Back to Journals » Eye and Brain » Volume 10
Optic nerve sheath meningiomas: prevalence, impact, and management strategies
Authors Parker RT, Ovens CA, Fraser CL, Samarawickrama C ![]()
Received 10 April 2018
Accepted for publication 10 July 2018
Published 24 October 2018 Volume 2018:10 Pages 85—99
DOI https://doi.org/10.2147/EB.S144345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Margaret Wong-Riley
Video abstract presented by Richard Parker.
Views: 4089
Richard T Parker,1 Christopher A Ovens,1 Clare L Fraser,2 Chameen Samarawickrama1,3
1Sydney Medical School, Discipline of Clinical Ophthalmology and Eye Health, University of Sydney, Sydney, NSW, Australia; 2Save Sight Institute, University of Sydney, Sydney, NSW, Australia; 3Department of Ophthalmology, Faculty of Medicine, South Western Sydney Clinical School, University of New South Wales, Sydney, NSW, Australia
Abstract: Optic nerve sheath meningiomas are rare benign neoplasms of the meninges surrounding the optic nerve. They are a significant cause of morbidity. While the mortality rate is practically zero, these tumors can blind or disfigure patients. Given that the clinical course can be variable, and treatment has the capacity to cause morbidity itself, the management of these patients can be difficult. We review the literature to discuss the prevalence of optic nerve sheath meningiomas, the association with neurofibromatosis type 2, natural history, and management options and strategies.
Keywords: meningioma, optic nerve neoplasms, neurofibromatosis 2, radiotherapy, imaging
Introduction
Optic nerve sheath meningiomas (ONSMs) are usually benign neoplasms of the meninges surrounding the optic nerve. Patients classically present in the fourth decade with a triad of vision loss, optic atrophy, and optociliary shunt vessels on fundoscopy.1 While optic nerve tumors are rare, ONSMs account for approximately one-third of all intrinsic tumors of the optic nerve.2 Importantly, they can be associated with neurofibromatosis type 2. While there is no mortality and little nonvisual morbidity associated with them, ONSMs often lead to vision loss in the affected eye. Treatments carry a risk of vision loss, so management of these patients becomes the careful task of balancing risk of progression and vision loss with the risk of treatment.
This review will discuss the prevalence of ONSMs and the association with neurofibromatosis type 2, natural history, and management options.
Search strategies
We searched MEDLINE for articles with subject headings *Optic Nerve Neoplasms/ and *Meningioma/. Review articles previously published3–13 were examined and relevant references were obtained and included in this review. Only English language documents were eligible for inclusion in this review.
Epidemiology
Orbital tumors are rare, and ONSMs represent a small subset of these tumors. Dutton3 conducted a comprehensive review, showing that ONSMs represent approximately 2% of all orbital tumors. ONSMs show a female predominance (61%–39% for males), and the mean age of patients at diagnosis is approximately 40 years. Males tend to present slightly earlier (36 years vs 42 years for females). Bilateral tumors occur in 5% of patients. Of meningiomas that involve the orbit, only 10% are of primary orbital origin, while the remainders are of intracranial origin. Tumors that arise from other intracranial or intraorbital locations and secondarily affect the optic nerve sheath are termed “secondary ONSMs”. Optic canal ONSMs, important due to the ease of compression of the optic nerve in this tight anatomical space, represent 8% of ONSMs.
Pediatric ONSMs are even more rare, comprising approximately 2%–4% of ONSMs3,14 and having an overall prevalence of between 1:95,000 and 1:525,000.15 Almost a third of pediatric patients with ONSMs are diagnosed with neurofibromatosis type 2 (NF2), and almost a third of children with NF2 are subsequently diagnosed with an ONSM.16
NF2
NF2 is strongly associated with the development of neural tumors, including ONSMs. It results from mutations in the NF2 tumor suppressor gene located on chromosome 22q12.17 It is inherited in an autosomal-dominant fashion, although there is a high spontaneous mutation rate.18 In addition, there is a high frequency of mosaicism in patients with sporadic mutations, which makes genetic diagnosis more difficult.17 The incidence of NF2 is approximately 1:25,000 to 1:40,000.17–19 Diagnosis can be challenging, with many children receiving a delayed diagnosis.18 Patients present with a variety of neural tumors, most commonly vestibular schwannomas and meningiomas, as well as cutaneous lesions and ophthalmic manifestations including cataract, retinal abnormalities (including retinal hamartomas), strabismus, and amblyopia.17,18
ONSMs are much more common in patients with NF2 than the general population. One study of patients with NF2 features described an incidence of 6.8% of ONSMs in patients with NF2.20 The authors make clear that this incidence is subjected to selection bias. Evans reports the incidence of ONSM in NF2 patients to be between 4.1% and 4.8%.19 Children who have meningiomas (not just ONSMs) are diagnosed with NF2 in 28% of cases.16,18,20 There may be a difference in the natural history of ONSMs in patients with and without NF2; however, the rarity of these disorders makes accurate assessment of this difficult (discussed below).
Clinical features
ONSMs classically present with a triad of visual loss, optic atrophy, and optociliary shunt vessels;1 however, this triad presenting in its entirety is rare.13 Vision loss is extremely common (97%) at presentation in the affected eye, but the degree is variable, with 45% of patients having acuity of 20/40 or better, and 24% having acuity of counting fingers or worse.3 Wright described a presenting acuity of “no perception of light” (NLP) in 24% of patients.21 Visual field defects are extremely common (83%) but again variable, most commonly manifesting as peripheral constriction (35%), central, centrocecal, and paracentral scotomas (together 29%), an enlarged blind spot (13%), and altitudinal defects (16%).3 Proptosis (2–5 mm) is common (59%), as is strabismus (47%), usually in attempted upgaze.3 An orbital compartment syndrome has been reported to develop as a result of an ONSM.22 The true incidence of pain and headache is difficult to quantify. The optic disc is almost always abnormal at presentation (98%) but may present as disc swelling and optic atrophy as well as the presence of optociliary shunt vessels, which are themselves less common (30%).
Optociliary shunt vessels are dilated normal anastomoses between the retinal venous system at the optic disc, and the choroidal venous circulation. It is hypothesized that they occur due to compression of the central retinal vein by tumor as it passes through the optic nerve.1 Direct evidence of altered central retinal venous flow has been obtained with Doppler imaging.23 Further supporting this hypothesis, shrinkage of optociliary shunt vessels has been observed when a tumor has been surgically removed24 and after radiotherapy treatment.25 Such vessels can be imaged in detail using indocyanine-green angiography.26 The shrinkage of optociliary shunt vessels suggests a reduction in central retinal venous pressure, which may be part of the mechanism behind vision improvement even when no macroscopic change in tumor volume is apparent.27
ONSMs can also coexist with other orbital disease. A case has been reported of simultaneous ONSM and an optic nerve glioma in the same optic nerve in a patient with neurofibromatosis type 1.28 A case of concomitant thyroid orbitopathy and bilateral ONSM has been reported.29
Diagnosis
The diagnosis of ONSM can be challenging due to their slow-growing nature and insidious vision loss. Optic nerve appearances can be variable, ranging from atrophy to normal to swollen in appearance, and the patients can be easily misdiagnosed.30 The clinical presentation of ONSMs can also be variable – in one case the presence of bilateral ONSMs was a surprise to clinicians treating a patient for presumed bilateral optic neuritis who had pain on eye movement.31 Careful follow-up and a high index of suspicion for retrobulbar pathology are, therefore, required to ensure that neuroimaging is appropriately ordered to enable the diagnosis to be made.
