Back to Journals » Open Access Journal of Clinical Trials » Volume 10
Open-label Phase II study of everolimus plus endocrine therapy in postmenopausal women with ER-positive and HER2-negative metastatic breast cancer (Chloe trial)
Authors Shimoi T ![]() , Shimomura A
, Shimomura A ![]() , Shien T
, Shien T ![]() , Uemura Y, Kato H, Kitada M, Toyama T, Aihara T, Mukai H
, Uemura Y, Kato H, Kitada M, Toyama T, Aihara T, Mukai H
Received 31 October 2017
Accepted for publication 15 December 2017
Published 16 February 2018 Volume 2018:10 Pages 13—18
DOI https://doi.org/10.2147/OAJCT.S155706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Tatsunori Shimoi,1 Akihiko Shimomura,1 Tadahiko Shien,2 Yukari Uemura,3 Hiroaki Kato,4 Masahiro Kitada,5 Tatsuya Toyama,6 Tomohiko Aihara,7 Hirofumi Mukai8
1Department of Breast and Medical Oncology, National Cancer Center Hospital, Tokyo, Japan; 2Department of Breast and Endocrine Surgery, Okayama University Hospital, Okayama, Japan; 3Division of Biostatistics, Clinical Research Support Center, Tokyo University Hospital, Tokyo, Japan; 4Department of Thoracic Surgery, Teine Keijinkai Hospital, Sapporo, Japan; 5Department of Surgery, Asahikawa Medical University, Asahikawa, Japan; 6Department of Oncology, Immunology and Surgery, Nagoya City University, Graduate School of Medical Science, Nagoya, Japan; 7Department of Breast Surgery, Keimeikai-Aihara Hospital, Osaka, Japan; 8Department of Breast and Medical Oncology, National Cancer Center Hospital East, Chiba, Japan
Background: This is a randomized, multicenter, open-label, Phase II study designed to evaluate the efficacy of everolimus added to continuous aromatase inhibitor (AI) administration in patients who had maintained stable disease or a better response for at least 5 months.
Patients and methods: Patients will be randomized to everolimus and standard therapy groups (1:1 ratio). In the everolimus group, patients will receive everolimus in addition to the AI agent. The standard therapy group will continue AI alone treatment. The primary endpoint is progression-free survival. Target accrual is 130 patients with a two-sided type I error rate of 10% and 80% power to detect 35% risk reduction.
Conclusion: The Chloe trial will provide important information about the efficacy and safety of adding everolimus to AI in patients with estrogen receptor-positive and human epidermal growth factor 2-negative metastatic breast cancer.
Keywords: hormone receptor-positive, breast cancer, aromatase inhibitor, everolimus
Introduction
Breast cancer is the most common malignant tumor in women. In Japan, the estimated number of women with breast cancer in 2015 was 89,400.1 Also, 13,800 women were thought to have died from the disease in the same year. Tumors reportedly metastasize to remote organs in 40% of patients diagnosed with breast cancer, and the median life expectancy after metastasis is 24–30 months.
According to the Japanese Breast Cancer Society, hormone receptor (HR)-positive breast cancer accounted for 80% of cases with this disease in 2011.2
As to pharmacotherapy for breast cancer, endocrine therapy causes milder adverse events than chemotherapy. Therefore, for patients with metastatic or recurrent HR-positive breast cancer who are not in a life-threatening state, it is common to administer endocrine therapy before chemotherapy with the aim of preserving quality of life (QOL).3 Since drug resistance develops during endocrine therapy in patients with HR-positive breast cancer, several strategies for overcoming such resistance have been investigated. Current counter-measures to these drug resistance phenomena include switching to chemotherapy while also continuing hormone therapy. Regarding the latter, endocrine therapies which utilize different mechanisms of action, along with agents that suppress altered signaling pathways causing drug tolerance, are being considered at present. In targeting these altered signaling pathways, modification of transcriptional pathways not mediated by estrogen receptor (ER) is preferred, with the PI3K/AKT/mTOR pathway being regarded as important in this respect. Indeed, it was previously reported that application of an mTOR inhibitor (everolimus) to endocrine therapy-resistant breast cancer cells restored their sensitivity to endocrine agents.4 The BOLERO-2 trial was an international randomized Phase III study of ~700 patients with breast cancer resistant to treatment with a nonsteroidal aromatase inhibitor (AI) agent; the aim of that clinical trial was to examine whether concomitant administration of everolimus restored sensitivity to exemestane that was being continuously administered.5 The study showed a statistically significant difference in progression-free survival (PFS) between the group treated with the combination of everolimus and exemestane (7.8 months) and that given exemestane alone (3.2 months; P<0.0001); the hazard ratio (HR) was 0.45 with a 95% CI of 0.38–0.54. Accordingly, both preclinical and clinical studies have demonstrated everolimus to be effective for overcoming resistance to endocrine therapy. Furthermore, a recent preclinical study demonstrated amelioration of the dependency of ERs on the genomic pathway with the application of a PI3K inhibitor or an mTOR inhibitor (rapamycin) to HR-positive breast cancer cells, raising the possibility that mTOR inhibitors potentiate the effects of endocrine therapy.6 Since endocrine therapy causes few adverse events, the patient’s QOL might be maintained for a longer time if the introduction of chemotherapy can be postponed. Therefore, we planned this study to examine whether concomitant administration of everolimus with endocrine therapy further prolongs PFS in patients with breast cancer whose hormone sensitivity remains intact.
