Back to Journals » Journal of Blood Medicine » Volume 5
Optimal management of hemophilic arthropathy and hematomas
Authors Lobet S, Hermans C, Lambert C
Received 19 June 2014
Accepted for publication 31 July 2014
Published 17 October 2014 Volume 2014:5 Pages 207—218
DOI https://doi.org/10.2147/JBM.S50644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Sébastien Lobet,1,2 Cedric Hermans,1 Catherine Lambert1
1Hemostasis-Thrombosis Unit, Division of Hematology, 2Division of Physical Medicine and Rehabilitation, Cliniques Universitaires Saint-Luc, Brussels, Belgium
Abstract: Hemophilia is a hematological disorder characterized by a partial or complete deficiency of clotting factor VIII or IX. Its bleeding complications primarily affect the musculoskeletal system. Hemarthrosis is a major hemophilia-related complication, responsible for a particularly debilitating chronic arthropathy, in the long term. In addition to clotting factor concentrates, usually prescribed by the hematologist, managing acute hemarthrosis and chronic arthropathy requires a close collaboration between the orthopedic surgeon and physiotherapist. This collaboration, comprising a coagulation and musculoskeletal specialist, is key to effectively preventing hemarthrosis, managing acute joint bleeding episodes, assessing joint function, and actively treating chronic arthropathy. This paper reviews, from a practical point of view, the pathophysiology, clinical manifestations, and treatment of hemarthrosis and chronic hemophilia-induced arthropathy for hematologists, orthopedic surgeons, and physiotherapists.
Keywords: hemophilia, arthropathy, hemarthrosis, hematoma, physiotherapy, target joint
Introduction to hemophilia treatment
Hemophilia is an X-linked hereditary bleeding disorder caused by deficiency in coagulation factor VIII (FVIII), for hemophilia A, and factor IX (FIX), for hemophilia B. Approximately 85% of patients with hemophilia (PwH) suffer from hemophilia A, and 15% from hemophilia B. The varying degrees of FVIII or FIX deficiency define the disease form as mild, moderate, or severe. Severe hemophilia is characterized by FVIII or FIX basal levels of less than 1%, moderate hemophilia by basal levels between 1% and 5%, and mild hemophilia by basal levels between 6% and 40%. The prevalence of hemophilia A is estimated at 1 in 5,000 live male births, and that of hemophilia B at 1 in 30,000.1 The deficient clotting factor activity results in inadequate thrombin generation, with a tendency for bleeding. Severe hemophilia patients are particularly symptomatic and display spontaneous bleeding from early childhood, primarily (80%–90% of bleeding episodes) in the musculoskeletal (MSK) system (muscles and large joints) and, less frequently, mucosal or cerebral hemorrhages. Intra-articular bleeding (hemarthrosis) causes synovial hypertrophy and cartilage damage, with gradual but inexorable joint destruction (hemophilic arthropathy).2
Hemophilia treatment is based on the intravenous injection of the deficient clotting factor. This is also referred to as replacement therapy. The treatment can be administered on-demand in more or less frequent sessions in order to treat or prevent occasional bleeding episodes. In severely affected patients, replacement therapy is administered in a prophylactic (or preventive) method, typically consisting of two to three administrations per week.3 Prophylaxis is used with the aim of maintaining minimal residual FVIII or FIX levels (>1%) and preventing spontaneous bleeding. Primary prophylaxis, which is initiated prior to or immediately following the first hemarthrosis, is used to prevent joint destruction or to halt its progression.
In the absence of primary prophylaxis, most severe PwH will sooner or later develop a first hemarthrosis, typically manifesting between the ages of 1 and 5 years. Repeated hemarthrosis is responsible for arthropathy in adulthood. Prophylaxis should therefore be initiated early and be continued throughout childhood, adolescence, and, often, adulthood. Given the variability of hemophilia’s phenotypic expression, replacement therapy should be individualized, with respect to dosing and injection frequency. The use of continuous FVIII or FIX infusion allows stable plasma concentration coagulation factor to be achieved and maintained. This treatment modality is recommended prior to and during surgical procedures or significant hemostatic challenges.4
The most challenging complication of therapy is the development of inhibitory alloantibodies directed against FVIII or FIX. These antibodies, also termed inhibitors, usually appear at the beginning of treatment, representing an immune response directed against FVIII or FIX, which is recognized as a foreign protein. These inhibitors develop in approximately 25%–30% of severe hemophilia A patients, yet only in 3%–5% of hemophilia B patients. They render replacement therapies ineffective, and limit patient access to a safe and effective standard of care, with increased morbidity and mortality risk.1 The consequences are dramatic, as hemarthroses are more frequent and more difficult to control, resulting in increased treatment costs and decreased quality of life (QoL).5 The introduction of bypassing agents, such as activated prothrombin complex concentrates (APCC) (Factor eight inhibitor bypassing activity or FEIBA®) or recombinant activated factor VII (rFVIIa or NovoSeven®), has dramatically improved the management of acute bleeding episodes in inhibitor patients, enabling them to be treated at home and resulting in substantially enhanced QoL.
Musculoskeletal complications of hemophilia
From hemarthrosis to chronic arthropathy
Unlike primary hemostasis disorders, such as functional platelet disorders, thrombocytopenia, or von Willebrand factor (VWF) deficiency, essentially characterized by cutaneous and mucosal bleeding, PwH develop deep hemorrhages mainly affecting muscles and large synovial joints. In severe hemophilia, 90% of bleeding episodes involve the MSK system, and in 80% of cases, the joints are particularly affected.6 These bleedings primarily consist of spontaneous hemarthroses that occur without any clearly identified cause and usually affect one joint at a time. In severe hemophilia patients, the initial hemarthrosis occurs at the time when the locomotor system is first solicited, namely when the child starts to walk. If left untreated, severe hemophilia patients may present more than 30 hemarthroses per year. Bleeding frequency and location tend to vary according to the patient’s age at consultation. While the knees and elbows are most commonly affected in patients older than 30 years of age, adolescents and young adults usually present bleedings affecting the ankles, despite adequate replacement therapy.7
Based on clinical manifestations, three stages can be distinguished: acute hemarthrosis that resolves with well-conducted replacement therapy and rehabilitation, usually without clinically detectable sequelae; subacute hemarthrosis occurs after repeated hemarthrosis episodes in the same joint – at this stage, the joint and surrounding soft tissues do not fully recover, while clinical signs of joint damage persist and are detectable between bleeding episodes; chronic arthropathy develops following numerous repeated bleeding episodes in joints, resulting in a significant loss of muscle function and muscle–tendon contractures.8
Acute hemarthrosis
For PwH, pain onset and local discomfort are the most common signs indicating that bleeding has started. If replacement therapy is not initiated quickly after the onset of joint bleeding, hemarthrosis will progress rapidly within a few hours, and the related transient functional impairment will evolve into a painful, hot, and swollen joint (Figure 1). In order to reduce intracapsular pressure, the affected limb tends to adopt an antalgic posture, usually in flexion, while weight-bearing becomes impossible, resulting in immediate loss of mobility. Administering coagulation factors rapidly reduces the pain, although inflammation and functional impairment may persist for some time, depending on the amount of blood present in the joint.