Imaging
Magnetic resonance imaging (MRI)
Diagnosis of ONSMs is best made with orbital and cranial imaging studies. The standard investigation for investigation and diagnosis of ONSM is MRI. ONSMs are best demonstrated on T1-weighted, gadolinium-enhanced, fat-suppressed sequences.32 Some examples are included in Figures 1–3. Certain subtypes of ONSMs, such as those that arise within the optic canal, are more challenging to diagnose, and for these, high-resolution MRI is essential.33 The superior soft tissue contrast of MRI, compared to computed tomography (CT), enables more effective differentiation of meningiomas from other optic nerve enlargements such as optic nerve gliomas, inflammatory conditions such as sarcoidosis, or other orbital lesions that do not arise from the optic nerve. Such differentiation is not perfect, and lesions can prove on biopsy to be of different histologic composition to that expected from imaging.34
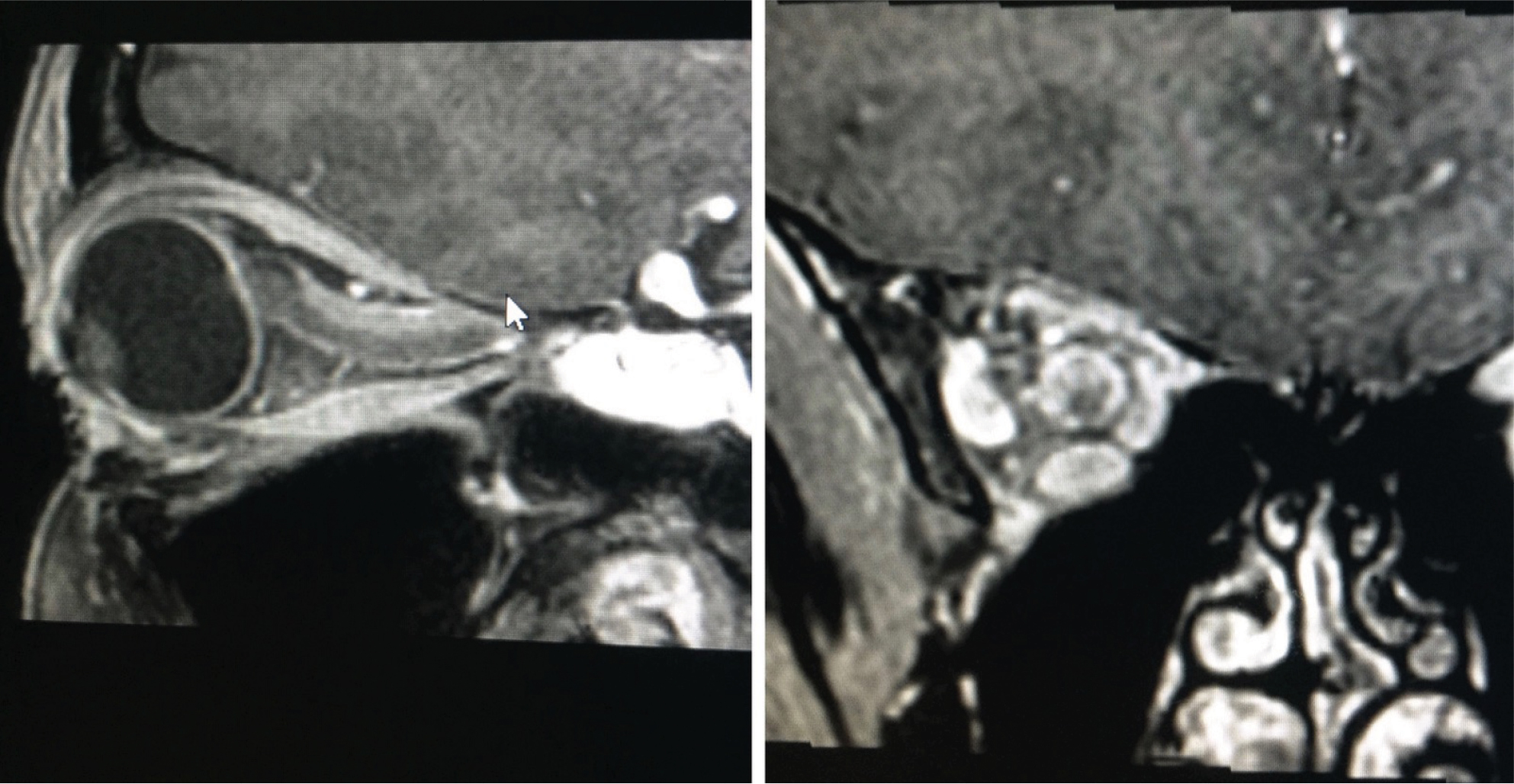 | Figure 1 A typical appearance of a tubular optic nerve sheath meningioma on magnetic resonance imaging (gadolinium contrast-enhanced T1-weighted sequence with fat suppression). The tumor surrounds the nerve and shows the typical “tram track” appearance on the sagittal image with the hypointense optic nerve tissue lying between the enhancing tumor. |
 | Figure 2 A large exophytic globular optic nerve sheath meningioma demonstrated on gadolinium contrast-enhanced T1-weighted fat suppressed magnetic resonance imaging. |
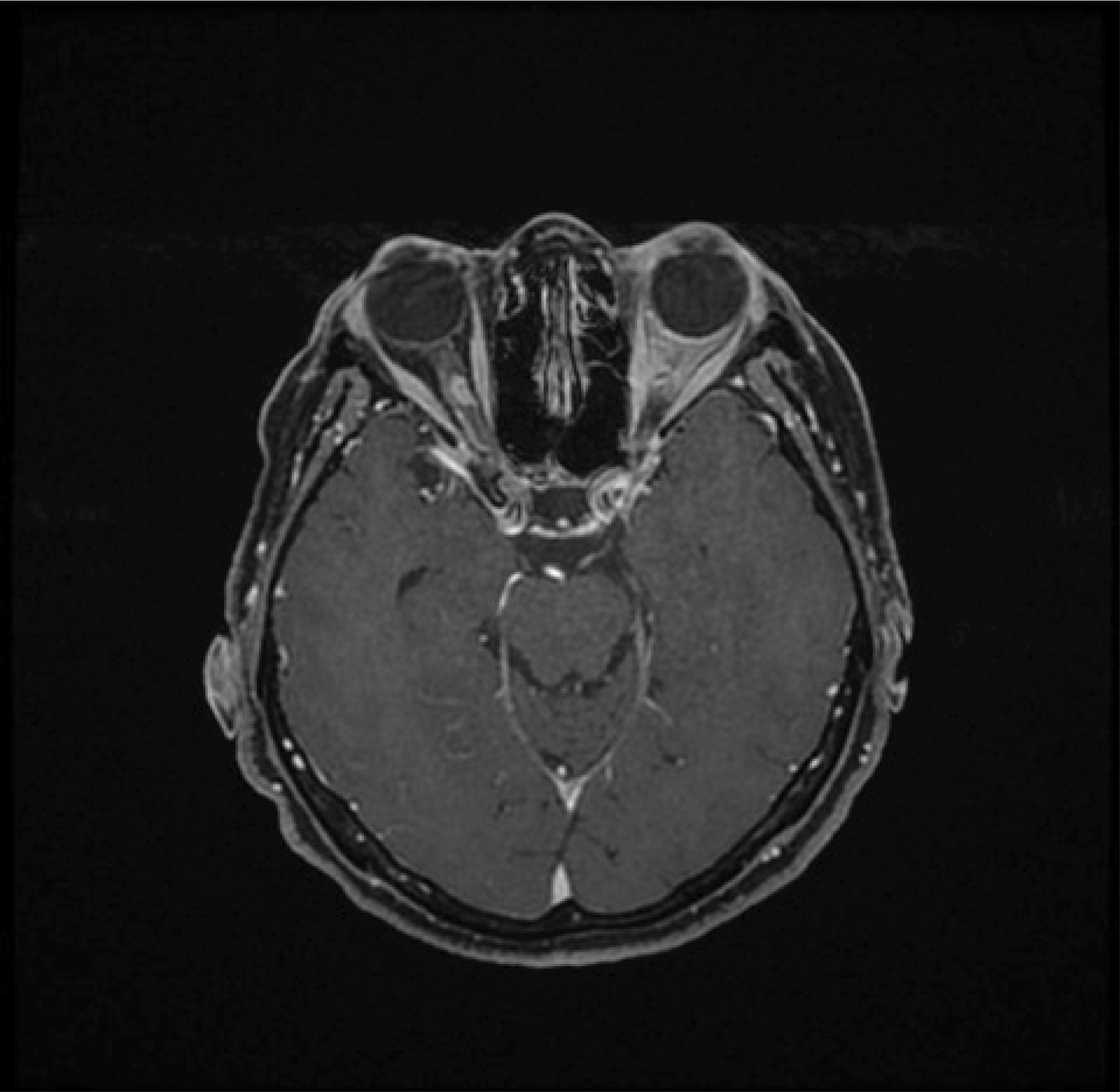 | Figure 3 Gadolinium contrast-enhanced T1-weighted magnetic resonance imaging with fat suppression demonstrating a small right optic nerve sheath meningioma postradiotherapy that has remained clinically and radiologically stable. Incomplete fat suppression artifact is present in the left orbit. |
Typical appearances of ONSMs on imaging32 are tubular expansion of the meninges surrounding the optic nerve (most common, 62%), globular (23%), fusiform (11%), and focal enlargement of the optic nerve (4%). “Tram tracking”, with the meningioma hyperdense (or hyperintense) on either side, relative to the optic nerve in the center is a classic sign and is demonstrated in 24% of tumors. Most tumors have smooth margins (80%). Some tumors, however, demonstrate alternative growth patterns to these.35 The presence of perioptic cysts associated with ONSMs is also well demonstrated on MRI.36
MRI is particularly superior to other modalities in the assessment of lesions near the orbital apex, as the soft tissue effacement, as a marker of the degree of compressive optic neuropathy, is well demonstrated. MRI is also superior to other modalities at demonstrating soft tissue involvement in the remainder of the orbit and intracranially.32
CT
Contrast-enhanced CT scans can also demonstrate the classic tram track sign,30,37 in contrast to an intrinsic optic nerve tumor such as an optic nerve glioma, which will demonstrate relatively uniform enhancement of the optic nerve itself. The calcification of ONSMs can give a tram track appearance on CT in the absence of the administration of intravenous contrast. While soft tissue contrast of CT scanning is inferior to MRI, CT remains superior for the assessment of calcification and of bony anatomy. Because bony anatomy is well demonstrated, CT is better to assess overall lesion configuration and relations.32
Ultrasound
Ultrasound can be used to demonstrate the tumor if it is located anteriorly,38 and blood flow within it can also be demonstrated with Doppler scanning.23 This imaging modality is noninvasive and does not require the prolonged immobilization of an MRI scan, and thus may be more appropriate for monitoring known lesions in children. Because posterior orbital tumors and intracranial ONSMs are not visible with ultrasound, this modality is not appropriate for primary screening and diagnosis.
Multifocal visual-evoked potential
A newer technique to monitor objective optic nerve function in patients is multifocal visual-evoked potential (mfVEP). In patients with known ONSMs, mfVEP can be used to monitor for functional compromise and progression of visual decline without the need for MRI.39 This has particular applicability in a pediatric population, where MRI scanning can require general anesthetic,40 with its associated risks.41 Although this modality will not replace MRI, evidence of progressive optic neuropathy, even the absence of visible MRI changes, could result in earlier treatment for patients who will eventually require treatment.