Patients and methods
This study will examine whether the efficacy of everolimus added to an AI is superior to that of the AI alone. Patients with ER-positive, human epidermal growth factor 2 (HER2)-negative metastatic breast cancer who have maintained stable disease or a better response for at least 5 months following AI administration, as primary endocrine therapy, will be enrolled (Figure 1).
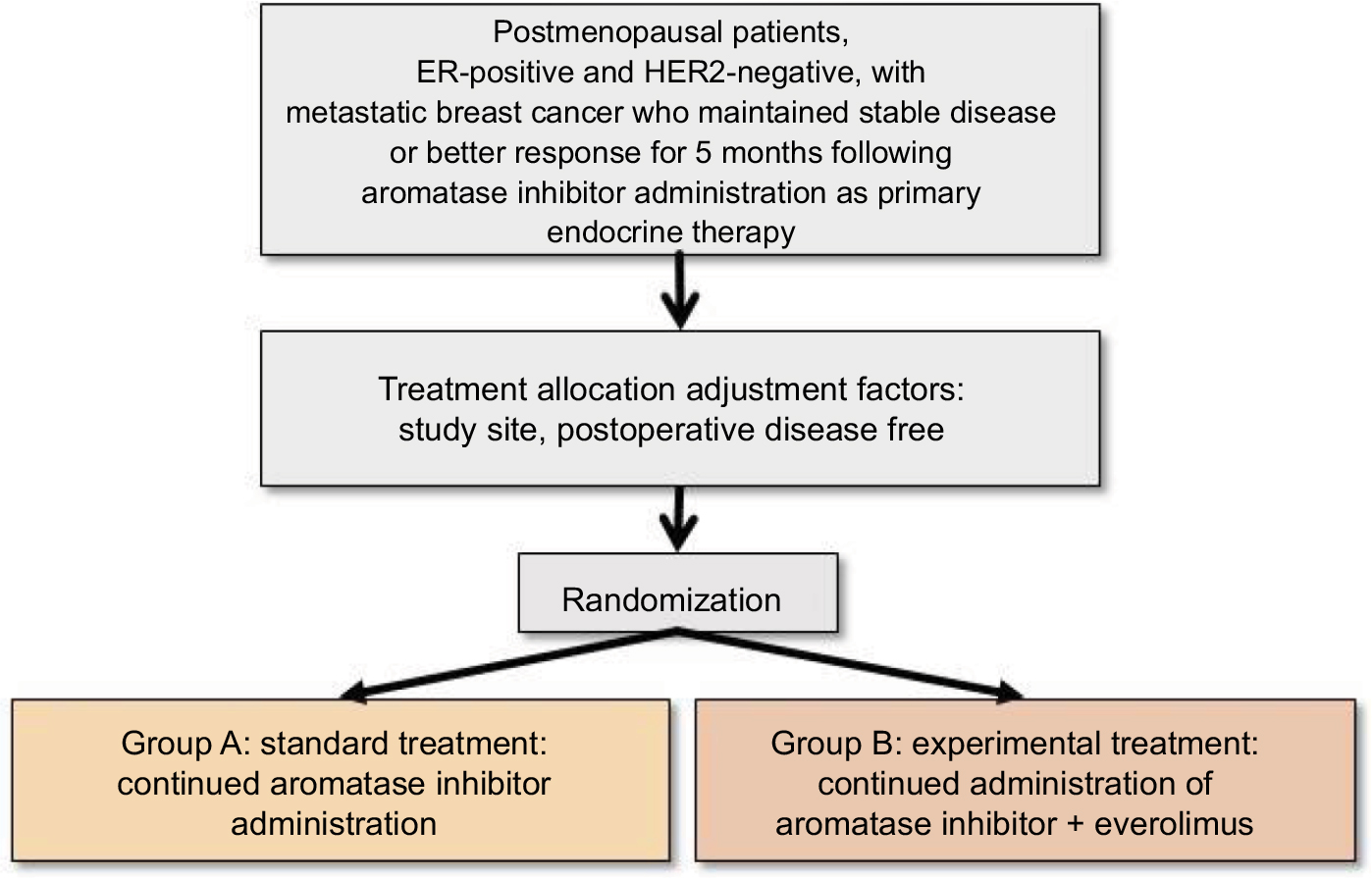 | Figure 1 Chloe trial design. Abbreviations: ER, estrogen receptor; HER2, human epidermal growth factor 2. |
Purpose
This study will be conducted on patients with recurrent metastatic cancer, focusing on the following three objectives:
- To examine whether additional administration of everolimus significantly prolongs the PFS period in postmenopausal patients with ER-positive, HER2-negative metastatic breast cancer who showed a positive response to an AI agent as primary endocrine therapy.
- To examine the effects of administration of everolimus in addition to an AI agent on the incidence of adverse events.
- To explore biomarkers related to the efficacy of everolimus.
Endpoints
- Primary endpoint: PFS
- Secondary endpoints
- Overall survival
- Response rate
- Disease control rate
- Adverse events
- Time to treatment failure
- Ratio of patients who continued the initial AI therapy for a year
Eligibility criteria
Inclusion criteria
- Histologically diagnosed breast cancer
- Immunohistochemically ER-positive (>10%) and HER2-negative (0.1%) cancer (or HER2-negative as determined by in situ hybridization).
- One or more measurable lesions based on Response Evaluation Criteria In Solid Tumors (Ver 1.1). As for bone lesions, patients with measurable osteolytic or osteolytic–osteoblastic lesions on computed tomography or magnetic resonance imaging (≥1 cm in largest diameter) will be eligible for enrollment.
- Patients with metastatic breast cancer satisfying at least one of the following two conditions:
