in a number of studies in a variety of cell subtypes.24–27 Knock-in mice, in particular, promote endogenous expression patterns which regulate the magnitude of opsin expression. Further, opsins expressed in genetic models have early onset expression which is useful in developmental studies.28 Bacterial artificial chromosome (BAC) transgenic or knock-in lines have been widely successful for expressing Cre or opsins using numerous cell-type specific promoters. This success stems from the large genetic capacity of BAC clones (~150 kilobases), which contain many of the genetic elements necessary to drive cell-specific expression or in the case of knock-ins having the opsin driven under the endogenous promoter and regulatory elements.21,29–35
Viral systems are widely used in optogenetic studies. Adeno-associated viruses (AAVs) or lentiviruses driven by neuron-specific promoters are injected directly in the brain region of interest (Figure 1A). Since opsins infect somas, dendrites, and axons, axonal terminals can be targeted in brain regions down-stream of the injection site. Indirect, binary expression systems, such as Cre-driven systems, provide high specificity for cell subtypes and permit the use of multiple viral constructs.3 One commonly used viral Cre system utilizes a double inverted open (DIO) reading frame construct to express opsins in a cell-type specific manner (Figure 1B). In a DIO system, the non-coding, opsin gene, in reverse, is flanked by two pairs of distinct lox sites specifically recognized and cleaved by Cre recombinase. In the presence of Cre, splicing occurs between one set of lox sites causing the gene to flip into the correct coding orientation. A second splicing event leads to the removal of two lox sites leaving the two incompatible lox sites, which prevents the gene from flipping back into the non-coding reverse direction, thus allowing expression of the opsin. The DIO opsin construct is often delivered via an AAV injection into a Cre expressing animal or in conjunction with a Cre expressing virus.3 Other binary expression systems, such as the tetracycline regulatory system, have also successfully been used in optogenetic studies.36–38 For instance, studies examining a fear memory engram, induced by fear conditioning, used a transgenic mouse line expressing a c-Fos specific promoter coupled to a gene encoding tetracycline transactivator (tTA) in the hippocampus to selectively target activated cells with ChR2 (Figure 1C).37,38 Doxycycline blocks the activity of tTA and when animals no longer receive doxycycline, ChR2 is expressed in c-Fos positive neurons using a tTA-ChR2-EYFP virus.
 | Figure 1 Common schemes utilized for optogenetic manipulation of neuronal subtypes. |
Trans-synaptic constructs to target opsins to presynaptic axonal afferents and upstream brain regions have been successfully used in optogenetic studies. A trans-neuronal retrograde pseudorabies virus39–41 or an AAV serotype that will infect terminals is used for this purpose.42 Retrograde Cre viruses are injected into the terminal site of specific projection neurons while a Cre-inducible opsin virus is targeted to the cell body region of the projection neurons (Figure 1D).40,41 Additionally, a pseudorabies virus-ChR2 approach was recently used to dissect the role of lateral habenula (LHb) and laterodorsal tegmentum inputs to the ventral tegmental area (VTA) in motivational behavior.39 More recently, herpes simplex virus or canine adenoviruses were utilized to deliver Cre to afferent axons and brain regions to allow for subsequent injection of opsin vectors in upstream cell bodies.24,43 These viruses provide a novel way of localizing and dissecting region specific afferent activity into a brain area of interest.
Temporal precision, light response reliability, wave-length specificity, kinetics, and hyperpolarization/depolarization characteristics have all led to the development of a variety of opsins (see Mattis et al44 and Yizhar et al3 for a more detailed comparison of opsins). ChR2(H134R), activated with 473 nm light, is the most commonly used ChR2 variant (Tables 1 and 2). Fast kinetics, glutamate mutated opsins such as ChR2(E123A), also known as ChETAA, have been developed for high fidelity at higher frequency photo-activation,35,44,45 while other constructs, such as the bistable step function opsins, were developed and selected for their slow open channel kinetics and long-lasting depolarization.3,5,46 Because heating of tissue can be an issue,47 step function opsins hold the selective advantage of activation with a short pulse of light, reducing the potential for tissue damage or unwanted artifacts.46 Red-shifted (590–630 nm) opsins can be combined with blue light activated opsins to control different neuronal subtypes in the same animal.3 Recently, a new red-shifted ChR2, ReaChR, was developed for deep transcranial stimulation of neurons without the need of skull thinning, optical windows, or implantable fibers.5,20 Inhibitory opsins, ( halorhodopsins) or protons, (bacteriorhodopsins), are designed to pass anions. Halorhodopsins such as NpHR, and the enhanced third generation NpHR (eNpHR3.0), pass chloride when activated with 590 nm light.3,48,49 The newer generation archeorhodopsin, ArchT (activated by 575 nm green/yellow light) is a useful alternative to halorhodopsin. ArchT has the added benefit of reduced post-inhibitory spiking following inhibition in comparison to eNpHR3.0 induced inhibition.49 Recently, based on structural analysis of ChR2,50 chloride-conducting ChR2 was developed for equivalent temporally precise inhibition at moderate light intensities, comparable to ChR2 stimulation.51 Similarly, the crystal structure of the ChR C1C2 (a chimera of ChR1 and ChR2) was resolved.50,52 Using site-directed mutations of C1C2, a blue-light activated, inhibitory chloride-pump C1C2 (iC1C2) was developed with a comparable reversal potential to eNphR3.0.52 Exciting alternatives to laser or light emitting diode illumination of opsins have been produced to avoid the use of implantable fibers. The luciferase-halorhodopsin system has been constructed for activation of ChR2 ex vivo or eNpHR3.0 by in vivo luciferase luminescence.53,54 This system utilizes the luciferin-induced luminescence of luciferase to drive activation of opsins in vivo through an intraperitoneal injection of luciferin. Opsins have been used to indirectly alter the excitability of cells through activation or inhibition of ion channels.55 Finally, development of novel light-sensitive tools to directly alter endogenous ion channel function provides a novel means to control cellular activity. For example, photo-switchable ion channel blockers, termed lumitoxins, allow for direct regulation of voltage dependent ion channels, particularly potassium channels, in order to regulate cell function and excitability.56 Light illumination alters the structure of the lumitoxin allowing the channel to be unblocked and function normally, subsequently altering cell excitability.