Biopsy
Biopsy of an optic nerve neoplasm is indicated when there is uncertainty regarding the diagnosis and careful observation is not clinically reasonable. Reasons for this may include a malignant or aggressive disease course, progressive vision loss, or a clinically and radiologically atypical lesion. Biopsy may also be useful if there are medicolegal concerns.42 It is worth considering that although the most common optic nerve tumors are gliomas and ONSMs, other neoplastic and inflammatory conditions can have a similar appearance on imaging, yet the management of these conditions is often entirely different from gliomas or ONSMs. Several techniques have been described for obtaining incisional biopsy specimens, including medial transconjunctival approach42 conceptually similar to an optic nerve sheath fenestration, and fine-needle aspiration biopsy under CT guidance.43 Biopsy carries similar risks to surgery (discussed below under “treatment options, surgery”; page 7) with a high risk of vision loss, and given modern neuroimaging is typically diagnostic, biopsy is no longer commonly indicated.
Natural history
The natural history of ONSM, concisely reviewed by Shapey,13 is of slow, progressive visual loss in the affected eye. The clinical course and effect on vision are variable. Some patients demonstrate long-term clinical stability despite tumor growth, and some patients have rapid vision loss even without measurable increase in tumor size. Good acuity at presentation is an indicator that the clinical course will be more benign; however, 85% of patients will lose vision over time without treatment.3 As a benign neoplasm, there is no significant systemic morbidity and no mortality associated with ONSMs.3,13 However, some ONSMs can exhibit aggressive behavior44 and cause profound orbital disfigurement. This is particularly true of tumors diagnosed in pediatric patients. Spontaneous improvement in vision is uncommon (18%) in one study,45 but possible.
A direct comparison of the natural history of ONSMs in patients with and without NF2 has to our knowledge not been made. Observations can be made about the difference in clinical features between the eight patients with NF2 reported by Bosch20 and Dutton’s review of 380 patients both with and without NF2. Bosch reported bilateral ONSMs in two (25%) of the reported NF2 patients, vs the Dutton’s reported rate of 5% for the population (accepting selection bias). Presenting vision in NF2 patients was NLP in three of ten eyes (30%), and in five of ten eyes (50%) visual acuity was “count fingers” (CF) or poorer. This is worse than Dutton’s population rate of 24% for CF or worse. Bosch reported in NF2 patients seven eyes with impaired ductions, vs Dutton’s population rate of 47%. Proptosis was only mentioned specifically for one patient in Bosch’s review, which is below Dutton’s reported population rate of 59%; however, data were not specifically tabulated for proptosis and thus may be incomplete. It should be emphasized that the low number of patients in this comparison makes drawing wider conclusions impossible. Meningiomas in other locations have been shown to be more aggressive in patients with NF2 compared to sporadic meningiomas.17
Treatment options
Observation
Observation of ONSMs historically has resulted in poor patient outcomes. Dutton3 reported that 86% of patients demonstrated decline in vision, with the remaining 14% demonstrating stable vision. No patients in this review demonstrated spontaneous improvement. Turbin46 reported on 13 patients in whom the most significant visual decline was in the observation group. Interestingly, patients with good vision at presentation seem to have better long-term stability32 than those with poor vision, perhaps reflecting that small amounts of tumor growth can have severe effects if the nerve is already compromised, whereas a healthy nerve can tolerate a similar insult without vision loss.
Despite clear evidence ONSMs overwhelmingly lead to progressive vision loss, certain cases can demonstrate remarkable stability. One patient with bilateral ONSMs (but not NF2) was observed for 27 years, from age 48 to 75, without progression.47 As previously mentioned, spontaneous improvement is possible but uncommon, with one study of 42 patients observed showing a spontaneous improvement rate of 7%.45 It is this variability in presentation that introduces the difficulty with when and how to commence treatment for these lesions, if at all.