- Remote metastasis judged not to be indicated for surgical resection at the first visit.
- Metastatic breast cancer except for local recurrence (the term local herein refers to the chest wall surrounded by the following areas: upward to the subclavian margin, downward to the costal arch, inward to the medial sternal margin and outward to the frontal margin of the latissimus dorsi muscle). Patients with local recurrence not indicated for surgical resection due to a diffuse lesion will also be eligible for enrollment.
- In patients receiving postoperative endocrine therapy, a 12-month or longer period must have passed since the end of the last administration. History of postoperative chemotherapy and elapsed time from chemotherapy, as well as a regimen of postoperative endocrine therapy, will not be considered when enrolling subjects.
- Among patients starting treatment with an AI agent within 5–7 months prior to study enrollment, those who have received no or only one regimen of chemotherapy will be eligible.
- No history of treatment with everolimus.
- Women with postmenopausal status.
- Eastern Cooperative Oncology Group performance status of 0 or 1. Patients with performance status 2 due to bone metastasis will also be eligible for study enrollment.
- In patients who have received radiation therapy, a 14-day or longer period must have passed since the end of the last radiation session.
- As to organ functions (within 14 days after study registration), patients must satisfy the following criteria:
- Neutrophil count 1,500/mm3 or more, or white blood cell count 3,000/mm3 or more
- Platelet count 100,000/mm3 or more
- Hemoglobin content 8.0 g/dL or higher
- Total bilirubin concentration no more than 1.5 mg/dL
- Aspartate transaminase (glutamic oxaloacetic transaminase) and alanine transaminase (glutamic pyruvic transaminase) activity no more than 100 U/L
- Serum creatinine concentration no more than 1.5 mg/dL
- Provision of written informed consent.