 | Table 2 Viral constructs utilized in optogenetic manipulation of behavior |
There are limited but promising uses of opsins to control neuronal processes within the cell or nucleus including intracellular signaling and transcription. These include G-protein coupled receptor (GPCR) opsins, termed OptoXRs; intracellular signaling opsins; and even opsins that can alter gene transcription through epigenetic mechanisms or activation of transcription factors.6–10,12,13,48,57,58 A small number of these opsins have been used in vivo and have reliably altered signaling properties in a temporal and region specific manner leading to changes in behavioral output.6–8,12,58
Neurobehavioral studies utilizing optogenetics
In vivo optogenetics has provided insight into the function of specific neural circuits underlying motor function and movement,59,60 motivation, reward and emotion,61–65 anxiety and fear learning,37,38,63 sleep and circadian rhythms,66 pain and sensation,67–70 and epilepsy (Tables 3 and 4).71–74
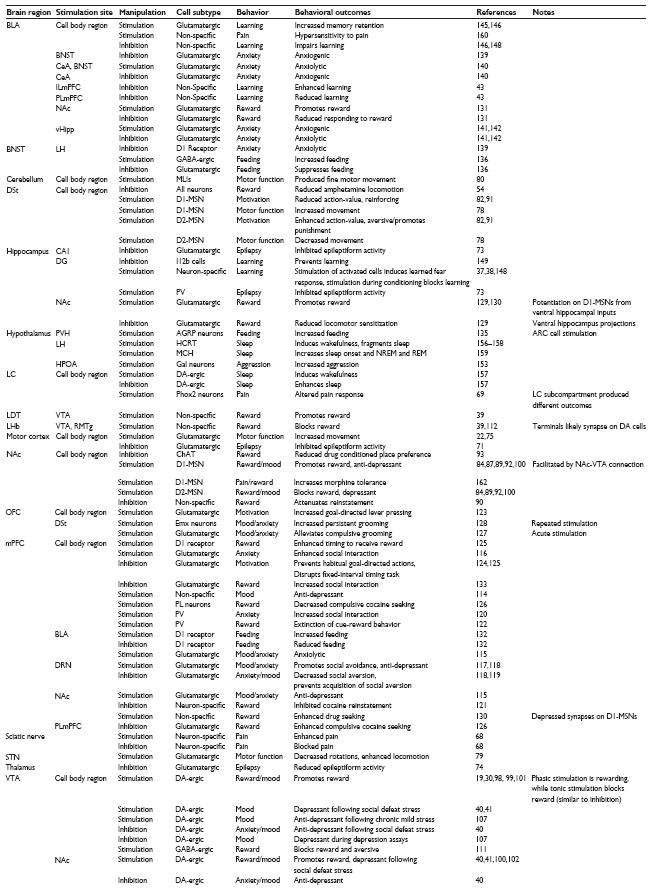 | Table 3 Behavioral outcomes of in vivo optogenetic manipulation with channel opsins |
 | Table 4 Behavior outcomes of in vivo optogenetic manipulation with signaling opsins |
Motor function
Motor function was one the first behavioral outputs explored using in vivo optogenetics. Aravanis et al75 demonstrated temporally precise whisker movement could be induced by stimulating the primary motor cortex using ChR2. Gradinaru et al22 directly stimulated the motor cortex unilaterally in Thy1 (thymus cell antigen 1)-ChR2 mice to induce motor-driven rotations in an open field.48,75 These studies proved that direct in vivo optogenetic manipulation of neurons was a viable method for altering behavioral output. Subsequent studies investigated deeper brain structures and their role in motor movement and control. Basal ganglia (BG) circuitry, particularly the striatum, has long been implicated in motor dysfunction in a number of disorders such as Parkinson’s disease.60 Medium spiny neurons (MSNs), the projection neurons of the striatum, are differentiated into two major subtypes based on dopamine (DA) receptor expression, dopamine 1 (D1) or dopamine 2 (D2) receptors, and their pathway projections.76,77 Optogenetic interrogation of these two parallel BG pathways demonstrated that bilateral D2-MSN stimulation in dorsal medial striatum produces a reduction in movement, as observed through freezing, and decreased ambulation, whereas unilateral stimulation yielded ipsilateral rotations.78 In contrast, unilateral D1-MSN stimulation produces contralateral rotations, while bilateral stimulation increases ambulation and decreases freezing allowing for recovery from parkinsonian bradykinesia outcomes after 6-hydroxydopamine lesioning of dopaminergic innervation to striatum. In comparison, high frequency stimulation of cortical terminals in the subthalamic nucleus, using Thy1-ChR2 mice, was able to rescue the motor deficits in the 6-hydroxydopamine lesion model.79 Although the BG circuitry was known to mediate Parkinson’s disease and other motor behavior, these studies were the first to specifically probe the BG sub-circuits and demonstrate their importance in motor function and dysfunction.
Optogenetic motor movement studies are not limited to the motor cortex and BG structures. One study, examining the role of cerebellar purkinje cells in fine motor movement, demonstrated that targeting stimulation to distinct subregions of the cerebellum results in distinct fine motor movements.80 Using neural nitric oxide synthase (nNOS)-ChR2 BAC transgenic mice, interneurons in the molecular layer of the cerebellum were stimulated to suppress purkinje cell activity. Stimulation of ChR2 expressing interneurons was sufficient to induce eye blinking, among other facial movements, and kinematics of the blink was controlled by intensity and duration of photo-stimulation, suggesting interneuron activity of the cerebellum is precisely tuned to control fine motor movement through inhibition of purkinje cells. Another study used vertebrate rhodopsin, a light sensitive Gi/o linked GPCR, to examine cerebellar motor behavior.58 Light-driven vertebrate rhodopsin activation of purkinje cells decreased latency to fall in a rotarod task, increased time to descend a pole in a pole test, and increased time to cross a beam in a beam walk test, indicating that Gi/o signaling in purkinje cells significantly influenced coordination and motor control.
Motivation, reward, and emotion – striatal neurons
The limbic system and the BG circuit are critical mediators of reward, motivation, reinforcement, and emotion. Along with motor dysfunction observed in dorsal striatal MSN circuits, the balance of MSN subtype tone is predicted to control motivational behavioral outcomes.76,81–83 Optogenetic activation of nucleus accumbens (NAc) D1-MSNs drives conditioned place preference (CPP) to cocaine and morphine,83–85 indicating that D1-MSN stimulation promotes reward. These results are likely due to the connection of D1-MSNs to VTA gamma-aminobutyric acid (GABA) interneurons, which, in part, disinhibit VTA-DA neurons.86–88 This disinhibition hypothesis was further investigated with optogenetic methods. High frequency stimulation of the D1-MSN terminals disinhibits VTA-DA neurons and high frequency stimulation optical stimulation of D1-MSN terminals prior to cocaine treatment subsequently results in an increase in cocaine locomotor sensitization and an occlusion of CPP.87 Conversely, we recently observed that D1-MSN inhibition using eNpHR3.0 decreases cocaine-induced locomotion.89 Together these results indicate that heightened activity of D1-MSNs on VTA inhibitory interneurons enhances reward. In contrast, cocaine84 and morphine85 place preference and reward are blunted by optogenetic stimulation of D2-MSNs. Further, ArchT inhibition of NAc core terminals in the ventral pallidum, which includes terminals of both MSN subtypes, attenuates reinstatement for cocaine seeking.90 In a non-drug paired paradigm, self-stimulation of D1-MSNs in the dorsomedial striatum causes persistent reinforcement whereas stimulation of D2-MSNs results in transient avoidance or punishment.82 A similar effect was observed in a goal-directed selection task, which demonstrated that optogenetic stimulation of distinct MSN subtypes oppositely mediates changes in action value.91 In line with these studies, we found positive effects of driving D1-MSN activity. High frequency optogenetic stimulation of NAc D1-MSNs with a ChETAA ChR2 promotes anti-depressant responses after chronic social defeat stress, while optogenetic activation of NAc D2-MSNs promotes depression-like behavior to subthreshold defeat stress.92 Collectively, optogenetic interrogation of striatal circuits demonstrates opposing behavioral functions for D1-MSNs and D2-MSNs, where heightened D1-MSN activity drives reward and reinforcement, while D2-MSN activity blocks reward and reinforcement or promotes negative outcomes. Optogenetic manipulation of striatal neurons is not limited to MSNs. Despite the low percentage of cholinergic interneurons in the striatum (~1%), direct inhibition of NAc cholinergic interneurons diminishes cocaine CPP.93 This effect may be mediated by diminished DA within the striatum, since increased DA release from VTA neurons occurs after synchronous stimulation of cholinergic interneurons.94–96
Optogenetic signaling tools are to date sparsely used in behavioral neuroscience. OptoXR proteins, GPCR chimeras with photo-activatable domains that allow for light-induced G-protein coupled signaling have been developed for the Gq-coupled α1 adrenergic receptor (a1AR-Gq).6 Light stimulation of this receptor induced calcium influx, activated Gq signaling including mitogen-activated protein (MAP) kinase pathway activation, and increased firing rates in NAc neurons.6 Photo-activation of this receptor was sufficient to produce enhanced place preference for a chamber associated with α1AR-Gq signaling. Optogenetic-induced signaling methods are not strictly limited to membrane-bound receptors. For instance, a constitutively active rho-GTPase (Rac1) fused to a photo- activatable light oxygen voltage construct was used to transiently activate Rac1 in the NAc. This decreased cocaine-induced CPP by increasing phosphorylation of the Rac1 target cofilin leading to decreased cocaine-induced structural plasticity in NAc MSNs.12
Motivation, reward, and mood – VTA
The VTA is a critical mediator of motivational behavior. With the advent of optogenetics, researchers can carefully unravel the complex mechanisms by which the VTA and its associated circuits modulate such behavior. Distinct behavioral outcomes observed from VTA manipulation are based on a number of factors: 1) cellsubtypes of the VTA and their projections, 2) location of the cells in the VTA, 3) firing frequency (Figure 2), and 4) temporally sensitive firing synchrony. One study conducted early on in the course of optogenetic research demonstrated that optogenetic induction of phasic, but not tonic firing of VTA DA neurons induced CPP.30 In fact, optical stimulation of VTA-DA neurons in TH-Cre rats or mice promotes intracranial self-stimulation.19,97 Furthermore, TH-Cre mice, with selective expression of ChR2 in VTA-DA neurons, were shown to rapidly learn to lever press in order to receive phasic photo self-stimulation.98 This stimulation requires temporally specific VTA-DA activation in reward learning. Cue-elicited responding to a reward is driven by temporally precise VTA-DA neuron activation at the time a reward is delivered or expected.99 Similarly, phasic VTA-DA and substantia nigra DA optogenetic stimulation produces operant place preference whereas eNpHR3.0 inhibition produces aversion and avoidance of the paired chamber.98 To add to this mounting evidence, phasic stimulation of VTA terminals in NAc reverses the diminished place preference for morphine caused by brain-derived neurotrophic factor (BDNF) in VTA.100 Additionally, phasic optogenetic stimulation of VTA-DA neurons facilitates food-seeking behavior and reinitiates lever pressing for food following extinction of the trained behavior.101 Intriguingly, tonic but not phasic VTA-DA signaling attenuates ethanol self-administration.102 Taken together, these findings support the hypothesis that phasic firing of DA neurons promotes positive rewarding outcomes while inhibition of phasic firing attenuates reward. However, this may be oversimplified as DA neurons have been demonstrated to encode aversive outcomes.103,104 An earlier observation noted that phasic firing of VTA-DA neurons was important in encoding depression-like behavior in mice that had previously undergone chronic social defeat stress.105,106 Optogenetic induction of phasic activity of VTA-DA neurons in mice during a subthreshold form of the social defeat paradigm rapidly induces a depressive phenotype.40 These studies directly confirmed the functional importance of VTA-DA neurons in encoding for the depressive phenotype. However, a concurrent study reported that induction of phasic firing in VTA-DA neurons reverses the expression of the depressive phenotype in mice that underwent chronic unpredictable stress.107 These two findings coincide with a series of studies showing that exposing rats to either strong or weak stress paradigm induces increased or decreased firing respectively in VTA-DA neurons.103 It is feasible that the magnitude of the stress used induces differing firing dynamics in the VTA neurons103 which could be mediated by time-dependent activation and synchronization of specific cell types and projection regions.108 Furthermore, differential VTA projections and neuronal subpopulations may mediate differing outcomes to stress. For instance, activity of VTA neurons projecting to NAc versus medial prefrontal cortex (mPFC) differentially encodes for susceptibility to depression-related behavior.40 These factors demonstrate the overall complexity of an integrative brain region and demonstrate the utility of optogenetics in the dissection of complex circuit function.