Observation can, therefore, be advocated when the vision in the affected eye is normal. Once demonstrated visual decline is observed, vision loss becomes expected with observation and the case for treatment becomes much stronger.
Radiotherapy
The difficulty with management of ONSMs that confronted clinicians approximately 20 years ago has been elegantly summarized previously:48–50 observation usually leads to visual deterioration, medical therapy is generally inadequate, and surgical therapy usually leads to vision loss, which, in a tumor that rarely causes local disfigurement, rarely causes bilateral vision loss, and never causes mortality, is precisely the outcome that treatment is aiming to prevent.
From a historical perspective, Turbin’s review of long-term outcomes of patients with ONSM in 200246 proved influential. Visual outcomes for 64 patients were reported comparing surgery, observation, radiotherapy, and combination surgery and radiotherapy. The visual outcomes in the radiotherapy-only group were superior to all others, with this group only showing no significant decline in visual acuity from diagnosis to last follow-up. Furthermore, the radiation group demonstrated a favorable complication rate of 33.3% (including radiation retinopathy, vascular occlusion, persistent iritis, or temporal lobe atrophy), compared to the complication rate of surgery of 66.7%. Similar results have been demonstrated in other series.32,51,52 Thus, radiotherapy, with superior outcomes and a favorable side effect profile, has emerged as a better treatment option for ONSMs.
Various modalities of radiotherapy have been used to treat ONSMs.53 Fractionated external beam radiotherapy delivers radiation to the target area over a number of sessions, whereas radiosurgery delivers the radiation in a single session. Three-dimensional conformal radiotherapy (3D-CRT) uses beam-forming technology and specialized software to accurately model the target tissue, and then administer radiation conforming to the target tissue volume, minimizing the amount of radiation delivered to nontarget tissues. Intensity-modulated radiotherapy (IMRT) is a specialized kind of 3D-CRT that further modifies the dose distribution within the target volume. Stereotactic methods use fixed markers, usually invasively fixed to the patient, for registration of treatment machines with images, providing highly accurate delivery of radiation where critical structures lie close to target tissues. Image-guided radiation therapy reacquires images at the time of therapy to ensure precise alignment of target and treated tissue volumes. This is mainly useful when there may be interval change in the target tissue between image acquisition and treatment delivery. This is useful with rapidly changing tumor volumes, or indeed with mobile organs such as the eye.
A summary of recent studies is provided in Table 1 for 3D-CRT,54–61 stereotactic 3D-CRT,32,51,55,58,62–76 IMRT,58,77,78 and stereotactic radiosurgery.79–81 There appears to be no major differences between the type of radiotherapy used to treat ONSM. Stability or improvement of visual function is obtained in more than 80% of patients.10,12 A total radiation dose of less than 54 Gy over 30 fractions is typically used, as this is the ceiling for optic nerve tolerance.66,82 There is a concern that the larger dose fractions used in radiosurgery, compared to fractionated radiotherapy, may exceed the dose limits of surrounding tissues leading to more visual complications. As such, radiosurgery is generally used in patients with very poor vision.
 | Table 1 Summary of radiotherapy treatment for ONSM Note: Where results for V/A of visual fields were reported for the cohort including but not exclusively limited to ONSMs, a “%” result was given. Abbreviations: CRAO, central retinal artery occlusion; 3D-CRT, three-dimensional conformal radiotherapy; 2D-CRT, two-dimensional conformal radiotherapy; conj, conjunctivitis; FSRT, fractionated stereotactic radiotherapy; ICP, intracranial pressure; IMRT, intensity modulated radiotherapy; ONSM, optic nerve sheath meningioma; SRS, stereotactic radiosurgery; V/A, visual acuity; VF, visual field. |
Toxicity
Choice of the optimal radiation dose is currently based on maximum safe levels for each structure involved (optic nerve, retina). A significant predictor of toxicity is the presence of other risk factors, such as age, smoking, diabetes, hypertension, and hypercholesterolemia.83 Overall, radiation-associated complications following radiotherapy for ONSMs are mostly mild and self-limited,66 and those that are not are uncommon.46 Acute toxicities include local effects such as erythema, alopecia, or orbital pain, inflammation of the anterior segment of the eye (blepharitis, conjunctivitis, keratitis, or iritis), orbital edema, and systemic effects including nausea and vomiting, headache, and fatigue; their incidence is summarized in Table 2. Of note, one case of obstructive hydrocephalus was reported, presumably due to tumor swelling.66 Chronic toxicities include dry eye from lacrimal gland irradiation, cataract formation, pituitary dysfunction, radiation retinopathy, and radiation optic neuropathy. These are rare at currently accepted dose ranges.
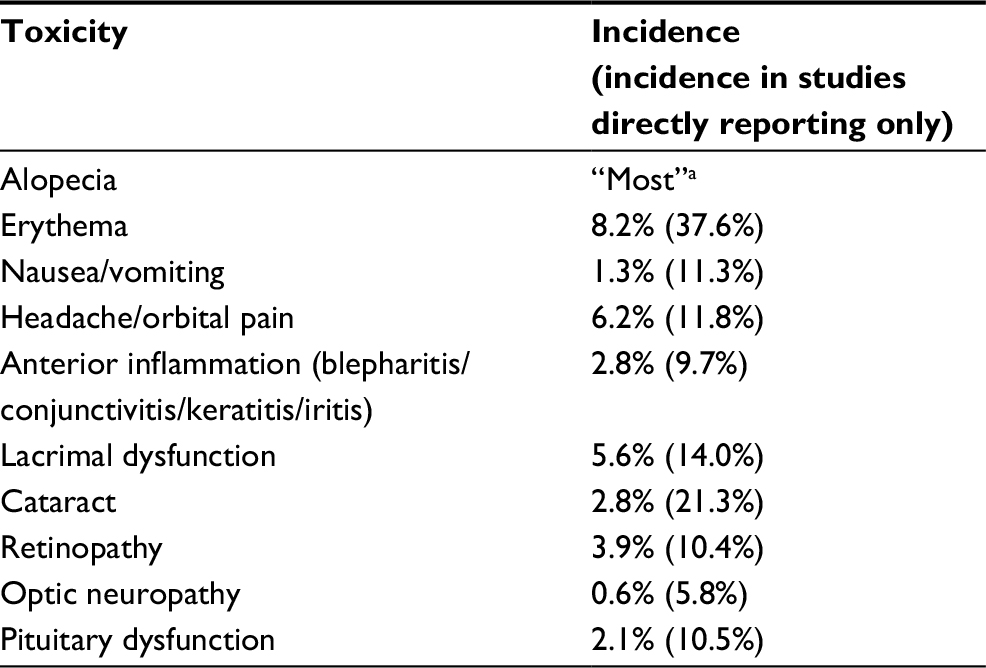 | Table 2 Incidence of radiotherapy side effects Notes: Total incidence of reported side effects based on all available studies and patients (n=466). It should be noted that these results are subjected to significant reporting bias, as where studies have not reported a specific side effect, it is unclear whether this side effect was absent or merely not commented upon (particularly for common or minor toxicity). The (X%) results indicate incidence only in those studies where the side effect was specifically commented upon. It is likely that the true incidence lies between these two values. aSeveral larger studies reported alopecia in most or all patients, without giving a numerical value. |
Abouaf et al observed that patients with decreased visual acuity at follow-up had higher mean eye dose levels (64 Gy vs 57.6 Gy).59 As expected, complication rates increase with higher delivered doses.83
Radiation retinopathy is more common when tumors are more anterior, and when patients have other predisposing microvascular risk factors as mentioned above. An eye dose of >50 Gy predisposes to retinal injury.84 If identified, radiation retinopathy can be treated with intravitreal antivascular endothelial growth factor antibodies.74,85,86
Radiation optic neuropathy is a late toxicity, developing between 3 months and 8 years after radiation therapy. Radiation doses of >50 Gy to the anterior visual pathway, or fractions >10 Gy, are generally required for radiation optic neuropathy to develop.82 Treatment is with corticosteroids, hyperbaric oxygen, and possibly anticoagulation.82 Unfortunately, the success of these measures is generally limited and highly time-critical.82 Radiation optic neuropathy has also been reported 27 months after proton beam therapy,87 which was successfully stabilized with intravenous methylprednisone.