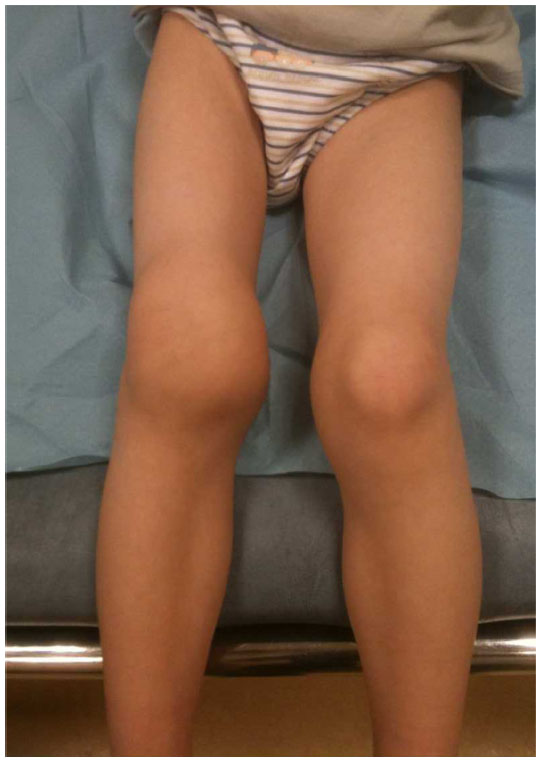 | Figure 1 Acute hemarthrosis of the right knee in a young boy with severe hemophilia A. |
Subacute hemarthrosis
Subacute hemarthroses arise after repeated episodes of hemarthrosis have occurred in the same joint. At this stage, the joint becomes a “target joint” due to its incomplete recovery. Clinical signs of joint damage are detectable between bleedings, consisting of decreased mobility, joint swelling due to joint effusion or synovial hypertrophy identifiable by palpation, and muscle, ligament, and capsular contractures.
Chronic arthropathy
Repeated hemarthrosis episodes engender a proliferative and destructive chronic synovitis with progressive joint destruction, known as hemophilic arthropathy. In the first stage, chronic knee arthropathy is characterized by joint hypertrophy due to chronic synovitis and effusion, in contrast to quadriceps atrophy (Figure 2A). Following this, an irregular epiphyseal hypertrophy deforms the joint and limits the range of motion (ROM), with a trend toward flexion contracture. In severe forms, we can observe static disorders such as genu valgum, lateral and posterior tibia subluxation and deformation in rotation, which lead to severe disability. Chronic ankle arthropathy primarily involves the tibio-talar and/or subtalar joint. Weight-bearing becomes painful, and joint stiffness is noted, particularly when getting out of bed in the morning. The patient’s movements in dorsiflexion and plantarflexion, as well as movements of inversion-eversion, become limited.9 Vicious attitudes in equine or plano-valgus deformation tend to develop (Figure 3). Chronic elbow arthropathy develops insidiously, leading to a loss of flexion-extension and prosupination, joint instability, and sometimes neurological complications like ulnar nerve impingement caused by osteophytes. In more advanced stages, a narrowing of the epiphyseal surfaces with loss of joint congruence, instability, and degradation of the remaining articular surfaces can be observed. Advanced bone changes, such as flattening of the femoral condyles or aseptic talus dome necrosis, as well as the presence of geodes and subchondral cysts are characteristic features of hemophilic arthropathy.
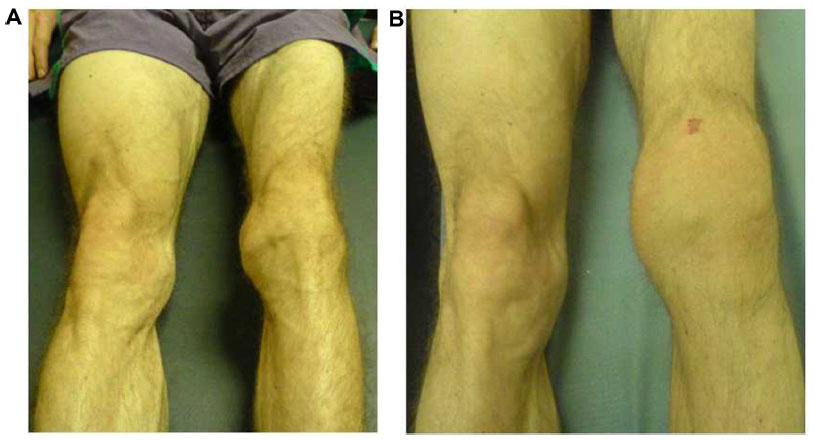 | Figure 2 (A) Patient with severe arthropathy of the left knee. The clinical Gilbert score was calculated to be 8/12, comprising presence of extension lag, limited flexion, severe muscular atrophy, crepitus on motion, and instability. (B) The clinical score could not be calculated in the same patient at the time of an acute hemarthrosis. |
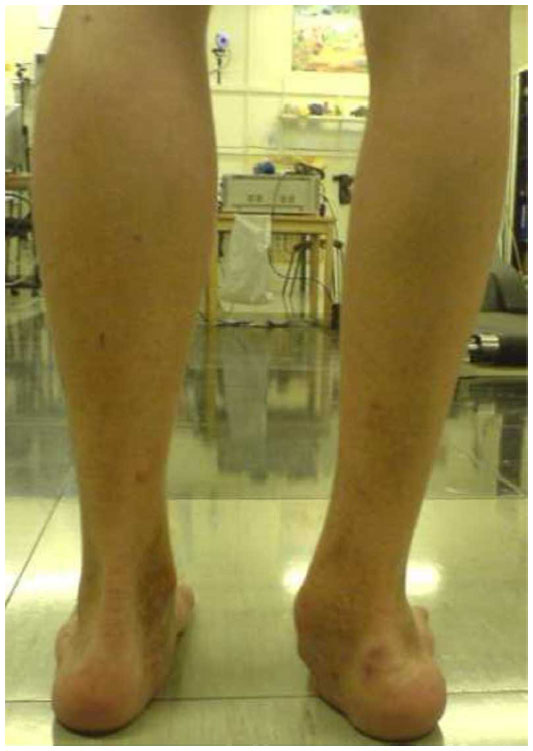 | Figure 3 Severe arthropathy of the right tibio-talar and subtalar joints, characterized by a plano-valgus deformation. |
Pathophysiology of hemophilic arthropathy
Hemarthrosis is the hallmark symptom of severe hemophilia. These joint bleedings are not usually observed in patients with impaired primary hemostasis, such as thrombopathy, thrombocytopenia, or von Willebrand disease (VWD). Only patients suffering from a complete deficiency of the VWF (Type 3 VWD) are at risk of hemarthrosis, which is enhanced by the FVIII deficiency resulting from VWF deficiency.