 | Figure 2 Optogenetically mimicking naturalistic neuronal firing differentially regulates cellular and behavioral outcomes. |
As noted before, VTA-GABA neuronal function plays an important role in reward behavior and optogenetics provides the means to dissect local and global VTA circuits. ChR2 activation of VTA-GABA neurons projecting to NAc cholinergic interneurons causes enhanced discrimination between non-motivational and motivational stimuli.109 Optical activation of VTA-GABA interneurons reduces reward consummatory behavior but not conditioned anticipatory behavior following reward-predictive cues.110 Taken together, these results implicate that activation of VTA-GABA interneurons is essential for driving adaptive responses to stimuli. Using a combination of glutamic acid decarboxylase (GAD)-Cre mice, in order to express and specifically stimulate GABA-ergic VTA interneurons with ChR2 and TH-Cre animals to inhibit VTA-DA neurons with eNpHR3.0, Tan et al111 discovered that inhibition of VTA-DA neurons through activation of inhibitory VTA-GABA interneurons produces an aversion to a conditioned chamber. This finding and previously discussed findings indicate that bidirectional modulation of VTA-DA neuron activity can promote opposite reward outcomes.101
Circuit specific modulation of different inputs to the VTA further highlights the complexity of VTA circuits in differential behavioral outcomes. For example, optogenetic induction of phasic activity in LHb cells specifically projecting to the VTA was shown to induce conditioned place aversion, while induction of phasic activity of laterodorsal tegmentum neurons projecting to VTA induced CPP.39 The same study reported that the VTA sends putative GABA-ergic projections to the LHb and that optical activation of the VTA-LHb circuit decreases post-synaptic LHb activity resulting in downstream increased activity of VTA-DA neuron activity. Furthermore, at the behavioral level, optical activation of the VTA-LHb circuit induces reward-related behavior as evidenced by increased CPP.112 Similarly, activation of LHb inputs to another midbrain, VTA-projecting region, the rostromedial tegmental nucleus, produces avoidance to a light-paired stimulation chamber and disrupts positive reinforcement.113 Collectively, these midbrain optogenetic studies demonstrate a more complex role, than previously thought, for midbrain neuronal populations and circuits in mediating both reward and aversion.
Mood, reward, and reinforcement – mPFC, ventral hippocampus, amygdala, and beyond
Similar to VTA manipulation, driving activity in distinct neuronal subpopulations of glutamatergic brain regions produces unique behavioral outcomes. High frequency ChR2 activation of PFC neurons produces anti-depressant responses after social defeat stress, as shown by increased social interaction and sucrose preference.114 Similarly, negative behavioral outcomes to social defeat stress, following administration of cholecystokinin to the mPFC, can be reversed by optical activation of mPFC terminals. At a circuit specific level, social avoidance is reversed by high frequency optogenetic stimulation of mPFC terminals in the NAc, while anxiety responses are blocked by activation of cortical terminals in basolateral amygdala (BLA), indicating the circuit specificity of these behavior.115 Using Thy1-ChR2 mice to optically activate PFC projection neurons, Kumar et al116 were able to reverse anxiety responses produced by chronic social stress and cause an anti-depressant response in forced swim test. The authors further showed that optical activation of PFC projection neurons entrains neural oscillatory activity and drives synchrony across limbic brain areas that regulate affect.116 Another study demonstrated that 20 Hz optogenetic stimulation of mPFC inputs into the dorsal raphe nucleus (DRN) increased kick frequency in the forced swim test.117 These studies promote the hypothesis that global activation of PFC circuits drive anxiolytic and anti-depressant outcomes which is in part mediated by the amygdala, NAc, and DRN. In contrast, driving mPFC terminals to the DRN with ChR2 activation during a sensory interaction period between social defeat episodes increases aversive behavior while inhibition with ArchT decreases aversive behavior.118 In addition, inhibition of DRN GABA-ergic interneurons, which receive direct glutamatergic connections from the mPFC, prevents acquisition but not expression of social avoidance behavior.119 These differing effects may be due to 1) the frequency of stimulation, 2) time-dependent stimulation effects or 3) activity alterations of differing mPFC sub-circuits and projections. Finally, optogenetic evidence suggests that imbalance of excitatory and inhibitory function in the mPFC mediates social behavior disruption. Using a step-function opsin to depolarize pyramidal cells in the mPFC to increase the excitation to inhibition ratio, social interaction was diminished in a three chambered social interaction test.120 Step-function opsin stimulation, with a redshifted ChR (C1V1) in parvalbumin interneurons, in order to restore the excitatory inhibitory balance through increased inhibition, partially restored social interaction in the same test.120
Though the frontal cortex is divided into many regions that have a diverse set of functions, optogenetic investigation into frontal cortex function in reward and motivation supports the idea that indiscriminate enhancement of frontal cortex activity promotes rewarding and positive outcomes, while inhibition blocks these effects. Inhibition of the prelimbic cortex of the mPFC, the NAc core itself, and prelimbic fibers to the NAc core inhibited cocaine reinstatement.121 Inhibition of mPFC pyramidal neurons through photo-stimulation of mPFC parvalbumin interneurons promotes extinction of cue-reward behavior.122 Frontal cortex stimulation is associated with positive rewards. Optogenetic orbitofrontal cortex (OFC) activation increases goal-directed lever pressing.123 In contrast, optogenetic inhibition of the infralimbic cortex during overtraining prevents learning necessary to develop habitual goal-directed actions.124 Optical inhibition of D1 expressing mPFC neurons, which are associated with heightened DA release in and enhanced activation of the mPFC, disrupts a fixed-interval timing task while temporally precise stimulation of these neurons during the interval enhances timed responses to receive rewards in the same task,125 suggesting that temporally specific stimulation is an important factor in frontal cortex mediated rewarding behavior.
Harmful and negative outcomes produced by compulsions are prevented by driving the frontal cortex. Optogenetic stimulation of the prelimbic cortex suppresses compulsive cocaine seeking when paired with a noxious stimulus (shock) while inhibition enhances compulsive seeking behavior even when the aversive shock stimulus is present.126 These outcomes suggest that enhancement of frontal cortex activity drives adaptive responses. Further evidence from optogenetic manipulation of OFC circuits supports this point. Acute 10 Hz optogenetic stimulation of OFC terminals in the striatum or of OFC projection neurons in a mouse model of obsessive grooming, alleviates compulsive grooming.127 However, in one study, 5 days of repeated 10 Hz stimulation of the OFC-striatal pathway increased persistent grooming behavior in mice, which was reversed by chronic fluoxetine treatment.128 Together, these two OFC studies indicate the behavioral importance of stimulation timing and stimulation frequency.