Exclusion criteria
- Active double cancer (simultaneous double cancer or metachronous double cancer within 5 years of the disease-free period). Carcinoma in situ (intraepithelial carcinoma and mucosal carcinoma) judged to already have been cured by local treatment will not be classified as active double cancer.
- Any history of serious drug hypersensitivity.
- Serious concomitant diseases (including pulmonary fibrosis or interstitial pneumonia, uncontrollable diabetes, serious cardiac dysfunction, renal failure, hepatic insufficiency, cerebrovascular disease and ulcer[s] requiring a blood transfusion).
- Active infectious disease requiring systemic treatment.
- Includes patients confirmed to be HBs antigen positive, and HBs and/or HBc antibody positive.
- Patients known to be infected with hepatitis C virus or with a history of hepatitis C virus infection.
- Active hemorrhagic diathesis or being treated with an oral vitamin K antagonist.
- Cerebral metastasis that is symptomatic or requires treatment.
- History of being administered medications known to be potent cytochrome P450 3A inhibitors or cytochrome P450 3A inducers (rifabutin, rifampicin, clarithromycin, ketoconazole, itraconazole, voriconazole, ritonavir and telithromycin).
- Currently receiving hormone replacement therapy.
- Any mental disorder potentially impacting the informed consent process.
- Any judgment that a patient would not be an appropriate candidate for participation in this study based on a physician’s assessment.
Pre-registration eligibility criteria
- Histologically confirmed breast cancer.
- Primary lesion(s) or metastatic lesion(s) that are immunohistochemically ER positive (>10%) and HER2 negative (0, 1 or negative by in situ hybridization).
- One or more measurable lesions according to Response Evaluation Criteria In Solid Tumors (Ver 1.1).
As for bone lesions, patients with measurable osteolytic or osteolytic–osteoblastic lesions on computed tomography or magnetic resonance imaging (≥1 cm in length) will be eligible for enrollment.
- Metastatic breast cancers must satisfy at least one of the following conditions:
- Remote metastasis judged not to be indicated for surgical resection at the first visit.
- Breast cancer showing progression or recurrence after treatment (surgery or pre- and postoperative treatment) or during postoperative treatment, except for local recurrence (the term local refers to the chest wall surrounded by the following areas: upward to the subclavian margin, downward to the costal arch, inward to the medial sternal margin and outward to the frontal margin of the latissimus dorsi muscle). Patients with local recurrence not indicated for surgical resection due to diffuse lesions will also be eligible for study enrollment.
- In patients who have received postoperative endocrine therapy, a 12-month or longer period must have passed since the end of the last administration. History of postoperative chemotherapy and elapsed time from chemotherapy, as well as the regimen of postoperative endocrine therapy, will not be considered when enrolling subjects.
- No or only one course of chemotherapy for the aforementioned bone lesions (3).
- No history of prior everolimus administration.
- Menopausal status, female.
- Provision of written informed consent.
Patient assignment
The Comprehensive Support Project for Oncology Research (CSPOR) data management center will randomly allocate study subjects to protocol treatments via a dynamic allocation procedure employing a minimization method using the following allocation adjustment factors. The exact treatment allocation algorithm will be decided by the person responsible for biostatistics and will involve the following parameters:
- Study sites (each site)
- Disease-free interval (<5 years/5 years or longer)
Treatment
Protocol treatment will be started within 2 weeks after the registration process has been completed.
Group A (standard treatment group)
The AI agent used in the initial pharmacologic therapy will be continuously administered. The patients will be administered one AI tablet per day. One treatment cycle will consist of 3 weeks, and administration will be continued until tumor progression occurs.
Group B (test treatment group)
In addition to the AI agent used in the initial pharmacologic therapy, everolimus will be administered at 10 mg/day (starting within 2 weeks after registration). One treatment cycle will consist of 4 weeks, and administration will be continued until tumor progression occurs.
Statistical analysis
Main analysis and assessment criteria
The purpose of the major analyses in this study is to examine whether the test treatment (continuous administration of an AI agent + everolimus) yields results superior to those of the standard treatment (continuous administration of an AI agent alone) regarding PFS, which is the primary endpoint of this study.