Based on the available data, it can be concluded that the absence of tissue factor in joints and muscles accounts for the articular tropism of bleeding in PwH.10 Tissue factor is a protein present on the surface of extravascular cells. When there is a breach in a blood vessel, FVIIa, present at low concentrations in the blood and primarily involved in initiating the process of coagulation, is brought into contact with tissue factor (the equivalent of matchbox). This connection causes the “spark” that triggers the coagulation process, principally comprising thrombin generation and the conversion of fibrinogen to fibrin. An amplification loop involving FVIII, FIX, and FXI leads to an explosive amount of thrombin being generated. While it is critical that this amplification loop functions correctly in tissues containing low tissue factor levels, the loop is ineffective in PwH, resulting in insufficient thrombin generation, with hemorrhages of varying severity. Hemorrhages occur principally in the large synovial joints, such as the knees, elbows, and ankles, which are at increased risk of traumatism.
The pathological mechanism underlying the development of hemophilic arthropathy is rather complex and not yet fully understood. This mechanism is likely multifactorial, involving cartilage degradation and inflammation of the synovial membrane due to the noxious effects related to blood effusions.11,12 Repeated episodes of hemarthrosis lead to an inflammatory and hypertrophic synovitis, with hemosiderin being deposited in synovial macrophages. This hyperemic and hypertrophic synovium is more sensitive to microtraumatisms, thus causing a vicious circle of bleeding, synovitis, and new hemorrhage. The enzymes and cytokines involved in degrading the hemosiderin released into the joint further contribute to synovial inflammation and cartilage destruction, among other factors, such as increased intracapsular pressure. Firstly, fibrous tissue develops within the synovium, then intra-articular and capsular fibrosis manifests with joint stiffening, also termed joint ankylosis. In addition to this inflammatory response, a second pathogenic mechanism comes into play. The presence of blood within the joint has a direct corrosive effect on the cartilage, inducing chondrocyte apoptosis and thus altering the cartilage matrix. The joint bleeds thus provoke a hemophilic arthropathy, with all the characteristics of both inflammatory joint diseases, like rheumatoid arthritis and degenerative diseases like osteoarthritis.
While all the joints of the upper and lower limbs may be affected by hemarthrosis, it is much rarer to see hip, shoulder, carpus, or small hand or foot joint involvement. Over 80% of hemarthroses occur in the knee, elbow, or ankle. Given that the intra-articular bleeding originates in the synovial plexus, a biomechanical hypothesis can be advanced. These types of trochlear or hinge joints are subject to significant constraints in rotation and present a convex surface fitting into a concave surface, potentially promoting bleedings by impingement of the enlarged synovium. Joints that tend to bleed frequently (ie, target joints) are those that are most frequently affected by chronic arthropathy at a later stage. This potentially causal relationship is still subject to frequent debate, as some patients with significant joint damage experience few episodes of clinically diagnosed hemarthrosis, despite having developed first arthropathy signs revealed by magnetic resonance imaging (MRI).13 It is likely that hemophilia is associated with repeated subclinical intra-articular bleeding, even in intensively-treated patients.
Muscular complications: hematomas
Hematomas are another major complication of hemophilia.8 These hematomas typically do not occur spontaneously but as a result of trauma, even if minimal. The hematoma develops gradually, causing an often-delayed diagnosis. Hematoma severity depends on the size and location. Hematoma size is often related to the anatomical lesion site. Subcutaneous hematomas rarely grow to a large size, due to the limited space available. Conversely, hematomas located in a large muscle, such as the gluteal region or calf, can reach a large size because bleeding occurs in an expandable space, tolerant of large volume increases, before clinical signs develop.
Bleeding in large sheath muscles, such as the thigh or gluteal region, can lead to severe anemia. In small sheath muscles, like the anterior surface of the forearm, palm, or calf, bleeding may cause neurovascular compression. Some hematomas can be dangerous, owing to their location. Hematomas located near a nerve root ending or a vascular bundle may compromise the functional prognosis. Hematomas of the gluteal region or popliteal fossa can, for instance, lead to sciatic nerve compression. Hematomas of the anterior compartment of the forearm are dangerous, given their potential to compress the median or ulnar nerve, resulting in a Volkmann’s contracture affecting the tendons. Hematomas of the iliopsoas muscle are common, initially causing moderate groin pain associated with a vicious attitude in hip flexion. Clinical signs may be misleading, further delaying the diagnosis. As a consequence, the diagnosis is often made when femoral nerve compression or severe anemia is observed. In rare cases, hematoma may present as an encapsulated blood collection referred to as hemophilic pseudotumor.