Various glutamatergic inputs into NAc can influence motivational behavior. Optogenetic stimulation of ventral hippocampal (vHipp), mPFC, and BLA glutamatergic terminals in the NAc shell produce rewarding effects and promote place preference.129 Cocaine selectively potentiates vHipp inputs to the NAc shell and ChR2 activation of this pathway enhances cocaine induced locomotor sensitization while NpHR inhibition reduces sensitization.129 Cocaine seeking is driven by potentiation of vHipp synapses on D1-MSNs.130 In contrast, depressing mPFC synapses on D1-MSNs attenuates cocaine seeking,130 suggesting cocaine can differentially modify glutamatergic synapses on MSN subtypes. Another study examined distinct BLA glutamatergic projections specifically innervating the NAc. Terminal BLA, but not mPFC ChR2 stimulation in the NAc promotes self-stimulation.131 Furthermore, NpHR inhibition of BLA terminals reduces behavioral responding to sucrose.131 Optogenetic inhibition of BLA to NAc core by the ArchT opsin prevents cocaine reinstatement and seeking.121 These findings indicate that glutamatergic signaling to the NAc facilitates reward and reinforcement in a projection specific manner.
Feeding behavior has been examined with optogenetics in a number of studies.27 Heterogeneous effects on motivation, food consumption, and feeding behavior are observed with PFC lesions132 and optogenetics provides a means of dissecting feeding behavior through bidirectional alteration of neuronal activity. D1 receptor expressing mPFC projection neurons are activated during feeding behavior, an effect that was mimicked by optogenetic activation of these neurons,132 while inhibition of D1 mPFC neurons reduced feeding. These optogenetic-driven behavioral effects are localized to medial BLA neurons as stimulation of D1 mPFC terminals in the medial BLA enhances feeding while inhibition of medial BLA cell somas during terminal stimulation restores normal feeding.132 Similarly, studies in female rats demonstrate that eNpHR3.0 mediated inhibition of mPFC blocks stress-induced reinstatement to food seeking.133 Currently, there is a lack of optogenetic studies in female rodents, but given the role of sex differences in behavior and CNS disease,134 we will likely see more sex specific studies in the future. Sex differences are likely to be more apparent in well-studied sexually dimorphic regions such as the hypothalamus. The hypothalamus is heavily involved in basal states and behavior including feeding. Activation of agouti-related peptide (AGRP) positive neurons and their axon terminals in the paraventricular hypothalamus (PVH) evokes feeding behavior, which is mediated by AGRP inhibition of PVH cells. This effect is mimicked by chemogenetic inhibition of PVH cells.135 These experiments identified that AGRP neurons from the arcuate nucleus (ARC) suppresses PVH oxytocin neurons to evoke feeding. However, it has been found that acute stimulation of AGRP axons in multiple brain regions was sufficient to enhance feeding,26 including areas that are not normally associated with a rapid enhancement of feeding behavior. Dissection of specific circuits arising from the lateral hypothalamus has distinguished the bed nucleus of the stria terminalis (BNST), a region that integrates motivational information and interacts with the VTA and the lateral hypothalamus, as a particularly important region in feeding behavior.136 Optogenetic activation of BNST GABA-ergic projections to the lateral hypothalamus enhance feeding while inhibition is aversive and suppresses feeding. In conjunction with these findings, stimulation of glutamatergic lateral hypothalamic neurons suppresses feeding, indicating the BNST in part controls lateral hypothalamic control of feeding behavior,136 distinguishing the complexity of circuits involved in natural, primal behavior.
Anxiety, fear, and aggression
The amygdala is well-known for its role in emotion, as lesions of this structure produce profound deficits in emotion-related behavior.137–139 Tye et al examined the bidirectional effects of stimulating and inhibiting the amygdala in anxiety behavioral tasks.140 Activity of glutamatergic projection neurons from the BLA, expressing either ChR2 or eNpHR3.0, was bidirectionally manipulated during the elevated plus maze test and open field test. Somatic stimulation of the BLA produced a robust anxiolytic effect. This result was mediated through BLA to central amygdala (CeA) synapses since stimulation of BLA fibers in the CeA produced an anxiolytic effect while inhibition of these fibers was anxiogenic.140 Further interrogation of anxiety-related circuits demonstrated that eNpHR3.0 inhibition of BLA inputs to the vHipp decreases anxiety-related behavior in the elevated plus maze and open field while stimulation produces anxiety.141 Stimulation of this pathway is also sufficient to drive avoidance to a resident intruder while inhibition reversed this effect.142 These studies suggest BLA to vHipp connections drive an adaptive form of anxiety, allowing for avoidance of possible harmful stimuli. The extended amygdala including the BNST is also involved in fear and anxiety. Optogenetic mapping and behavioral analysis of the BNST and BLA circuit indicates BLA inputs in part aid in distinguishing locations that promote anxiety.136,139 Stimulation of BNST glutamatergic projections to VTA-GABA-ergic cells promoted anxiogenic and aversive effects while activation of BNST GABA-ergic projections to these same VTA neurons produced anxiolytic and rewarding effects.136 These results shed light on non-reward related functions of VTA circuits. However, other midbrain nuclei involved in reward and mood such as the DRN are also involved in anxiety-like behavior. Using light sensitive 5-HT1A Gi/o serotonin receptor in the DRN, Masseck et al8 examined anxiety behavior in the open field and in novelty suppressed feeding.8 Indirect photo-inhibition of DRN neurons, with this light sensitive 5-HT1A variant, induced anxiogenic behavior. A subsequent study from this same group used a vertebrate melanopsin coupled with a 5-HT2C receptor, a Gq linked receptor, in GABA-ergic neurons of the DRN.7 They found that activation of the light sensitive 5-HT2C receptor in GABA-ergic neurons decreased activity in serotonergic neurons, and produced an anxiolytic response. The same response was reproduced with ArchT inhibition of DRN serotonergic neurons. These outcomes demonstrate that differential modes of GPCR signaling can produce similar effects on neuronal activity and ultimately behavior.
Well-established fear learning paradigms allow for simple characterization of fear-related behavior with optogenetics in order to better define the circuitry that mediates fear. Particularly interesting are optogenetic studies examining mPFC, vHipp, and amygdala circuits in fear conditioning and extinction. It was observed that specific connections from both the mPFC and the vHipp monosynaptically innervate both principle excitatory neurons and inhibitory neurons in the BLA.143 These connections recruit feed-forward inhibition of excitatory neurons in the amygdala. mPFC to BLA connections are associated with retrieval of fear extinction memory. Using optogenetic and electrophysiological methods, the strength of mPFC to BLA connections was observed to be depressed following extinction of conditioned fear.144 However, extinction is not associated with feed-forward inhibition of excitatory amygdala mPFC to BLA circuits.144 These results implicate a role for mPFC in controlling BLA output in relation to fear memories. However, reciprocal connections to the mPFC also control fear behavior. Connections from the basal amygdala to subregions of the mPFC, specifically the prelimbic and infralimbic subdivision, are oppositely involved in retrieval of a fear memory and direct inhibition produces opposite freezing responses in conjunction with a conditioned stimulus.43 Local manipulation of the amygdala also produces similar learning outcomes. Optogenetic activation of the lateral amygdala during a conditioned auditory stimulus is capable of enhancing fear stimulus learning.145 In conjunction with these findings, gamma-frequency stimulation (40 Hz) of BLA glutamatergic pyramidal cells with a ChETAA ChR2 enhanced retention of an inhibitory avoidance cue while inhibition using ArchT impairs retention of the learned avoidance.146 These results demonstrate that driving excitatory cells of lateral subregions of the amygdala promote fear learning.