Surgery
Surgery was the historical treatment of choice for ONSMs.3 Surgical treatment is typically associated with loss of vision in the affected eye, as the tumor shares a blood supply from the pial vessels with the optic nerve. Therefore, complete removal of the tumor also strips the blood supply of the nerve, leading to vision loss.3 The alternative, incision of the optic nerve sheath to provide decompression, was associated with seeding of the tumor into the orbital tissues.21,88 Sacrifice of the tumor and nerve en bloc has also been reported, usually in eyes with no vision,21,88 but also unfortunately in eyes with useful remaining vision.89
Dutton’s review3 showed progression to no perception of light with surgical treatment in 78% of cases, with less severe visual decline in a further 16%. Wright88 reported that all 27 patients with surgery performed via lateral orbitotomy lost vision within 18 months of surgery. Wright later reviewed a series of 50 patients and noted that younger patients harbored more aggressive disease with more frequent intracranial involvement, and recommended sacrifice of the optic nerve in these cases.21 With reference to 57 primary and secondary ONSMs, Cristante investigated visual outcomes following surgery and concluded that surgical treatment of primary ONSMs was unlikely to be of benefit.90 Delfini91 reported that eleven of 13 patients with ONSM managed surgically suffered loss of vision; however, they were still recommended surgery when vision loss became apparent, as, in their opinion at the time, radiotherapy was ineffective. Saaed reported poor outcomes in patients with both partial resections and decompressions.32 Roser reported that 50% of patients with rapid visual decline, treated surgically, had improved or stabilized vision, advocating surgical intervention in cases of intracranial extension, disfiguring orbitopathy, or rapid progression of visual decline.92 Surgical intervention was also recommended in patients with no useful vision to enable en bloc resection of the tumor and optic nerve, with complete histologic clearance. Indeed, transection of the optic nerve and meninges from globe to pre-chiasm in unilaterally blind patients with ONSM has been proposed as a strategy for preventing contralateral spread.93 Other summaries have been more conservative in their approach given the benefit of surgery is only clear when there is evidence of posterior tumor growth, which may threaten vision in the contralateral eye.7,94,95
In light of these poor results, with the demonstration of the efficacy of radiotherapy and tumor control, and with its more favorable visual outcomes, surgical therapy is no longer recommended for patients with intraorbital ONSM who still have vision. Surgical therapy can be considered in patients who require tissue diagnosis, with posterior extension of the tumor, in patients with complete vision loss in whom en bloc resection is possible and desired, or in patients who have significant orbital disfigurement. These conditions preclude the majority of patients with ONSM. Surgical therapy is generally of limited use and careful consideration should be given to alternatives before proceeding.
Local chemotherapy
In some limited cases, surgery combined with local chemotherapeutic agents can be effective. A cystic ONSM, present in a patient with good vision in the affected eye, refractory to initial cyst excision and subsequent radiotherapy, was successfully treated with further surgical drainage of the cyst and application of mitomycin C 0.04% for 5 minutes directly to the cyst opening. No complications were observed either at the time of surgery or on follow-up over 19 months, and the patient’s vision stabilized in the affected eye.96
Systemic chemotherapy
Prior to the routine application of radiotherapy for ONSMs, there were no intermediate treatments between observation and surgery. Several reports, therefore, were made of attempts to treat these tumors with systemic medical therapy. Nevertheless, despite occasional reported successes, systemic medical therapy has not been demonstrated to adequately treat ONSMs.50
Hydroxyurea
A single case report of treatment with hydroxyurea has been published with encouraging results (visual acuity improvement from 20/400 to 20/25),97 with the authors commenting that hydroxyurea has been used for treatment of untreatable meningiomas in other locations.
Hormonal therapy
Meningiomas express a wide range of hormone and growth factor receptors. Of particular note, progesterone receptors are frequently expressed by these tumors. One study reported 64% of tumors examined to be positive for progesterone receptors.98 This may explain the higher incidence of these tumors in females, and their progression during pregnancy.99 On this basis, therapy with hormonal antagonists has been trialled in cases of unresectable meningioma. One study of 14 patients used mifepristone, an antiprogesterone medication, with five patients showing tumor shrinkage and three further patients showing visual improvement.100 Another case has been reported of unresectable bilateral ONSMs (not associated with NF2) that was successfully stabilized by treatment with a progesterone antagonist medication (gestrinone) after biopsy confirmation that this particular tumor expressed high levels of progesterone receptors.101 Such therapy is usually inappropriate for adolescent patients14 given the possible effects on growth, and in patients contemplating pregnancy or who are currently pregnant.
Initiation of treatment
The critical issue in the management of ONSM is when to initiate treatment. On the basis of the evidence to date, we concur that once vision is demonstrated to decline, treatment should be commenced.
Patients with poor initial vision may also be appropriate for initial treatment, without waiting for demonstration of progression. Several observations support this hypothesis. Multiple studies support that poorer vision at the commencement of treatment results in a poorer outcome.71,77,90 Other sensory tumors, for example acoustic neuromas, also have better outcomes when treated earlier.62 The duration of symptoms has been negatively correlated with final visual acuity in a surgical treatment cohort of one study,92 and in another radiotherapy study.59 Patients who demonstrate stable or improved vision seem to be those who are treated earlier.59,60 Unfortunately, without a direct comparison of early vs delayed treatment it is difficult to draw accurate conclusions about causality from these observations.
Treatment of blind eyes
It remains controversial whether or not to treat eyes that have extremely poor vision, or no vision remaining. There are reports of patients with light perception vision recovering to CF vision following radiotherapy.46,73 In contrast, once patients have reached no light perception vision, visual recovery is very unlikely. Multiple reports of such patients having radiotherapy have shown no improvement.57,69,73,74,79 In such patients, en bloc surgical resection has been reported92 and can be appropriate to prevent growth and recurrence, particularly if the tumor demonstrates posterior spread or if there is significant orbital disfigurement.
Responses to therapy
Location of ONSM, response to therapy, and outcomes
Historically ONSMs with intracranial extension were favored for surgical management to decrease the risk of extension to the optic chiasm and the risk of bilateral visual loss.3 With the increased use of radiotherapy, this approach is now in need of reconsideration. The location of the meningioma has not been shown to alter its response to radiotherapy,77 but meningiomas spread across multiple regions have lower rates of local control after radiotherapy.102 Skull base meningiomas respond well to radiotherapy.103 Opinion is divided on whether primary or secondary ONSMs show different visual acuity improvements with radiotherapy, with some71 reporting improved outcomes for secondary ONSMs, and others66 reporting no difference.
Optical coherence tomography (OCT) as a prognostic indicator
A newer predictor of response to therapy is the retinal nerve fiber layer thickness, as measured by OCT. It has been demonstrated that low nerve fiber layer thickness on OCT predicts poor outcomes, along with duration of symptoms.104 Poor nerve fiber layer thickness is a quantifiable marker of optic atrophy, and clearly once atrophy has occurred from compression from an ONSM the chance of recovery is low. These findings reflect similar findings in pituitary lesions causing optic chiasm compression.