Clinical and radiological diagnosis of hemophilic arthropathy
A complete MSK assessment of joint and muscle function should be performed in any PwH, with an evaluation of the joints conducted every 6 months in children and, at least, every year in adults.4 Several clinical scores have been proposed. The Gilbert score constitutes an additive score used to quantify joint defects.14 The Gilbert score is based on different functional and structural aspects, including joint pain, bleeding frequency, deformation in the frontal plane, presence of a flexion contracture, ROM limitation, instability, muscular atrophy, presence of crepitus on motion, and joint swelling (Figure 2A and B). Despite lacking psychometric properties, such as reproducibility, sensitivity, and responsiveness, the Gilbert score is still used in clinical practice to evaluate patients with evident arthropathy signs, especially as it offers the possibility of systematizing the clinical examination.15 The Hemophilia Joint Health Score (HJHS), which was recently developed by a consensus of experts, is more sensitive to early changes; while originally created for the pediatric population, it may also be recommended in adults.16,17
Radiography has been used for several decades to evaluate hemophilic arthropathy. Two classification systems of the ankles, knees, and elbows, both based on conventional radiography, have been widely used to quantify hemophilic arthropathy in clinical trials: the Arnold–Hilgartner (A-H) scale and the Pettersson score.18,19 The A-H scale is a progressive scoring method including six stages (Stages 0–5), with Stage 5 corresponding to the worst finding (ie, end-stage arthropathy). The A-H scale is based principally on radiographic findings and attempts to categorize joint changes into stages that have surgical significance. The Pettersson score is an additive scale where each abnormality is graded from 0 to 2, and the points of each category are then summed in order to obtain an individual joint score (Figure 4A and B). There are some differences between the two scoring systems. While the A-H scale is simple and easy to use by everyone, the Pettersson score is more meticulous and more efficiently discriminates between the different stages of hemophilic arthropathy. In addition, the Pettersson score is not designed to evaluate soft tissue changes, as these are inherently difficult to assess through plain radiography. The major limitation of radiography is that it is able only to depict gross joint alterations, and cannot detect early changes in soft tissues, synovium, or cartilage, and is therefore insensitive to the early changes of hemophilic arthropathy.20 In this case, once radiographic changes become apparent, the arthropathy has usually already reached an advanced and irreversible clinical stage. More refined and sensitive tools are required to monitor subtle changes in joints, to improve therapy.
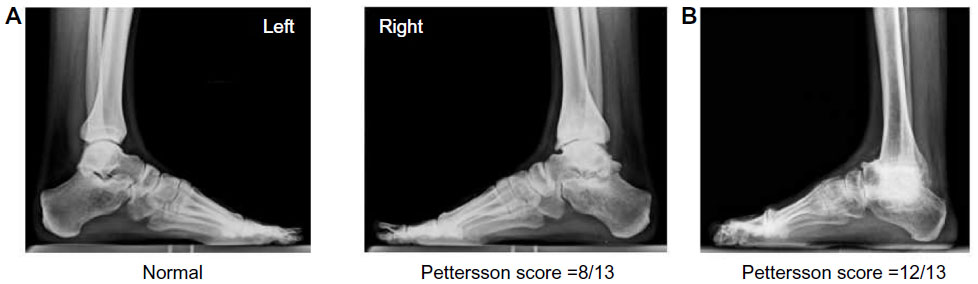 | Figure 4 Pettersson radiological score (the maximum of 13 points representing most severe arthropathy) in a 27-year-old severe hemophilia A patient showing (A) a normal ankle (left) and a moderate arthropathy (right). (B) End-stage hemophilic arthropathy (avascular necrosis of the talar dome, multiple osteophytes, severe joint space narrowing, and natural fusion of the joint). |
Ultrasonography (US) is a low-cost, noninvasive, and real-time imaging technique.21,22 US is ideally suited for determining the extent and degree of soft tissue inflammation, detecting the presence of intra-articular fluid, carrying out follow-up, and fine-tuning treatment for patients with hemarthrosis or chronic synovitis, so as to adjust the dosage of coagulation factor. Moreover, this examination is easy to perform in children.
MRI is currently the most sensitive imaging technique for early detection of joint lesions.23 In contrast to conventional radiography and US, MRI enables multitissue imaging that includes the bones as well as the surrounding ligaments and tendons. MRI is able to visualize all hemophilic arthropathy lesions and may thus be used to detect and monitor less-advanced joint damage, particularly common at the early stages of arthropathy. It is therefore considered as the gold standard for identifying early changes in joints. Nonetheless, MRI is costly, less accessible, and more time-consuming than other imaging techniques. Furthermore, it often requires sedation in children.
An overview of advantages and drawbacks of main diagnostic approaches assessing MSK in PwH is summarized in Table 1.
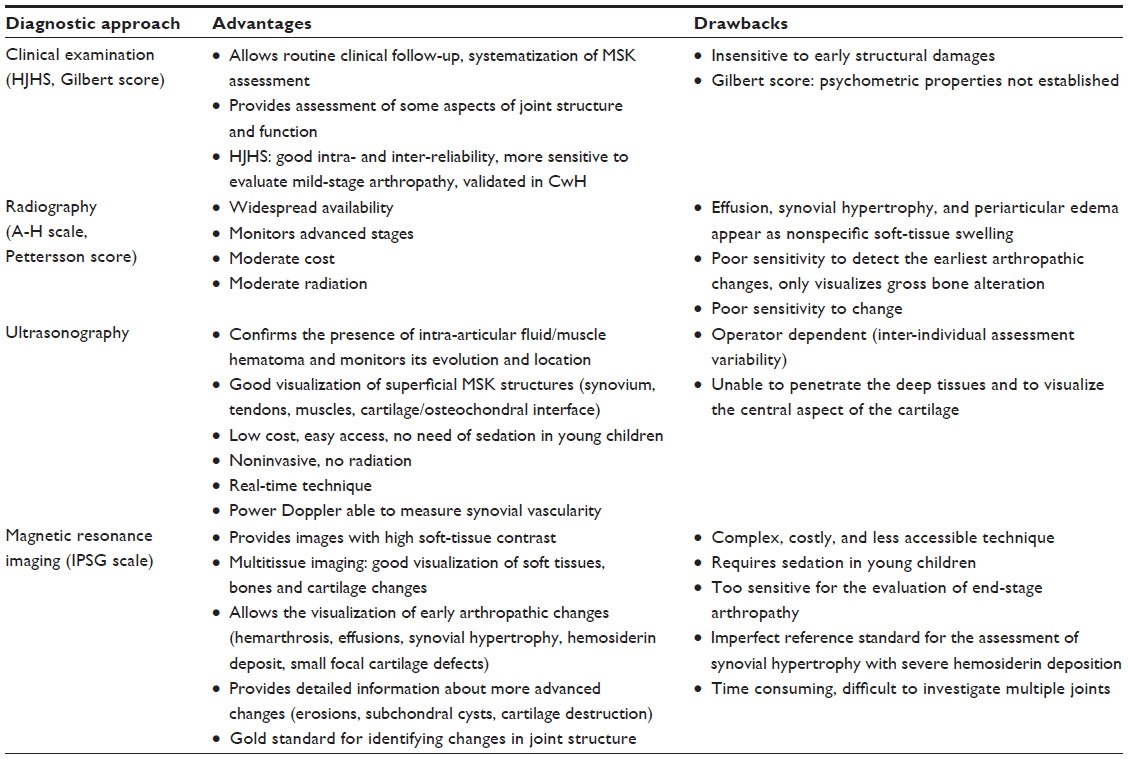 | Table 1 Advantages and drawbacks of main diagnostic approaches assessing MSK impairments in patients with hemophilia |
In addition to the structural damage caused to joint cartilage by recurrent bleeding episodes, PwH may experience functional limitations, as well as difficulties in performing and participating in everyday activities. The International Classification of Functioning, Disability, and Health (ICF) describes the functioning of a person as the perception of the body by the individual and society.24 Clinical scores, such as the Gilbert score and the HJHS, integrate both aspects of body function and body structure. Activity limitations (eg, the inability to climb or go down stairs or difficulty shaving due to elbow arthropathy) can be measured using two hemophilia-specific scores: a self-reported score, the Hemophilia Activities List (HAL); and a performance-based score, the Functional Independence Score for Hemophilia (FISH).25,26 In conjunction with clinical evaluation, it is also recommended that the impact of joint damage on QoL be assessed (eg, CHO-KLAT, Haemo-QoL scores).27
Management of acute hemorrhagic episodes
The management of acute bleeding episodes is based currently on immediate replacement therapy and the RICE (Rest, Ice, Compression, and Elevation) regime. In the acute phase, the first therapeutic measure consists in injecting clotting factor within 2 hours of bleeding onset. The classically recommended dose is 25 to 40 IU/kg of FVIII, or 50 IU/kg in cases of severe hemarthrosis.4 Several injections, spaced out over time, are generally required.