To understand how contextual fear memory is encoded and stored, optogenetics has been used in the hippocampus of rodents.147 Inhibition of dentate gyrus (DG) cells during encoding of a contextual fear memory blocks learning as shown by poor retrieval.148 Interestingly, stimulation of the same cells produces a similar effect, suggesting any alteration of DG cell firing during learning prevents encoding of a fear memory. Furthermore, inhibition of DG cells during training in a spatially driven, active avoidance place task attenuates learning of the avoidance zone.148 Inhibition of DG GABA-ergic neurons prevents acquisition in a morris water maze task.149 Taken together, these results demonstrate that alteration of naturalistic neuronal firing in the DG prevents normal learning. A recent study used a complex genetically-driven optogenetic paradigm to selectively activate neuronal ensembles important for memory engrams. A c-fos induced ChR2 was used to control activated cells (see Techniques and technological advances section and Figure 1C) and reactivate cell populations of the DG that were previously activated during fear memories in a fear conditioning paradigm.37,38 Stimulating DG neurons involved in the memory engram recapitulated freezing behavior that occur during this conditioning. This optogenetic system is underutilized in rodent behavior and has yet to be used in other brain regions. However, similar, non-optogenetic methodologies to inactivate neuronal ensembles using other methodologies have also provided insight into context-induced behavior.150,151
Aggression studies involving optogenetic manipulations have provided insights into overlapping circuitry mediating attacking and mating behavior. Neuron-specific optogenetic stimulation of the ventrolateral subdivision of the ventromedial hypothalamus (VMHvl) enhances male mice aggression and leads to indiscriminate attacking behavior.152 Interestingly, activity of these aggression neurons are suppressed when a distinct population of VMHvl neurons involved in mating are active. Inhibition of VMHvl firing can attenuate aggressive behavior.152 The medial preoptic area (MPOA) has overlapping functions in both mating and aggression similar to that of the VMHvl. Two subsets of galanin-expressing neurons of the MPOA mediate female parental responses and mating.153 Activating the MPOA optogenetically reduces male aggression behavior to other males and pups. Further, activation of this region can induce pup grooming. These results provide evidence that sex, aggression, and parenting behavior are produced in overlapping brain regions, but are distinguished neurobiologically by cell type and region specific neuronal activity.
Circadian, sleep, alertness
The circadian and sleep-wake regulator cycles are independent, but closely linked systems. The suprachiasmatic nucleus, a pair of nuclei in the hypothalamus, governs the rhythms of the sleep-wake cycle. The suprachiasmatic nucleus in-turn coordinates the activity of other self-sustained clocks and brain regions implicated in mood disorders such as the hippocampus, VTA, mPFC, LHb, and NAc.154 Since these neurons exhibit daily rhythms in firing, inhibition of a circuit at the period of time an animal is most active may yield more robust behavioral effects compared to inhibition when the neural circuit is the least active. Furthermore, since circadian rhythms exhibit rhythms that last from minutes to hours within a 24 hour cycle, it would be beneficial to use chronic optogenetic stimulation paradigms lasting minutes to hours in order to better elucidate naturalistic diurnal and circadian rhythmicity of neural circuits and the subsequent changes in animal behavior.
There is a great prevalence of published studies using optogenetics in the modulation of the sleep-wake cycle. The circuitry of arousal involves various neurotransmitter systems from differing nuclei such as the hypocretin (HCRT), noradrenergic (NOR), serotonergic, cholinergic neurons from the lateral hypothalamus, locus coeruleus (LC), DRN, and pedunculopontine/laterodorsal tegmental nuclei, respectively. Previous pharmacological evidence suggested that HCRT and NOR circuitry are important in promoting arousal in the animal.155 However, with optogenetics, it is possible to selectively, reversibly, and rapidly modulate these circuits in order to study the effects on arousal and determine the causal link between circuit activity and sleep or arousal states. Optogenetically mimicking naturalistic activity of HCRT neurons increases behavioral transitions from non-rapid eye movement (NREM) sleep or REM sleep to wakefulness.156 Furthermore, optogenetic stimulation of NOR, LC neurons induces rapid wakefulness from both NREM and REM sleep.157 It should be noted that both HCRT and LC neurons are also implicated in a wide variety of behavior such as depression, stress, attention, learning and memory, and addiction. For example, in vivo optogenetic manipulation of HCRT neurons, resulting in sleep fragmentation, impairs memory consolidation.158 A compelling hypothesis posits that differing firing dynamics of HCRT and LC neurons may result in differing states of arousal. Future experiments involving optogenetic induction of distinct firing patterns in HCRT and LC neurons may help elucidate the subtle role of differing firing dynamics in behavioral output. Melanin concentrating hormone neurons located in the hypothalamus project widely throughout the brain, including to arousal neurons, and have been proposed to have a role in promoting sleep. Recent optogenetic studies show that specific activation of melanin concentrating hormone neurons increases both sleep onset and subsequent time animals spent in NREM and REM sleep.159 Elucidating the roles of specific neural circuits in sleep regulation prove potentially useful for the future development of treatments for insomnia. Another intriguing possibility is that optogenetics will help elucidate the circuitry involved in, the interesting yet counterintuitive observations that, sleep deprivation induces rapid reversal of depression.
Pain
Very few studies use optogenetics to directly study pain behavior.67 One study used non-specific opsins injected into the sciatic nerve to trans-dermally produce pain responses through stimulation, while preventing pain responses through inhibition.68 This technique holds the potential for relatively non-invasive and direct therapeutic strategies to treat peripheral pain. Other optogenetic studies, focused on the CNS, led to insights about subcortical and brain stem regions that mediate pain. The NOR system mediates in part analgesic outcomes to peripheral pain and alters spinal nociception.67 Optogenetic ChR2 excitation of the LC enhanced the withdrawal threshold for a normally noxious heat stimulus.69 Similarly excitation in dorsal LC regions reduced the withdrawal threshold, while stimulation in ventral LC regions enhanced withdrawal threshold, demonstrating that different subpopulations within the LC mediate opposing nociception outcomes. Optogenetic studies have explored atypical pain-related regions of the brain such as the CeA. Hypersensitivity to bladder distention can increase visceromotor responses to painful stimuli when the CeA is optogenetically stimulated.160 Further, optogenetic investigation of central brain regions may be important to assess mechanisms of pain-associated tolerance and addiction. Analgesic tolerance to opioid medications for a number of diseases, such as chronic pain, has produced problems for long-term treatment strategies. One study addressed morphine analgesia and tolerance by activating NAc MSNs expressing regulator of G-protein signaling 9 (Rgs9) complexes. Rgs9 complexes, known to modulate opioid GPCR responsiveness and desensitization161,162 are important in morphine analgesia and tolerance in the NAc. This study demonstrated that optogenetic activation of Rgs9 expressing cells of the NAc, which should include all MSNs, enhanced the development of morphine tolerance, an effect that was mediated through D1-MSNs.162
Epilepsy
Epilepsy is characterized by abnormal neuronal activity promoted by enhanced connectivity, synaptic transmission, and intrinsic excitability, which can lead to spontaneous generation of seizures.71 Due to a wide array of inhibitory tools, optogenetics holds great potential in preventing seizure generation or silencing seizures before their onset. It was first observed that stimulation train induced bursting can reliably be inhibited by NpHR in organotypic culture, indicating that halorhodopsin held the potential to stifle seizure induced activity.72 Later, in vivo studies supported this approach as a valid and reliable model for the treatment of epilepsy. NpHR reduced seizure activity produced by tetanus toxin-mediated generation of spontaneous seizures.71 Halorhodopsin inhibition of seizures was observed to halt seizure activity induced by kainic acid in the hippocampus.73 This strategy has been further extended to a more naturalistic model of seizures in which a spontaneous seizure disorder was produced by cortical injury.74 In this study, thalamic seizures were interrupted by light stimulation of eNpHR3.0 containing cells. However, inhibition of seizure activity has not been limited to direct inhibition of primary excitatory cells. Stimulation of parvalbumin interneurons and thus an enhancement in inhibitory GABA-ergic signaling has been observed to attenuate kainic acid induced seizure activity and behavioral seizures.73
The future of optogenetics
In the past decade optogenetics has rapidly advanced, leading to a large toolbox of available technologies. These tools continue to progress and improve researchers’ ability to dissect complex brain circuitry. Viral and transgenic optogenetic constructs may be used for mapping large parts of the neural connectome with both ex vivo slice and in vivo electrophysiology techniques. Coupling these methodologies with microscopy techniques including in vivo calcium imaging, optogenetics could provide cell-specific investigation of neuronal firing in awake, behaving animals while maintaining optical control over specific cell subtypes.163 The ability to exert fine temporal control of cell firing using optogenetics, will make it possible to elucidate the functional role of subtle changes in both firing frequency and patterns in specific cell types and neural circuits and their effects on behavior. Using structural information and mutagenesis, development of alternative, wavelength specific opsins would allow for various wavelengths of light to be used in one region for simultaneous optical control of different cell subtypes, to dissect discreet connections and firing patterns in specific circuits through combinatorial experiments with multiple light-driven constructs. Future studies will likely use activity-dependent promoters allowing for manipulation of previously activated neuronal ensembles that mediate behavioral outcomes. Activity-dependent activation of opsins in combination with luminescent proteins (eg, luciferase) could potentially prove a useful therapeutic tool to prevent negative outcomes of neural hyperactivity through inhibition (eg, seizure activity) or hypoactivity through stimulation, while bypassing invasive optic fiber implants. Lastly, many more promising applications will be developed for optogenetics in higher animals considering opsins and optogenetics are now currently being used in primates.164
Conclusion
In vivo stimulation and inhibition of specific circuits is essential for understanding the emergence of behavioral phenotypes and region specific functions, particularly in neurological and psychological diseases and disorders. As treatments for many of these diseases are few and often ineffective, optogenetics holds the potential to unearth new therapeutic strategies. Optogenetics is currently used for circuit mapping, temporal specific activation or inhibition of cell subtypes, signaling alterations, and functional discovery of brain circuits and their behavioral correlates. Using optogenetics, under the control of cell-type specific promoters or Cre rodent lines, has led to a wealth of information about the connectivity and activity among brain structures and has thus provided researchers with the knowledge of microcircuit level dysfunction in rodent behavior. Such findings have enlightened potential treatment strategies for many CNS diseases.