Disc swelling
Another novel predictor of outcomes is the presence of optic disc edema at diagnosis. With increasing atrophy of the optic nerve head, disc swelling becomes less pronounced, while vision typically deteriorates. The presence of optic disc edema could, therefore, be expected to indicate a threatened, rather than atrophied, nerve. The presence of disc edema at the time of treatment has been strongly associated with a significantly improved visual response to primary radiotherapy.55
Visual improvement despite stable radiographic appearance
Another peculiarity regarding treated ONSMs is demonstrated in patients who have improved visual function despite stable tumor size on MRI scanning. In most cases, tumors demonstrate stabilization of size, rather than macroscopic shrinkage (Table 1). With the purported mechanism of optic neuropathy being compression, due to tumor volume, or ischemic, due to interposition of the tumor between the optic nerve and its pial blood supply, it would be reasonable to expect that patients with visual improvement should show reduction in tumor volume. Nevertheless, patients with tumors that demonstrate no change in size can show improvements in vision.63,73 Tumors in these cases demonstrated reduced metabolic activity when measured by 111In-octreotide scintigraphy. This may suggest that active tumor, consuming available blood supply, contributes to optic neuropathy, rather than simply direct compression effects. 111In-octreotide scintigraphy can therefore be useful in determining if failure to improve vision after therapy is due to failure of the tumor to respond, or due to treatment-related radiation-induced optic neuropathy.73
Discordance between improvement in visual acuity and visual fields
It is also worth mentioning that visual acuity may not be the only factor that can improve with therapy. Visual fields also show change with treatment. More marked visual field improvement than visual acuity improvement with radiotherapy treatment has been demonstrated,67 and other cases have shown decreased central acuity but with improved visual fields.59 The improvement in visual fields with treatment may be due to the retinotopic organization of fibers within the optic nerve, with macular fibers in the nerve located centrally within the posterior nerve and less susceptible to extrinsic compression.105 Irrespective of visual acuity outcomes, peripheral field improvement remains important from a functional perspective.
Conclusion
ONSMs continue to present a management challenge to clinicians. Until a therapy is developed that directly targets tumor cells and leaves normal surrounding structures undamaged, treatment-induced morbidity remains a real constraint on the feasibility of delivering early therapy to prevent vision loss. Given the variable natural history of these lesions, there will always be uncertainty around when and how to initiate treatment. Nevertheless, with earlier detection with improved imaging technology, more accurate radiotherapy delivery, and improved case selection for surgical management, it is hoped that outcomes will continue to improve for these patients.
Acknowledgement
The authors would like to acknowledge Dr Yael Barnett and Dr Geoffrey Parker for providing the images used in this review.
Disclosure
The authors report no conflicts of interest in this work.
References
Frisèn L, Royt WF, Tengroth BM. Optociliary veins, disc pallor and visual loss. A triad of signs indicating spheno-orbital meningioma. Acta Ophthalmol. 1973;51(2):241–249. | ||
Cantore WA. Neural orbital tumors. Curr Opin Ophthalmol. 2000;11(5):367–371. | ||
Dutton JJ. Optic nerve sheath meningiomas. Surv Ophthalmol. 1992;37(3):167–183. | ||
Turbin RE, Pokorny K. Diagnosis and treatment of orbital optic nerve sheath meningioma. Cancer Control. 2004;11(5):334–341. | ||
Carrasco JR, Penne RB. Optic nerve sheath meningiomas and advanced treatment options. Curr Opin Ophthalmol. 2004;15(5):406–410. | ||
Melian E, Jay W. Primary radiotherapy for optic nerve sheath meningioma. Semin Ophthalmol. 2004;19(3–4):130–140. | ||
Kim JW, Rizzo JF, Lessell S. Controversies in the management of optic nerve sheath meningiomas. Int Ophthalmol Clin. 2005;45(4):15–23. | ||
Berman D, Miller NR. New concepts in the management of optic nerve sheath meningiomas. Ann Acad Med Singapore. 2006;35(3):168–174. | ||
Eddleman CS, Liu JK. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus. 2007;23(5):E4. | ||
Jeremic B, Pitz S. Primary optic nerve sheath meningioma: stereotactic fractionated radiation therapy as an emerging treatment of choice. Cancer. 2007;110(4):714–722. | ||
Gondi V, Tome WA, Mehta MP. Fractionated radiotherapy for intracranial meningiomas. J Neurooncol. 2010;99(3):349–356. | ||
Bloch O, Sun M, Kaur G, Barani IJ, Parsa AT. Fractionated radiotherapy for optic nerve sheath meningiomas. J Clin Neurosci. 2012;19(9):1210–1215. | ||
Shapey J, Sabin HI, Danesh-Meyer HV, Kaye AH. Diagnosis and management of optic nerve sheath meningiomas. J Clin Neurosci. 2013;20(8):1045–1056. | ||
Grob SR, Jakobiec FA, Rashid A, Macintosh P, Kelly H, Fay A. Pediatric optic nerve meningioma: diagnostic and therapeutic challenges. Ophthalmic Plast Reconstr Surg. 2016;32(6):e160–e164. | ||
Levin LA, Jakobiec FA. Optic nerve tumors of childhood: a decision-analytical approach to their diagnosis. Int Ophthalmol Clin. 1992;32(1):223–240. | ||
Harold Lee HB, Garrity JA, Cameron JD, Strianese D, Bonavolontà G, Patrinely JR. Primary optic nerve sheath meningioma in children. Surv Ophthalmol. 2008;53(6):543–558. | ||
Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet. 2009;373(9679):1974–1986. | ||
Ardern-Holmes S, Fisher G, North K. Neurofibromatosis Type 2. J Child Neurol. 2017;32(1):9–22. | ||
Evans DG. Neurofibromatosis type 2. Handb Clin Neurol. 2015;132:87–96. | ||
Bosch MM, Wichmann WW, Boltshauser E, Landau K. Optic nerve sheath meningiomas in patients with neurofibromatosis type 2. Arch Ophthal. 2006;124(3):379–385. | ||
Wright JE, Mcnab AA, Mcdonald WI. Primary optic nerve sheath meningioma. Br J Ophthalmol. 1989;73(12):960–966. | ||
Jaggi GP, Mironov A, Huber AR, Killer HE. Optic nerve compartment syndrome in a patient with optic nerve sheath meningioma. Eur J Ophthalmol. 2007;17(3):454–458. | ||
Jacquemin C, Bosley T, Mullaney P. Orbital color Doppler imaging of optic nerve tumors. Int Ophthalmol. 1999;23(1):11–15. | ||
Nichols C, Pincus DW, Bhatti MT. Optociliary shunt vessels. Neurology. 2005;64(8):1443. | ||
Mashayekhi A, Shields JA, Shields CL. Involution of retinochoroidal shunt vessel after radiotherapy for optic nerve sheath meningioma. Eur J Ophthalmol. 2004;14(1):61–64. | ||
Muci-Mendoza R, Arevalo JF, Ramella M, et al. Optociliary veins in optic nerve sheath meningioma. Indocyanine green videoangiography findings. Ophthalmology. 1999;106(2):311–318. | ||