The application of ice following a soft tissue injury is believed to help manage acute hemarthrosis-related pain by decreasing nerve conduction velocity, reducing edema formation through proinflammatory response reduction, and inducing vasoconstriction, thereby reducing blood flow to the injured tissue.28 However, Forsyth et al have recently suggested that any decrease in intra-articular temperature could interfere with coagulation in the presence of acute tissue lesions (in vitro) in both in animals and human subjects with normal hemostasis.29,30 Furthermore, applying ice actually delays healing by preventing macrophages from releasing insulin-like growth factor (IGF-1) within the damaged muscle or joint. Since applying ice to an injury has been shown to reduce pain, it is our view that cooling an injured joint or muscle for short periods, soon after the bleeding, is acceptable, but that this should not exceed 6 hours.
The rest imposed to a joint means immobilization and no weight-bearing. In cases of impaired hemostasis, synovial tissue may be predisposed to rebleeding during the fragile wound healing phases of angiogenesis and fibroblast proliferation.31 Joints should be rested in a functional position, thus favoring early, conservative, and gentle mobilization. In terms of the load that can be applied to a lower limb joint during the acute phase, a recent animal study indicated that weight-bearing on a bleeding joint, as compared against a non-weight-bearing one, resulted in more cartilage matrix damage.32 It is therefore advisable to avoid weight-bearing during the first week; the use of walking aids (crutches, walker) enables progressive weight-bearing from the seventh day onwards.
An appropriate balance should be established between rest, early mobilization, and weight-bearing so as to prevent unwanted complications associated with immobilization, while simultaneously minimizing the risk of rebleeding, synovitis, and cartilage damage. Externally applied compression helps to limit joint swelling by increasing external pressure and limiting joint capsule distension, thus leading to reduced bleeding through earlier tamponade.33 Elevation is also an effective method for reducing hemarthrosis-related swelling. Elevating the swollen area to above heart level reduces capillary hydrostatic pressure, and gravity helps the blood to flow back to the heart.30
An imaging assessment is not routinely required except in cases of trauma. Angiography with embolization is exceptionally carried out, when major hemarthrosis does not respond well to initial treatment.34 A local vascular abnormality that can be embolized is sometimes detected. It is necessary to administer analgesics, like paracetamol, but caution should be exercised in hepatitis C patients with hepatic insufficiency. In these cases, opioids and cyclooxygenase-2 (COX-2) selective non-steroidal anti-inflammatory drugs (NSAIDs) (with no effect on primary hemostasis) can potentially be used. As highlighted in a recent review, the management of hemarthrosis is based on low-level evidence, given the limited number of studies and the lack of randomized studies.4
In a small number of cases, often in post-traumatic context, hemarthrosis can be massive and expose the cartilage to a large amount of blood. The bleeding episode can, of course, be interrupted by the infusion of clotting factor, but there is an unavoidable time elapse before the synovial membrane has reabsorbed the large amount of blood. During this time, the cartilage is exposed to high concentrations of blood. Given the known correlation between the extent and time of exposure to blood on the one hand and the degree of chondrocyte apoptosis on the other hand, joint aspiration can be recommended as early as possible, ideally within 24 hours.35 This procedure is indicated in cases of major bleeding when the joint remains tight, swollen, and painful.2 It relieves pain and spasm, and accelerates functional rehabilitation. Aspiration with washing and local steroid infiltration presupposes that the patient is protected by coagulation factor infusion and is performed in strictly aseptic conditions, and is only recommended for severe hemarthrosis. The systematic use of joint aspiration should be avoided, owing to the risk of infection and other complications. Joint aspiration is formally indicated in case of acute hip hemarthrosis, though rare, with the aim of preventing aseptic osteonecrosis.2
The goal of physiotherapy in the initial acute phase of muscle hematoma is to decrease pain and swelling, support wound healing, and maintain hemostasis.36 Clinical guidelines recommend the RICE regime, with some supplementary restrictions imposed. A short resting period, consisting either of immobilization or non-weight-bearing, is advised for the first 48–72 hours, post injury, to prevent further hemorrhage. The use of crutches is recommended for patients with the most severe type of lower extremity hematoma, particularly when the injury is located at a site which poses difficulties for sufficient immobilization, such as in iliopsoas hematoma. In young children, compression is not recommended, given their inability to alert physicians of adverse symptoms such as paresthesia.36 In the acute phase, all therapies that impede the hemostatic process, such as massages and heat sources, are contraindicated.
Following the short immobilization period, low-intensity stretching exercises should be initiated, within the limits of pain, in order to distend the maturing scar while it is still plastic.36 Since any loss of muscle strength may increase the risk of muscle bleeds during physical activities, a specifically-tailored muscle strengthening program is advisable, involving firstly isometric contractions then concentric exercises. Any estimates of time to full recovery in PwH should be realistic and based on known timelines from nonhemophilic populations.36 Rehabilitation can be discontinued based on the patient’s ability to stretch the injured muscle to pre-bleed levels, as well as the pain-free use of the injured muscle. In cases where simple everyday movement has induced the hematoma, it is important to continue rehabilitation beyond pre-bleed levels in order to improve functional movement patterns.
The principles of management of an acute hemarthrosis in a PwH are summarized in Table 2. The steps and treatments indicated in the emergency room are summarized in Table 3.