Disclosure
The authors have no conflicts of interest to disclose.
References
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8(9):1263–1268. | |
Zhang F, Aravanis AM, Adamantidis A, de Lecea L, Deisseroth K. Circuit-breakers: Optical technologies for probing neural signals and systems. Nat Rev Neurosci. 2007;8(8):577–581. | |
Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71(1):9–34. | |
Nagel G, Szellas T, Huhn W, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100(24):13940–13945. | |
Lin JY. A user’s guide to channelrhodopsin variants: Features, limitations and future developments. Exp Physiol. 2011;96(1):19–25. | |
Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458(7241):1025–1029. | |
Spoida K, Masseck OA, Deneris ES, Herlitze S. Gq/5-HT2c receptor signals activate a local GABAergic inhibitory feedback circuit to modulate serotonergic firing and anxiety in mice. Proc Natl Acad Sci U S A. 2014;111(17):6479–6484. | |
Masseck OA, Spoida K, Dalkara D, et al. Vertebrate cone opsins enable sustained and highly sensitive rapid control of gi/o signaling in anxiety circuitry. Neuron. 2014;81(6):1263–1273. | |
Oh E, Maejima T, Liu C, Deneris E, Herlitze S. Substitution of 5-HT1A receptor signaling by a light-activated G protein-coupled receptor. J Biol Chem. 2010;285(40):30825–30836. | |
Stierl M, Stumpf P, Udwari D, et al. Light modulation of cellular cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium beggiatoa. J Biol Chem. 2011;286(2):1181–1188. | |
Ryu MH, Moskvin OV, Siltberg-Liberles J, Gomelsky M. Natural and engineered photoactivated nucleotidyl cyclases for optogenetic applications. J Biol Chem. 2010;285(53):41501–41508. | |
Dietz DM, Sun H, Lobo MK, et al. Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nat Neurosci. 2012;15(6):891–896. | |
Hahn KM, Kuhlman B. Hold me tightly LOV. Nat Methods. 2010;7(8): 595, 597. | |
Konermann S, Brigham MD, Trevino AE, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500(7463):472–476. | |
Motta-Mena LB, Reade A, Mallory MJ, et al. An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat Chem Biol. 2014;10(3):196–202. | |
Sparta DR, Stamatakis AM, Phillips JL, Hovelso N, van Zessen R, Stuber GD. Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat Protoc. 2011;7(1): 12–23. | |
Britt JP, McDevitt RA, Bonci A. Use of channelrhodopsin for activation of CNS neurons. Curr Protoc Neurosci. 2012;Chapter 2:Unit2.16. | |
Francis TC, Lobo MK. Optogenetic regulation of dopamine receptor-expressing neurons. In: Mario Tiberi WW, editors. Dopamine Receptor Technologies. Neuromethods ed. Springer Protocols. In Press 2014. | |
Kim TI, McCall JG, Jung YH, et al. Injectable, cellular-scale optoelectronics with applications for wireless optogenetics. Science. 2013;340(6129):211–216. | |
Lin JY, Knutsen PM, Muller A, Kleinfeld D, Tsien RY. ReaChR: A red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat Neurosci. 2013;16(10):1499–1508. | |
Madisen L, Mao T, Koch H, et al. A toolbox of cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci. 2012;15(5):793–802. | |
Gradinaru V, Thompson KR, Zhang F, et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27(52):14231–14238. | |
Arenkiel BR, Peca J, Davison IG, et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54(2):205–218. | |
Fenno LE, Mattis J, Ramakrishnan C, et al. Targeting cells with single vectors using multiple-feature boolean logic. Nat Methods. 2014;11(7):763–772. | |
Adrover MF, Shin JH, Alvarez VA. Glutamate and dopamine transmission from midbrain dopamine neurons share similar release properties but are differentially affected by cocaine. J Neurosci. 2014;34(9): 3183–3192. | |
Betley JN, Cao ZF, Ritola KD, Sternson SM. Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell. 2013;155(6):1337–1350. | |
Krashes MJ, Kravitz AV. Optogenetic and chemogenetic insights into the food addiction hypothesis. Front Behav Neurosci. 2014;8:57. | |
Lim DH, Ledue J, Mohajerani MH, Vanni MP, Murphy TH. Optogenetic approaches for functional mouse brain mapping. Front Neurosci. 2013;7:54. | |
Gerfen CR, Paletzki R, Heintz N. GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron. 2013;80(6):1368–1383. | |
Tsai HC, Zhang F, Adamantidis A, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324(5930):1080–1084. | |
Zhang F, Gradinaru V, Adamantidis AR, et al. Optogenetic interrogation of neural circuits: Technology for probing mammalian brain structures. Nat Protoc. 2010;5(3):439–456. | |
Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459(7247):698–702. | |
Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28(28):7025–7030. | |
Yang XW, Model P, Heintz N. Homologous recombination based modification in escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat Biotechnol. 1997;15(9): 859–865. | |
Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. | |
Zhu P, Narita Y, Bundschuh ST, et al. Optogenetic dissection of neuronal circuits in zebrafish using viral gene transfer and the Tet system. Front Neural Circuits. 2009;3:21. | |
Liu X, Ramirez S, Pang PT, et al. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012; 484(7394):381–385. | |
Ramirez S, Liu X, Lin PA, et al. Creating a false memory in the hippocampus. Science. 2013;341(6144):387–391. | |
Lammel S, Lim BK, Ran C, et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491(7423): 212–217. | |
Chaudhury D, Walsh JJ, Friedman AK, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493(7433):532–536. | |
Walsh JJ, Friedman AK, Sun H, et al. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci. 2014;17(1):27–29. | |
Rothermel M, Brunert D, Zabawa C, Diaz-Quesada M, Wachowiak M. Transgene expression in target-defined neuron populations mediated by retrograde infection with adeno-associated viral vectors. J Neurosci. 2013;33(38):15195–15206. | |
Senn V, Wolff SB, Herry C, et al. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron. 2014;81(2): 428–437. | |
Mattis J, Tye KM, Ferenczi EA, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods. 2011;9(2):159–172. | |
Gunaydin LA, Yizhar O, Berndt A, Sohal VS, Deisseroth K, Hegemann P. Ultrafast optogenetic control. Nat Neurosci. 2010;13(3):387–392. | |
Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat Neurosci. 2009;12(2):229–234. | |
Christie IN, Wells JA, Southern P, et al. fMRI response to blue light delivery in the naive brain: Implications for combined optogenetic fMRI studies. Neuroimage. 2012;66C:634–641. | |
Gradinaru V, Zhang F, Ramakrishnan C, et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141(1):154–165. | |
Raimondo JV, Kay L, Ellender TJ, Akerman CJ. Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat Neurosci. 2012;15(8):1102–1104. | |
Kato HE, Zhang F, Yizhar O, et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature. 2012;482(7385): 369–374. | |
Wietek J, Wiegert JS, Adeishvili N, et al. Conversion of channelrhodopsin into a light-gated chloride channel. Science. 2014;344(6182): 409–412. | |
Berndt A, Lee SY, Ramakrishnan C, Deisseroth K. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science. 2014;344(6182):420–424. | |
Berglund K, Birkner E, Augustine GJ, Hochgeschwender U. Light-emitting channelrhodopsins for combined optogenetic and chemical-genetic control of neurons. PLoS One. 2013;8(3):e59759. | |
Land BB, Brayton CE, Furman KE, Lapalombara Z, Dileone RJ. Optogenetic inhibition of neurons by internal light production. Front Behav Neurosci. 2014;8:108. | |
Masseck OA, Rubelowski JM, Spoida K, Herlitze S. Light- and drug-activated G-protein-coupled receptors to control intracellular signalling. Exp Physiol. 2011;96(1):51–56. | |