The null hypothesis of this study is that there is no difference in PFS between the two treatment groups, as examined by a stratified log-rank test with stratification of the subjects by PFS (<5 years/5 years or longer). If a situation occurs wherein the number of subjects or the number of events is too small to perform a stratified log-rank test, handling of allocation adjustment factors will be defined in the statistical analysis plan prepared without information on intergroup comparisons before the confirmatory analysis with the intergroup comparison.
As to PFS as the primary endpoint, cumulative survival curves, median survival time and annual survival rate are to be estimated by the Kaplan–Meier method. The CI for median survival time will be calculated by the method of Brookmeyer and Crowley,9 and that for annual survival rate will be calculated employing the equation of Kalbfleisch and Prentice.10 A stratified Cox proportional hazard model using PFS as strata will be applied to calculate the HR and 95% CI for comparisons between the groups as a means of estimating the relative therapeutic effect. To adjust for background factors deviating from the normal range, an adjusted HR will also be calculated, if necessary.
If the hypothesis that PFS is longer in the test treatment group than in the standard treatment group is verified in this Phase II study, we will proceed to the Phase III study with appropriate consideration of safety issues. In the Phase III study, we will examine whether the total period of endocrine therapy is prolonged, thereby improving the cost-effectiveness and cost-utility including QOL measures, based on the early introduction of everolimus rather than waiting to use this agent until the breast cancer has metastasized.
Sample size and follow-up period
The clinical hypothesis of this study is that the test treatment (continuous administration of an AI agent + everolimus) is superior to the standard treatment (continuous treatment with an AI agent alone) in terms of PFS. The expected median PFS in patients treated with AI agents is 8–12 months. Patients who have been continuously administered an AI agent for 5–7 months will be included in this study. Therefore, PFS is expected to range from 2 to 6 months from the time of allocation to this study. Since the study subjects will be patients who have maintained stable disease for at least 5 months, it is expected that longer PFS will be obtained in this study.
On the other hand, in the BOLERO-2 trial that was conducted in patients with breast cancer who had acquired resistance to nonsteroidal AI agents, PFS was longer by 4.6 months in the group treated with combination therapy consisting of exemestane and everolimus (7.8 months) than in the group treated with exemestane alone (3.2 months) (HR: 0.45, 95% CI: 0.38–0.54; P<0.0001).5 Since our study subjects will be patients with stable disease, in our view, a relative intergroup efficacy for the test treatment group yielding an HR of 0.6 or higher will be necessary.
The necessary sample size was calculated under the following assumptions: PFS will be 4–10 months, HR 0.65, one-sided α error 10%, β error 20% and the registration period 4–10 months, with the entire study period being 48 months (follow-up for 24 months from the day of the final patient registration).
If the mean survival time in the standard treatment group is assumed to be 10 months, the necessary sample size for both groups becomes 116 subjects (number of necessary events is 100). Therefore, we set the target sample size to 130 subjects for both groups (65 subjects in each group), assuming a ~10% rate of drop-outs or ineligible subjects.
Institutional review board or ethics committee and registration of the protocol
To conduct this study, the executive committee was organized. In addition, the steering committee, study review board and independent data monitoring committee organized by CSPOR of Breast Cancer (CSPOR-BC) performed the review of the study protocol, consideration of and decision on study policy, and management and supervision of study operation. Moreover, the institutional review board of the Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences and Okayama University Hospital, Ethics Committee approved this study (approval number 臨1702-008).
The study protocol was registered at the website of the University Hospital Medical Information Network, Japan (protocol ID 000025156), on 1 March 2017.
Discussion
This is a randomized, multicenter, open-label, Phase II study designed to evaluate the efficacy of everolimus added to continuous AI administration in patients who had maintained stable disease or a better response for at least 5 months.