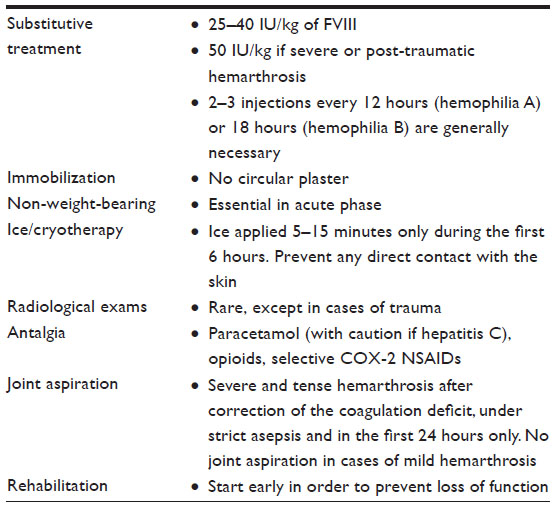 | Table 2 Principles of management of acute hemarthrosis in a hemophilia A patient |
 | Table 3 Hemophilia in emergency room – steps and treatment indicated |
Management strategies for chronic hemophilic arthropathy and acute hematoma
Conservative treatments: physiotherapy, rehabilitation, and orthoses
Weekly physiotherapy sessions, designed to improve global joint mobilization and prevent vicious attitudes and muscle atrophy, may be offered to PwH with chronic arthropathy.37 In individuals with advanced single- or multiple-joint arthropathy, maximizing function and adapting to limitations should be the focus of therapy, using appropriate strategies such as pacing activities, exercise programs, the provision of aids for everyday activities, and the avoidance of aggravating activities.
Adaptive and corrective splints and orthoses may be considered for joint instability and deformity. PwH with valgus/varus malalignment of the hindfoot and flat/cavus foot often experience discomfort while walking or standing for long periods. The use of insoles and specially-designed orthopedic shoes has been shown to reduce pain and improve ankle propulsion in patients with end-stage ankle arthropathy.38 While there is currently no consensus with respect to the best type of foot orthosis for managing foot pain in PwH, we proposed insoles as the first option for patients with moderate ankle arthropathy or partially correctable rear-foot (compared against orthopedic shoes) for those with more severe pain or poor ROM.38 Hydrotherapy is particularly recommended for patients with multiple joint arthropathies, who find land-based functional stretching and strengthening exercise difficult.36
Deeper hematomas can be difficult to diagnose, as visible signs may be lacking.36 In this context, US and MRI are also important diagnostic tools. The assessment of muscle structure and function following a muscle bleed is essential. Although the information on muscle integrity may not affect the immediate hematological input, this will guide the rehabilitation process and determine the timelines and outcome expected. Failure in this regard may lead to recurrent or new bleeding episodes at the site of injury or due to compensation strategies adopted as a result of the injury.36
Orthopedic surgery
Surgical procedures should be restricted to patients with severe joint damage and for whom conservative measures have failed. In addition, the benefits of surgery should outweigh potentially-related complications, such as infections, neuropathies, and bleeding. In many cases, surgery is delayed due to the young age of the patient, and taking into account the technical difficulties in hemostasis. When considering surgery in PwH, certain precautions must be taken. Operations must be performed under the coverage of replacement therapy, by an experienced surgeon, and in a center specifically specialized in hemophilia treatment, in order to manage and adapt to daily replacement therapy. This multidisciplinary approach is essential to ensure surgical success.
In cases of repeated hemarthrosis in a single joint, with the related risk of synovitis and chronic arthropathy, a synovectomy or synoviorthesis may be justified. These techniques are designed to partially or totally destroy the hypertrophic synovium refractory to conservative treatment, and are indicated when bleedings reoccur that replacement therapy fails to control. Nevertheless, these procedures must be performed before the development of excessive joint damage.39 Irrespective of which technique is chosen, none are capable of preventing the degradation of already damaged joints and cannot improve joint function. Synoviorthesis consists in injecting a chemical or radioisotope product into the joint. This technique prevents inflammation by facilitating the formation of a fibrous tissue that inhibits vessel proliferation and reduces the risk of intra-articular bleeding. It is minimally invasive and can be performed in outpatient clinics, requiring no major coverage with coagulation factors. Surgical synovectomy consists in conducting as complete a surgical excision as possible of the hypertrophic synovial membrane. This simultaneously enables foreign bodies to be washed and retrieved from the joint. This procedure can be performed either by open or arthroscopic approach. Arthroscopy avoids large incisions, is associated with less severe functional loss, and enables a larger excision of the synovial tissue and earlier rehabilitation, in comparison with the open procedure.40 Synovectomy, however, requires administration of more coagulation factors than that required in synoviorthesis.
Total knee replacement (TKR) has become the treatment of choice for patients with severe arthropathy of the knee (Figure 5A and B). TKR abolishes pain and corrects some deformations. Yet this procedure can be complicated by the presence of an articular fibrosis or severe bone deformities in the preoperative stage. The joint stiffness associated with hemophilia seems to be principally related to the presence of contractures. These contractures may result from recurrent hemarthrosis with subsequent fibroblastic proliferation and progressive arthropathy, or even extra-articular intramuscular bleeding leading to fibrosis.41 Arthrofibrosis and bone deformities in the preoperative stage are responsible for joint stiffness, which remains a relatively common complication for PwH undergoing TKR despite an intensive and tailored rehabilitation program. For some patients, however, the resulting slight improvements in ROM combined with reduced articular pain have a significantly positive impact on their QoL. A long-term rehabilitation program should be recommended for all PWH undergoing TKR. Lobet et al have proposed an algorithm for the physiotherapy management of end-stage hemophilic arthropathy of the knee.37 Combined with the use of clotting factor replacement, the rehabilitation program following TKR in PwH can, to some extent, be extrapolated from the program followed by patients with osteoarthritis of the knee.