de Alba Campomanes AG, Larson DA, Horton JC. Immediate shrinkage of optociliary shunt vessels after fractionated external beam radiation for meningioma of the optic nerve sheath. AJNR Am J Neuroradiol. 2008;29(7):1360–1362. | ||
Büyükkapu-Bay S, Akça A, Karadoğan M, Çorapçioğlu F, Anik Y. Concomitant meningioma and glioma within the same optic nerve in neurofibromatosis type 1. J Child Neurol. 2014;29(3):385–388. | ||
Garg A, Patel P, Lignelli A, Baron E, Kazim M. Coincidental optic nerve meningioma and thyroid eye disease. Ophthal Plast Reconstr Surg. 2015;31(4):e94–e95. | ||
Baehring JM. Tram track sign. J Neurooncol. 2007;85(1):75. | ||
Sawaya RA, Sidani C, Farah N, Hourani-Risk R. Presumed bilateral optic nerve sheath meningiomas presenting as optic neuritis. J Neuroophthalmol. 2008;28(1):55–57. | ||
Saeed P, Rootman J, Nugent RA, White VA, Mackenzie IR, Koornneef L. Optic nerve sheath meningiomas. Ophthalmology. 2003;110(10):2019–2030. | ||
Jackson A, Patankar T, Laitt RD. Intracanalicular optic nerve meningioma: a serious diagnostic pitfall. AJNR Am J Neuroradiol. 2003;24(6):1167–1170. | ||
Savignac A, Lecler A. Optic nerve meningioma mimicking cavernous hemangioma. World Neurosurg. 2018;110:301–302. | ||
Samarawickrama C, Frydenberg E, Wells M, Steel T, Ghabrial R. An unusual radiological presentation of optic nerve sheath meningioma. Saudi J Ophthalmol. 2016;30(2):137–139. | ||
Koolen M, Lambrechts I. Optic nerve sheath meningioma with perioptic cyst. JBR-BTR. 2009;92(4):228. | ||
Kanamalla US. The optic nerve tram-track sign. Radiology. 2003; 227(3):718–719. | ||
Garcia JPS, Finger PT, Kurli M, Holliday RA. 3D ultrasound coronal C-scan imaging for optic nerve sheath meningioma. Br J Ophthalmol. 2005;89(2):244–245. | ||
Jayanetti V, Klistorner AI, Graham SL, et al. Monitoring of optic nerve function in neurofibromatosis 2 children with optic nerve sheath meningiomas using multifocal visual evoked potentials. J Clin Neurosci. 2018;50:262–267. | ||
Cahoon GD, Davison TE. Prediction of compliance with MRI procedures among children of ages 3 years to 12 years. Pediatr Radiol. 2014;44(10):1302–1309. | ||
Mir Ghassemi A, Neira V, Ufholz L-A, et al. A systematic review and meta-analysis of acute severe complications of pediatric anesthesia. Paediatr Anaesth. 2015;25(11):1093–1102. | ||
Gündüz K, Catak E, Erden E. Optic nerve biopsy via a medial transconjunctival orbitotomy approach in the diagnosis of optic nerve and sheath tumors. Orbit. 2010;29(4):190–193. | ||
Ban Y, Kusaba K, Miyao A, Akimoto K, Maki K, Yoshida T. Biopsy of orbital meningioma by computed tomography-guided fine-needle aspiration. Jpn J Ophthalmol. 2005;49(4):336–338. | ||
Amoli FA, Mehrabani PM, Tari AS. Aggressive orbital optic nerve meningioma with benign microscopic features: a case report. Orbit. 2007;26(4):271–274. | ||
Egan RA, Lessell S. A contribution to the natural history of optic nerve sheath meningiomas. Arch Ophthal. 2002;120(11):1505–1508. | ||
Turbin RE, Thompson CR, Kennerdell JS, Cockerham KP, Kupersmith MJ. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109(5):890–900. | ||
Kothari NA, Kulkarni KM, Lam BL. Untreated bilateral optic nerve sheath meningiomas observed for 27 years. J Neuroophthalmol. 2013;33(1):45–47. | ||
Miller NR. The evolving management of optic nerve sheath meningiomas. Br J Ophthalmol. 2002;86(11):1198. | ||
Miller NR. Radiation for optic nerve meningiomas: is this the answer? Ophthalmology. 2002;109(5):833–834. | ||
Miller NR. New concepts in the diagnosis and management of optic nerve sheath meningioma. J Neuroophthalmol. 2006;26(3):200–208. | ||
Adeberg S, Welzel T, Rieken S, Debus J, Combs SE. Prior surgical intervention and tumor size impact clinical outcome after precision radiotherapy for the treatment of optic nerve sheath meningiomas (ONSM). Radiat Oncol. 2011;6(1):117. | ||
Smee RI, Schneider M, Williams JR. Optic nerve sheath meningiomas — non-surgical treatment. Clin Oncol. 2009;21(1):8–13. | ||
Stieber VW. Radiation therapy for visual pathway tumors. J Neuroophthalmol. 2008;28(3):222–230. | ||
Moyer PD, Golnik KC, Breneman J. Treatment of optic nerve sheath meningioma with three-dimensional conformal radiation. Am J Ophthalmol. 2000;129(5):694–696. | ||
Saeed P, Blank L, Selva D, et al. Primary radiotherapy in progressive optic nerve sheath meningiomas: a long-term follow-up study. Br J Ophthalmol. 2010;94(5):564–568. | ||
Narayan S, Cornblath WT, Sandler HM, Elner V, Hayman JA. Preliminary visual outcomes after three-dimensional conformal radiation therapy for optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2003;56(2):537–543. | ||
Metellus P, Kapoor S, Kharkar S, et al. Fractionated conformal radiotherapy for management of optic nerve sheath meningiomas: long-term outcomes of tumor control and visual function at a single institution. Int J Radiat Oncol Biol Phys. 2011;80(1):185–192. | ||
Lesser RL, Knisely JPS, Wang SL, Yu JB, Kupersmith MJ. Long-term response to fractionated radiotherapy of presumed optic nerve sheath meningioma. Br J Ophthalmol. 2010;94(5):559–563. | ||
Abouaf L, Girard N, Lefort T, et al. Standard-fractionated radiotherapy for optic nerve sheath meningioma: visual outcome is predicted by mean eye dose. Int J Radiat Oncol Biol Phys. 2012;82(3):1268–1277. | ||
Adams G, Roos DE, Crompton JL. Radiotherapy for optic nerve sheath meningioma: a case for earlier intervention? Clin Oncol. 2013;25(6):356–361. | ||
Lee AG, Woo SY, Miller NR, Safran AB, Grant WH, Butler EB. Improvement in visual function in an eye with a presumed optic nerve sheath meningioma after treatment with three-dimensional conformal radiation therapy. J Neuroophthalmol. 1996;16(4):247–251. | ||
Richards JC, Roden D, Harper CS. Management of sight-threatening optic nerve sheath meningioma with fractionated stereotactic radiotherapy. Clin Experiment Ophthalmol. 2005;33(2):137–141. | ||
Pacelli R, Cella L, Conson M, et al. Fractionated stereotactic radiation therapy for orbital optic nerve sheath meningioma – a single institution experience and a short review of the literature. J Radiat Res. 2011;52(1):82–87. | ||
Arvold ND, Lessell S, Bussiere M, et al. Visual outcome and tumor control after conformal radiotherapy for patients with optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2009;75(4):1166–1172. | ||
Paridaens ADA, van Ruyven RLJ, Eijkenboom WMH, Mooy CM, van den Bosch WA. Stereotactic irradiation of biopsy proved optic nerve sheath meningioma. Br J Ophthalmol. 2003;87(2):246–247. | ||
Paulsen F, Doerr S, Wilhelm H, Becker G, Bamberg M, Claßen J. Fractionated stereotactic radiotherapy in patients with optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2012;82(2):773–778. | ||
Pitz S, Becker G, Schiefer U, et al. Stereotactic fractionated irradiation of optic nerve sheath meningioma: a new treatment alternative. Br J Ophthalmol. 2002;86(11):1265–1268. | ||