Schmidt D, Tillberg PW, Chen F, Boyden ES. A fully genetically encoded protein architecture for optical control of peptide ligand concentration. Nat Commun. 2014;5:3019. | |
Strickland D, Moffat K, Sosnick TR. Light-activated DNA binding in a designed allosteric protein. Proc Natl Acad Sci U S A. 2008;105(31): 10709–10714. | |
Gutierrez DV, Mark MD, Masseck O, et al. Optogenetic control of motor coordination by gi/o protein-coupled vertebrate rhodopsin in cerebellar purkinje cells. J Biol Chem. 2011;286(29):25848–25858. | |
Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends Neurosci. 2012;35(9):557–564. | |
Vazey EM, Aston-Jones G. New tricks for old dogmas: Optogenetic and designer receptor insights for parkinson’s disease. Brain Res. 2013;1511:153–163. | |
Lobo MK, Nestler EJ, Covington HE 3rd. Potential utility of optogenetics in the study of depression. Biol Psychiatry. 2012;71(12):1068–1074. | |
Nieh EH, Kim SY, Namburi P, Tye KM. Optogenetic dissection of neural circuits underlying emotional valence and motivated behaviors. Brain Res. 2013;1511:73–92. | |
Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012; 13(4):251–266. | |
Lenz JD, Lobo MK. Optogenetic insights into striatal function and behavior. Behav Brain Res. 2013;255:44–54. | |
Lammel S, Tye KM, Warden MR. Progress in understanding mood disorders: Optogenetic dissection of neural circuits. Genes Brain Behav. 2014;13(1):38–51. | |
Sidor MM, McClung CA. Timing matters: Using optogenetics to chronically manipulate neural circuitry and rhythms. Front Behav Neurosci. 2014;8:41. | |
Carr FB, Zachariou V. Nociception and pain: Lessons from optogenetics. Front Behav Neurosci. 2014;8:69. | |
Iyer SM, Montgomery KL, Towne C, et al. Virally mediated optogenetic excitation and inhibition of pain in freely moving nontransgenic mice. Nat Biotechnol. 2014;32(3):274–278. | |
Hickey L, Li Y, Fyson SJ, et al. Optoactivation of locus ceruleus neurons evokes bidirectional changes in thermal nociception in rats. J Neurosci. 2014;34(12):4148–4160. | |
Browne LE, Woolf CJ. Casting light on pain. Nat Biotechnol. 2014; 32(3):240–241. | |
Wykes RC, Heeroma JH, Mantoan L, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med. 2012;4(161):161ra152. | |
Tonnesen J, Sorensen AT, Deisseroth K, Lundberg C, Kokaia M. Optogenetic control of epileptiform activity. Proc Natl Acad Sci U S A. 2009;106(29):12162–12167. | |
Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun. 2013;4:1376. | |
Paz JT, Davidson TJ, Frechette ES, et al. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci. 2013;16(1):64–70. | |
Aravanis AM, Wang LP, Zhang F, et al. An optical neural interface: In vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J Neural Eng. 2007;4(3):S143–S156. | |
Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366–375. | |
Gerfen CR, Engber TM, Mahan LC, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250(4986):1429–1432. | |
Kravitz AV, Freeze BS, Parker PR, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466(7306):622–626. | |
Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324(5925):354–359. | |
Heiney SA, Kim J, Augustine GJ, Medina JF. Precise control of movement kinematics by optogenetic inhibition of purkinje cell activity. J Neurosci. 2014;34(6):2321–2330. | |
Maia TV, Frank MJ. From reinforcement learning models to psychiatric and neurological disorders. Nat Neurosci. 2011;14(2):154–162. | |
Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15(6): 816–818. | |
Lobo MK, Nestler EJ. The striatal balancing act in drug addiction: Distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat. 2011;5:41. | |
Lobo MK, Covington HE 3rd, Chaudhury D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330(6002):385–390. | |
Koo JW, Lobo MK, Chaudhury D, et al. Loss of BDNF signaling in D1R expressing NAc neurons enhances morphine reward by reducing GABA inhibition. Neuropsychopharmacology. 2014;39(11):2646–2653. | |
Xia Y, Driscoll JR, Wilbrecht L, Margolis EB, Fields HL, Hjelmstad GO. Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J Neurosci. 2011;31(21): 7811–7816. | |
Bocklisch C, Pascoli V, Wong JC, et al. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science. 2013;341(6153):1521–1525. | |
Smith RJ, Lobo MK, Spencer S, Kalivas PW. Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol. 2013;23(4):546–552. | |
Chandra R, Lenz JD, Gancarz AM, et al. Optogenetic inhibition of D1R containing nucleus accumbens neurons alters cocaine-mediated regulation of Tiam1. Front Mol Neurosci. 2013;6:13. | |
Stefanik MT, Kupchik YM, Brown RM, Kalivas PW. Optogenetic evidence that pallidal projections, not nigral projections, from the nucleus accumbens core are necessary for reinstating cocaine seeking. J Neurosci. 2013;33(34):13654–13662. | |
Tai LH, Lee AM, Benavidez N, Bonci A, Wilbrecht L. Transient stimulation of distinct subpopulations of striatal neurons mimics changes in action value. Nat Neurosci. 2012;15(9):1281–1289. | |
Francis TC, Chandra R, Friend DM, et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry. Epub July 28, 2014. | |
Witten IB, Lin SC, Brodsky M, et al. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330(6011):1677–1681. | |
Cachope R, Mateo Y, Mathur BN, et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: Setting the tone for reward processing. Cell Rep. 2012;2(1):33–41. | |
Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75(1):58–64. | |
Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, Kreitzer AC. Striatal cholinergic interneurons drive GABA release from dopamine terminals. Neuron. 2014;82(1):63–70. | |
Witten IB, Steinberg EE, Lee SY, et al. Recombinase-driver rat lines: Tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron. 2011;72(5):721–733. | |
Ilango A, Kesner AJ, Broker CJ, Wang DV, Ikemoto S. Phasic excitation of ventral tegmental dopamine neurons potentiates the initiation of conditioned approach behavior: Parametric and reinforcement-schedule analyses. Front Behav Neurosci. 2014;8:155. | |
Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, Janak PH. A causal link between prediction errors, dopamine neurons and learning. Nat Neurosci. 2013;16(7):966–973. | |
Koo JW, Mazei-Robison MS, Chaudhury D, et al. BDNF is a negative modulator of morphine action. Science. 2012;338(6103):124–128. | |
Adamantidis AR, Tsai HC, Boutrel B, et al. Optogenetic interrogation of dopaminergic modulation of the multiple phases of reward-seeking behavior. J Neurosci. 2011;31(30):10829–10835. | |
Bass CE, Grinevich VP, Gioia D, et al. Optogenetic stimulation of VTA dopamine neurons reveals that tonic but not phasic patterns of dopamine transmission reduce ethanol self-administration. Front Behav Neurosci. 2013;7:173. | |
Valenti O, Gill KM, Grace AA. Different stressors produce excitation or inhibition of mesolimbic dopamine neuron activity: Response alteration by stress pre-exposure. Eur J Neurosci. 2012;35(8): 1312–1321. | |
Friedman AK, Walsh JJ, Juarez B, et al. Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science. 2014;344(6181):313–319. | |
Krishnan V, Han MH, Graham DL, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131(2):391–404. | |
Cao JL, Covington HE 3rd, Friedman AK, et al. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30(49):16453–16458. | |
Tye KM, Mirzabekov JJ, Warden MR, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493(7433):537–541. | |
Brischoux F, Chakraborty S, Brierley DI, Ungless MA. Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci U S A. 2009;106(12):4894–4899. | |
Brown MT, Tan KR, O’Connor EC, Nikonenko I, Muller D, Luscher C. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. 2012;492(7429): 452–456. | |
van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. 2012;73(6): 1184–1194. | |