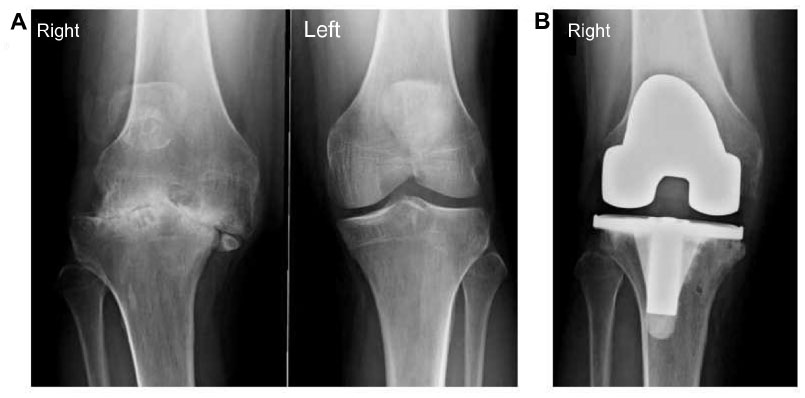 | Figure 5 (A) Severe right knee arthropathy in a 32-year-old severe hemophilia A patient. It should be noted that the left knee never experienced any hemarthroses, and therefore presents no articular damage. (B) The same subject following TKR on the right side. |
In cases of severe elbow arthropathy, bony deformations of the joint surfaces are commonly observed, and the enlargement of the radial head limits prosupination movement. For these patients, surgical resection of the radial head can be proposed, to be performed conjointly with a synovectomy and joint lavage.42 The procedure is easy to perform, has few complications, reduces the occurrence of bleeding episodes, and reduces painful symptoms. While ROM can consequently be improved immediately, primarily in the prosupination direction, movement in flexion-extension is not positively impacted. The indication for total elbow prosthesis should be considered when arthropathy impairs everyday activities. However, elbow joint disease is often tolerated for a long time. When a prosthesis is eventually indicated, the bone transformations are so significant that an effective placement of a prosthesis is impossible.
For severe end-stage ankle arthropathy associated with severe pain, deformities of the hindfoot in the frontal plane, and frequent bleeding, the recommended procedure is surgical arthrodesis of the tibio-talar and/or subtalar joint.43 To date, total ankle replacement (TAR) remains an uncertain therapeutic option with insufficient follow-up data. The first results of TAR in PwH do, however, seem satisfactory, both in terms of pain reduction and improvement of ankle ROM.44 Nevertheless, it seems that such a procedure can only be considered in PwH who still possess some ROM of the ankle.
Participation in physical activity and sport
For many years, people with hemophilia and other bleeding disorders were previously discouraged from practicing sports or physical activities in the attempt to minimize the occurrence of hemorrhagic incidents. Participation in appropriate sports and physical activity should, however, be encouraged as part of the global approach to hemophilia management as this provides numerous physical benefits, as well as improving the emotional and social well being in PwH. An active lifestyle is associated with the prevention of cardiovascular disease, diabetes mellitus, cancer, hypertension, obesity, depression, and osteoporosis, which also holds true for those suffering from chronic health conditions, as with PwH.45
Walking, swimming, and cycling, to name but a few, allow patients to strengthen their muscle mass, making them less vulnerable to bleeding joints. On the other hand, contact sports and competitions expose them to greater risks of bleeding, and patients should be informed accordingly.45 Physical activity for PwH is associated with specific challenges, such as the risk of injury, overloading, and potential bleeds; yet, correctly managed, participation in physical activity and exercise can improve strength, proprioception, joint ROM, as well as reducing frequency of bleeding.46 Prior to starting a new physical activity, a thorough MSK function assessment should be performed, and patients should be advised on appropriate, safe activities, on an individual basis. The physiotherapist can give recommendations on the adaptations needed in order for a PwH to safely participate in a sport. Sports and physical activities should ideally be performed under the cover of factor replacement, and more intense physical activity, particularly in competitions, should be matched to the day of prophylaxis administration.
Analgesia and the use of anti-inflammatory drugs
As explained above, the use of analgesics and anti-inflammatory drugs is recommended at the onset of acute hemarthrosis. The painful symptoms associated with chronic arthropathy can be relieved by NSAIDs. The COX-2 selective NSAIDs, which have the advantage of not provoking a disturbance of primary hemostasis, are particularly indicated in PwH.47
Conclusion: the multidisciplinary follow-up
Ideally, any patient with severe hemophilia should be followed-up by a hemophilia specialist, with one or two visits conducted per year. For patients with moderate or minor deficit, annual follow-ups are also essential. Hemophilia is a rare and complex disease. In order to offer comprehensive and coherent support, multiple skills are required, and must be coordinated simultaneously. Specialists in coagulation, qualified nurses, physiotherapists, and orthopedic surgeons must work together closely to support the hemophiliac patient and manage related orthopedic complications. For this reason, patient follow-up should ideally be centralized within multidisciplinary hemophilia centers working closely in collaboration with local hospitals and physicians. The evaluation of joint status, along with its evolution and the impact on QoL, represent an integral part of the hemophilia consultation.
Acknowledgment
The development of this review paper was made possible by a financial grant from the Bayer Hemophilia Awards Program. From this program, Sébastien Lobet received an Early Career Investigator Award in 2012.
Disclosure
The authors have stated that they have no interests that could be perceived as posing any conflict or bias.