Several randomized controlled studies that showed the additive effect of everolimus on endocrine therapy for postmenopausal, advanced or metastatic breast cancer cases resistant to AIs or nonsteroidal AIs have been reported. The randomized Phase III trial, BOLERO-2 trial, revealed that the addition of everolimus improved the PFS rates from 3.2 months on exemestane alone to 7.8 months with the combination (HR: 0.45, 95% CI: 0.35–0.54; P<0.0001).5 A randomized, Phase II trial, TAMRAD trial, investigated the everolimus plus tamoxifen combination versus tamoxifen alone in postmenopausal women with HR-positive, HER2-negative, AI-resistant metastatic breast cancer. The 6-month clinical benefit rate was 61.1% in the everolimus plus tamoxifen arm versus 42.1% in the tamoxifen alone arm (P=0.045). The median time to progression increased from 4.5 months on tamoxifen alone to 8.6 months with the combination (HR: 0.54, 95% CI: 0.36–0.81; P=0.002).7 Another Phase II trial, PrECOG 0102, investigated everolimus in combination with fulvestrant in postmenopausal women with HR-positive, HER2-negative metastatic breast cancer after progression on AI therapy. The median PFS increased from 5.1 months on fulvestrant alone to 10.4 months with the combination (HR: 0.60, 95% CI: 0.40–0.92; P=0.02).8 Although the addition of everolimus is shown in AI-resistant cases, the additional benefit of everolimus has not been studied for AI-susceptible cases.
This Chloe trial examines whether concomitant administration of everolimus with endocrine therapy further prolongs PFS in patients with breast cancer whose hormone sensitivity remains intact.
At present, an ongoing Phase II study (BOLERO-4 trial, NCT 01698918) is being conducted overseas to evaluate the efficacy and safety of combined use of everolimus with letrozole as the initial therapy for metastatic ER-positive, HER2-negative breast cancer. However, patients in that study are administered everolimus from the beginning of the trial period regardless of their hormone sensitivity. Since our study will be conducted with patients whose hormone sensitivity remains intact, the backgrounds of our patients will differ from those of the BOLERO-4 trial subjects. If our Phase II study supports the hypothesis that additional administration of everolimus to patients with intact hormone sensitivity can prolong the administration period of AI agents, through postponing the acquisition of drug resistance, we will undertake a subsequent Phase III study to confirm the usefulness of everolimus in terms of cost-effectiveness and cost utility. We will focus on prolongation of the entire period of hormone therapy by early administration of everolimus to ER-positive, aromatase-sensitive breast cancer patients. Therefore, we plan to assess QOL in this Phase III study after confirmation of the core hypothesis.
In summary, the Chloe trial will provide important information about the efficacy and safety of adding everolimus to AI in patients with ER-positive, HER2-negative metastatic breast cancer.
Acknowledgments
This study will be conducted under the framework of a Comprehensive Support Project for Oncological Research of Breast Cancer (CSPOR-BC) project.
Research funding is to be provided to CSPOR by Novartis Pharma. Novartis Pharma will not take part in this study other than providing information relevant to appropriate use of the study drug. The executive committee decided on all aspects of the planning, implementation, and publication of this study.
Author contributions
TS and AS contributed equally to writing this manuscript. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Statistics Japan. [Cancer statistics in Japan – 2015]. Available from: http://ganjoho.jp/reg_stat/statistics/brochure/backnumber/2015_jp.html. Accessed October 31, 2017. Japanese. | ||
Kurebayashi J, Miyoshi Y, Ishikawa T, et al. Clinicopathological characteristics of breast cancer and trends in the management of breast cancer patients in Japan: based on the Breast Cancer Registry of the Japanese Breast Cancer Society between 2004 and 2011. Breast Cancer. 2015;22(3):235–244. | ||
Hortobagyi GN. Treatment of breast cancer. N Engl J Med. 1998;339(14):974–984. | ||
Boulay A, Rudloff J, Ye J, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005;11(14):5319–5328. | ||
Pritchard KI, Lebrun F, Beck JT, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. | ||
Bosch A, Li Z, Bergamaschi A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci Transl Med. 2015;7(283):283ra51. | ||
Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30(22):2718–2724. | ||
Kornblum NS, Manola J, Klein P, et al. Abstract S1–02: PrECOG 0102: a randomized, double-blind, Phase II trial of fulvestrant plus everolimus or placebo in post-menopausal women with hormone receptor (HR)-positive, HER2-negative metastatic breast cancer (MBC) resistant to aromatase inhibitor (AI) therapy. Cancer Res. 2017;77(4):S1–S2. | ||
Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics. 1982;38(1):29–41. | ||
Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. New York: John Wiley & Sons, Inc.; 1980. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.