Tan KR, Yvon C, Turiault M, et al. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012;73(6):1173–1183. | |
Stamatakis AM, Jennings JH, Ung RL, et al. A unique population of ventral tegmental area neurons inhibits the lateral habenula to promote reward. Neuron. 2013;80(4):1039–1053. | |
Stamatakis AM, Stuber GD. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci. 2012;15(8):1105–1107. | |
Covington HE 3rd, Lobo MK, Maze I, et al. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010;30(48):16082–16090. | |
Vialou V, Bagot RC, Cahill ME, et al. Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: Role of DeltaFosB. J Neurosci. 2014;34(11):3878–3887. | |
Kumar S, Black SJ, Hultman R, et al. Cortical control of affective networks. J Neurosci. 2013;33(3):1116–1129. | |
Warden MR, Selimbeyoglu A, Mirzabekov JJ, et al. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature. 2012;492(7429):428–432. | |
Challis C, Beck SG, Berton O. Optogenetic modulation of descending prefrontocortical inputs to the dorsal raphe bidirectionally bias socioaffective choices after social defeat. Front Behav Neurosci. 2014;8:43. | |
Challis C, Boulden J, Veerakumar A, et al. Raphe GABAergic neurons mediate the acquisition of avoidance after social defeat. J Neurosci. 2013;33(35):13978–13988. | |
Yizhar O, Fenno LE, Prigge M, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477(7363):171–178. | |
Stefanik MT, Moussawi K, Kupchik YM, et al. Optogenetic inhibition of cocaine seeking in rats. Addict Biol. 2013;18(1):50–53. | |
Sparta DR, Hovelso N, Mason AO, et al. Activation of prefrontal cortical parvalbumin interneurons facilitates extinction of reward-seeking behavior. J Neurosci. 2014;34(10):3699–3705. | |
Gremel CM, Costa RM. Orbitofrontal and striatal circuits dynamically encode the shift between goal-directed and habitual actions. Nat Commun. 2013;4:2264. | |
Smith KS, Graybiel AM. A dual operator view of habitual behavior reflecting cortical and striatal dynamics. Neuron. 2013;79(2): 361–374. | |
Narayanan NS, Land BB, Solder JE, Deisseroth K, DiLeone RJ. Prefrontal D1 dopamine signaling is required for temporal control. Proc Natl Acad Sci U S A. 2012;109(50):20726–20731. | |
Chen BT, Yau HJ, Hatch C, et al. Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature. 2013;496(7445):359–362. | |
Burguiere E, Monteiro P, Feng G, Graybiel AM. Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science. 2013;340(6137):1243–1246. | |
Ahmari SE, Spellman T, Douglass NL, et al. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science. 2013;340(6137):1234–1239. | |
Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron. 2012;76(4):790–803. | |
Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, Luscher C. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509(7501):459–464. | |
Stuber GD, Sparta DR, Stamatakis AM, et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475(7356):377–380. | |
Land BB, Narayanan NS, Liu RJ, et al. Medial prefrontal D1 dopamine neurons control food intake. Nat Neurosci. 2014;17(2):248–253. | |
Calu DJ, Kawa AB, Marchant NJ, et al. Optogenetic inhibition of dorsal medial prefrontal cortex attenuates stress-induced reinstatement of palatable food seeking in female rats. J Neurosci. 2013;33(1):214–226. | |
McCarthy MM, Arnold AP, Ball GF, Blaustein JD, De Vries GJ. Sex differences in the brain: The not so inconvenient truth. J Neurosci. 2012;32(7):2241–2247. | |
Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012;488(7410):172–177. | |
Jennings JH, Sparta DR, Stamatakis AM, et al. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496(7444):224–228. | |
Adolphs R, Tranel D. Impaired judgments of sadness but not happiness following bilateral amygdala damage. J Cogn Neurosci. 2004;16(3): 453–462. | |
Adolphs R, Baron-Cohen S, Tranel D. Impaired recognition of social emotions following amygdala damage. J Cogn Neurosci. 2002;14(8): 1264–1274. | |
Kim SY, Adhikari A, Lee SY, et al. Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature. 2013;496(7444):219–223. | |
Tye KM, Prakash R, Kim SY, et al. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471(7338): 358–362. | |
Felix-Ortiz AC, Beyeler A, Seo C, Leppla CA, Wildes CP, Tye KM. BLA to vHPC inputs modulate anxiety-related behaviors. Neuron. 2013;79(4):658–664. | |
Felix-Ortiz AC, Tye KM. Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci. 2014;34(2):586–595. | |
Hubner C, Bosch D, Gall A, Luthi A, Ehrlich I. Ex vivo dissection of optogenetically activated mPFC and hippocampal inputs to neurons in the basolateral amygdala: Implications for fear and emotional memory. Front Behav Neurosci. 2014;8:64. | |
Cho JH, Deisseroth K, Bolshakov VY. Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron. 2013;80(6):1491–1507. | |
Johansen JP, Hamanaka H, Monfils MH, et al. Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci U S A. 2010;107(28):12692–12697. | |
Huff ML, Miller RL, Deisseroth K, Moorman DE, LaLumiere RT. Posttraining optogenetic manipulations of basolateral amygdala activity modulate consolidation of inhibitory avoidance memory in rats. Proc Natl Acad Sci U S A. 2013;110(9):3597–3602. | |
Goshen I. The optogenetic revolution in memory research. Trends Neurosci. 2014;37(9):511–522. | |
Kheirbek MA, Drew LJ, Burghardt NS, et al. Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron. 2013;77(5):955–968. | |
Andrews-Zwilling Y, Gillespie AK, Kravitz AV, et al. Hilar GABAergic interneuron activity controls spatial learning and memory retrieval. PLoS One. 2012;7(7):e40555. | |
Cruz FC, Koya E, Guez-Barber DH, et al. New technologies for examining the role of neuronal ensembles in drug addiction and fear. Nat Rev Neurosci. 2013;14(11):743–754. | |
Garner AR, Rowland DC, Hwang SY, et al. Generation of a synthetic memory trace. Science. 2012;335(6075):1513–1516. | |
Lin D, Boyle MP, Dollar P, et al. Functional identification of an aggression locus in the mouse hypothalamus. Nature. 2011;470(7333): 221–226. | |
Wu Z, Autry AE, Bergan JF, Watabe-Uchida M, Dulac CG. Galanin neurons in the medial preoptic area govern parental behaviour. Nature. 2014;509(7500):325–330. | |
Albrecht U. Circadian clocks and mood-related behaviors. Handb Exp Pharmacol. 2013;(217):227–239. | |
Burgess CR, Peever JH. A noradrenergic mechanism functions to couple motor behavior with arousal state. Curr Biol. 2013;23(18): 1719–1725. | |
Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450(7168):420–424. | |
Carter ME, Brill J, Bonnavion P, Huguenard JR, Huerta R, de Lecea L. Mechanism for hypocretin-mediated sleep-to-wake transitions. Proc Natl Acad Sci U S A. 2012;109(39):E2635–E2644. | |
Rolls A, Colas D, Adamantidis A, et al. Optogenetic disruption of sleep continuity impairs memory consolidation. Proc Natl Acad Sci U S A. 2011;108(32):13305–13310. | |
Konadhode RR, Pelluru D, Blanco-Centurion C, et al. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci. 2013;33(25): 10257–10263. | |
Crock LW, Kolber BJ, Morgan CD, et al. Central amygdala metabotropic glutamate receptor 5 in the modulation of visceral pain. J Neurosci. 2012;32(41):14217–14226. | |
Ballon DR, Flanary PL, Gladue DP, Konopka JB, Dohlman HG, Thorner J. DEP-domain-mediated regulation of GPCR signaling responses. Cell. 2006;126(6):1079–1093. | |
Gaspari S, Papachatzaki MM, Koo JW, et al. Nucleus accumbens-specific interventions in RGS9–2 activity modulate responses to morphine. Neuropsychopharmacology. 2014;39(8):1968–1977. | |
Cui G, Jun SB, Jin X, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494(7436): 238–242. | |
Gerits A, Vanduffel W. Optogenetics in primates: A shining future? Trends Genet. 2013;29(7):403–411. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.