Liu JK, Forman S, Hershewe GL, Moorthy CR, Benzil DL. Optic nerve sheath meningiomas: visual improvement after stereotactic radiotherapy. Neurosurgery. 2002;50(5):950–957. | ||
Milker-Zabel S, Huber P, Schlegel W, Debus J, Zabel-du Bois A. Fractionated stereotactic radiation therapy in the management of primary optic nerve sheath meningiomas. J Neurooncol. 2009;94(3):419–424. | ||
Landert M, Baumert BG, Bosch MM, Lutolf UM, Landau K. The visual impact of fractionated stereotactic conformal radiotherapy on seven eyes with optic nerve sheath meningiomas. J Neuroophthalmol. 2005;25(2):86–91. | ||
Becker G, Jeremic B, Pitz S, et al. Stereotactic fractionated radiotherapy in patients with optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2002;54(5):1422–1429. | ||
Baumert BG, Villà S, Studer G, et al. Early improvements in vision after fractionated stereotactic radiotherapy for primary optic nerve sheath meningioma. Radiother Oncol. 2004;72(2):169–174. | ||
Andrews DW, Faroozan R, Yang BP, et al. Fractionated stereotactic radiotherapy for the treatment of optic nerve sheath meningiomas: preliminary observations of 33 optic nerves in 30 patients with historical comparison to observation with or without prior surgery. Neurosurgery. 2002;51(4):890–894. | ||
Soldà F, Wharram B, Gunapala R, Brada M. Fractionated stereotactic conformal radiotherapy for optic nerve sheath meningiomas. Clin Oncol. 2012;24(8):e106–e112. | ||
Fineman MS, Augsburger JJ. A new approach to an old problem. Surv Ophthalmol. 1999;43(6):519–524. | ||
Subramanian P, Bressler NM, Miller NR. Radiation retinopathy after fractionated stereotactic radiotherapy for optic nerve sheath meningioma. Ophthalmology. 2004;111(3):565–567. | ||
Maclean J, Fersht N, Bremner F, Stacey C, Sivabalasingham S, Short S. Meningioma causing visual impairment: outcomes and toxicity after intensity modulated radiation therapy. Int J Radiat Oncol Biol Phys. 2013;85(4):e179–e186. | ||
Grant W, Cain RB. Intensity modulated conformal therapy for intracranial lesions. Med Dosim. 1998;23(3):237–241. | ||
Marchetti M, Bianchi S, Milanesi I, et al. Multisession radiosurgery for optic nerve sheath meningiomas--an effective option: preliminary results of a single-center experience. Neurosurgery. 2011;69(5):1116–1122. | ||
Klink DF, Miller NR, Williams J. Preservation of residual vision 2 years after stereotactic radiosurgery for a presumed optic nerve sheath meningioma. J Neuroophthalmol. 1998;18(2):117–120. | ||
Romanelli P, Bianchi L, Muacevic A, Beltramo G. Staged image guided robotic radiosurgery for optic nerve sheath meningiomas. Comput Aided Surg. 2011;16(6):257–266. | ||
Danesh-Meyer HV. Radiation-induced optic neuropathy. J Clin Neurosci. 2008;15(2):95–100. | ||
Farzin M, Molls M, Kampfer S, et al. Optic toxicity in radiation treatment of meningioma: a retrospective study in 213 patients. J Neurooncol. 2016;127(3):597–606. | ||
Brown GC, Shields JA, Sanborn G, Augsburger JJ, Savino PJ, Schatz NJ. Radiation retinopathy. Ophthalmology. 1982;89(12):1494–1501. | ||
Finger PT. Radiation retinopathy is treatable with anti–vascular endothelial growth factor bevacizumab (Avastin). Int J Radiat Oncol Biol Phys. 2008;70(4):974–977. | ||
Gupta A, Muecke JS. Treatment of radiation maculopathy with intravitreal injection of bevacizumab (Avastin). Retina. 2008;28(7):964–968. | ||
Siddiqui JD, Loeffler JS, Murphy MA. Radiation optic neuropathy after proton beam therapy for optic nerve sheath meningioma. J Neuroophthalmol. 2013;33(2):165–168. | ||
Wright JE, Call NB, Liaricos S. Primary optic nerve meningioma. Br J Ophthalmol. 1980;64(8):553–558. | ||
Fayaz I, Gentili F, Mackenzie IR. Optic nerve sheath meningioma. J Neurol Neurosurg Psychiatr. 1999;67(3):408–409. | ||
Cristante L. Surgical treatment of meningiomas of the orbit and optic canal: a retrospective study with particular attention to the visual outcome. Acta Neurochir (Wien). 1994;126(1):27–32. | ||
Delfini R, Missori P, Tarantino R, Ciappetta P, Cantore G. Primary benign tumors of the orbital cavity: comparative data in a series of patients with optic nerve glioma, sheath meningioma, or neurinoma. Surg Neurol. 1996;45(2):147–154. | ||
Roser F, Nakamura M, Martini-Thomas R, Samii M, Tatagiba M. The role of surgery in meningiomas involving the optic nerve sheath. Clin Neurol Neurosurg. 2006;108(5):470–476. | ||
Zweckberger K, Unterberg AW, Schick U. Pre-chiasmatic transection of the optic nerve can save contralateral vision in patients with optic nerve sheath meningioms. Clin Neurol Neurosurg. 2013;115(12):2426–2431. | ||
Schick U, Dott U, Hassler W. Surgical management of meningiomas involving the optic nerve sheath. J Neurosurg. 2004;101(6):951–959. | ||
Schick U, Jung C, Hassler W. Primary optic nerve sheath meningiomas: a follow-up study. Cent Eur Neurosurg. 2010;71(03):126–133. | ||
Dhoot DS, Shults WT, Ng JD, Jd N. Successful use of mitomycin C to prevent recurrence of the cystic component of an optic nerve sheath meningioma. Ophthal Plast Reconstr Surg. 2008;24(3):235–236. | ||
Paus S, Klockgether T, Urbach H, Schlegel U. Meningioma of the optic nerve sheath: treatment with hydroxyurea. J Neurol Neurosurg Psychiatr. 2003;74(9):1348–1350. | ||
Black P, Carroll R, Zhang J. The molecular biology of hormone and growth factor receptors in meningiomas. Acta Neurochir Suppl. 1996;65:50–53. | ||
Mafee MF, Goodwin J, Dorodi S. Optic nerve sheath meningiomas. Role of MR imaging. Radiol Clin North Am. 1999;37(1):37–58. | ||
Grunberg SM, Weiss MH, Spitz IM, et al. Treatment of unresectable meningiomas with the antiprogesterone agent mifepristone. J Neurosurg. 1991;74(6):861–866. | ||
Cassidy LM, Moriarty PA, Griffin JF, Kennedy SM. Hormonal treatment of bilateral optic nerve meningioma. Eye. 1997;11 (Pt 4):566–568. | ||
Fokas E, Henzel M, Surber G, Hamm K, Engenhart-Cabillic R. Stereotactic radiation therapy for benign meningioma: long-term outcome in 318 patients. Int J Radiat Oncol Biol Phys. 2014;89(3):569–575. | ||
Combs SE, Adeberg S, Dittmar JO, et al. Skull base meningiomas: long-term results and patient self-reported outcome in 507 patients treated with fractionated stereotactic radiotherapy (FSRT) or intensity modulated radiotherapy (IMRT). Radiother Oncol. 2013;106(2):186–191. | ||
Loo JL, Tian J, Miller NR, Subramanian PS. Use of optical coherence tomography in predicting post-treatment visual outcome in anterior visual pathway meningiomas. Br J Ophthalmol. 2013;97(11):1455–1458. | ||
Snell RS, Lemp MA. Clinical Anatomy of the Eye. 2nd ed. New Jersey: Wiley-Blackwell; 1997. | ||
Subramanian PS, Bressler NM, Miller NR. Radiation retinopathy after fractionated stereotactic radiotherapy for optic nerve sheath meningioma. Ophthalmology. 2004;111(3):565–567. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.