References
Franchini M, Mannucci PM. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis. 2012;7:24. | |
Rodriguez-Merchan EC, Jimenez-Yuste V, Aznar JA, et al. Joint protection in haemophilia. Haemophilia. 2011;17 Suppl 2:1–23. | |
Blanchette VS. Prophylaxis in the haemophilia population. Haemophilia. 2010;16 Suppl 5:181–188. | |
Hermans C, De Moerloose P, Fischer K, et al. Management of acute haemarthrosis in haemophilia A without inhibitors: literature review, European survey and recommendations. Haemophilia. 2011;17(3):383–392. | |
Brown TM, Lee WC, Joshi AV, Pashos CL. Health-related quality of life and productivity impact in haemophilia patients with inhibitors. Haemophilia. 2009;15(4):911–917. | |
Rodriguez-Merchan EC. Musculoskeletal complications of hemophilia. HSS J. 2010;6(1):37–42. | |
Stephensen D, Tait RC, Brodie N, et al. Changing patterns of bleeding in patients with severe haemophilia A. Haemophilia. 2009;15(6):1210–1214. | |
Schved JF. Physiopathologie et bases moléculaires. Hematologie. 2008;13-021-B-10. | |
Lobet S, Hermans C, Bastien GJ, Massaad F, Detrembleur C. Impact of ankle osteoarthritis on the energetics and mechanics of gait: the case of hemophilic arthropathy. Clin Biomech (Bristol, Avon). 2012;27(6):625–631. | |
Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24(6):1015–1022. | |
Roosendaal G, Lafeber FP. Pathogenesis of haemophilic arthropathy. Haemophilia. 2006;12 Suppl 3:117–121. | |
Valentino LA. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Haemost. 2010;8(9):1895–1902. | |
Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. | |
Gilbert MS. Prophylaxis: musculoskeletal evaluation. Semin Hematol. 1993;30(3 Suppl 2):3–6. | |
Silva M, Luck JV Jr, Quon D, et al. Inter- and intra-observer reliability of radiographic scores commonly used for the evaluation of haemophilic arthropathy. Haemophilia. 2008;14(3):504–512. | |
Feldman BM, Funk S, Lundin B, Doria AS, Ljung R, Blanchette V. Musculoskeletal measurement tools from the International Prophylaxis Study Group (IPSG). Haemophilia. 2008;14 Suppl 3:162–169. | |
Hilliard P, Funk S, Zourikian N, et al. Hemophilia joint health score reliability study. Haemophilia. 2006;12(5):518–525. | |
Arnold WD, Hilgartner MW. Hemophilic arthropathy. Current concepts of pathogenesis and management. J Bone Joint Surg Am. 1977;59(3):287–305. | |
Pettersson H, Ahlberg A, Nilsson IM. A radiologic classification of hemophilic arthropathy. Clin Orthop Relat Res. 1980;(149):153–159. | |
Jelbert A, Vaidya S, Fotiadis N. Imaging and staging of haemophilic arthropathy. Clin Radiol. 2009;64(11):1119–1128. | |
Aznar JA, Abad-Franch L, Perez-Alenda S, Haya S, Cid AR, Querol F. Ultrasonography in the monitoring of management of haemarthrosis. Haemophilia. 2011;17(5):826–828. | |
Muca-Perja M, Riva S, Grochowska B, Mangiafico L, Mago D, Gringeri A. Ultrasonography of haemophilic arthropathy. Haemophilia. 2012;18(3):364–368. | |
Lundin B, Babyn P, Doria AS, et al. Compatible scales for progressive and additive MRI assessments of haemophilic arthropathy. Haemophilia. 2005;11(2):109–115. | |
World Health Organisation. International Classification of Functioning, Disability and Health. Geneva: WHO; 2001. | |
Van Genderen FR, Westers P, Heijnen L, et al. Measuring patients’ perceptions on their functional abilities: validation of the Haemophilia Activities List. Haemophilia. 2006;12(1):36–46. | |
Poonnoose PM, Manigandan C, Thomas R, et al. Functional independence score in haemophilia: a new performance-based instrument to measure disability. Haemophilia. 2005;11(6):598–602. | |
Young NL, St-Louis J, Burke T, Hershon L, Blanchette V. Cross-cultural validation of the CHO-KLAT and HAEMO-QoL-A in Canadian French. Haemophilia. 2012;18(3):353–357. | |
Zourikian N, Forsyth AL. Physiotherapy evaluation and intervention in the acute hemarthrosis: challenging the paradigm. In: Rodriguez-Merchan EC, Valentino LA, editors. Current and Future Issues in Hemophilia Care. 1st ed. Hoboken, NJ. Wiley-Blackwell; 2011. p.156–161. | |
Forsyth AL, Rivard GE, Valentino LA, et al. Consequences of intra-articular bleeding in haemophilia: science to clinical practice and beyond. Haemophilia. 2012;18 Suppl 4:112–119. | |
Forsyth AL, Zourikian N, Valentino LA, Rivard GE. The effect of cooling on coagulation and haemostasis: Should “Ice” be part of treatment of acute haemarthrosis in haemophilia? Haemophilia. 2012;18(6):843–850. | |
Hoffman M. Animal models of bleeding and tissue repair. Haemophilia. 2008;14 Suppl 3:62–67. | |
Hakobyan N, Kazarian T, Valentino LA. Synovitis in a murine model of human factor VIII deficiency. Haemophilia. 2005;11(3):227–232. | |
Charalambides C, Beer M, Melhuish J, Williams RJ, Cobb AG. Bandaging technique after knee replacement. Acta Orthop. 2005;76(1):89–94. | |
Rodriguez-Merchan EC, Jimenez-Yuste V. The role of selective angiographic embolization of the musculo-skeletal system in haemophilia. Haemophilia. 2009;15(4):864–868. | |
Roosendaal G, Jansen NW, Schutgens R, Lafeber FP. Haemophilic arthropathy: the importance of the earliest haemarthroses and consequences for treatment. Haemophilia. 2008;14 Suppl 6:4–10. | |
Sorensen B, Benson GM, Bladen M, et al. Management of muscle haematomas in patients with severe haemophilia in an evidence-poor world. Haemophilia. 2012;18(4):598–606. | |
Lobet S, Pendeville E, Dalzell R, et al. The role of physiotherapy after total knee arthroplasty in patients with haemophilia. Haemophilia. 2008;14(5):989–998. | |
Lobet S, Detrembleur C, Lantin AC, Haenecour L, Hermans C. Functional impact of custom-made foot orthoses in patients with haemophilic ankle arthropathy. Haemophilia. 2012;18(3):e227–e235. | |
Rodriguez-Merchan EC. Radionuclide synovectomy (radiosynoviorthesis) in hemophilia: a very efficient and single procedure. Semin Thromb Hemost. 2003;29(1):97–100. | |
Verma N, Valentino LA, Chawla A. Arthroscopic synovectomy in haemophilia: indications, technique and results. Haemophilia. 2007; 13 Suppl 3:38–44. | |
Atkins RM, Henderson NJ, Duthie RB. Joint contractures in the hemophilias. Clin Orthop Relat Res. 1987;(219):97–106. | |
Silva M, Luck JV Jr. Radial head excision and synovectomy in patients with hemophilia. Surgical technique. J Bone Joint Surg Am. 2008; 90 Suppl 2 Pt 2:254–261. | |
Pasta G, Forsyth A, Merchan CR, et al. Orthopaedic management of haemophilia arthropathy of the ankle. Haemophilia. 2008;14 Suppl 3:170–176. | |
Barg A, Elsner A, Hefti D, Hintermann B. Haemophilic arthropathy of the ankle treated by total ankle replacement: a case series. Haemophilia. 2010;16(4):647–655. | |
Negrier C, Seuser A, Forsyth A. The benefits of exercise for patients with haemophilia and recommendations for safe and effective physical activity. Haemophilia. 2013;19(4):487–498. | |
Souza JC, Simoes HG, Campbell CS, Pontes FL, Boullosa DA, Prestes J. Haemophilia and exercise. Int J Sports Med. 2012;33(2):83–88. | |
Rattray B, Nugent DJ, Young G. Celecoxib in the treatment of haemophilic synovitis, target joints, and pain in adults and children with haemophilia. Haemophilia. 2006;12(5):514–517. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.