Back to Archived Journals » Oncolytic Virotherapy » Volume 7
Oncolytic virotherapy in upper gastrointestinal tract cancers
Authors Yokoda R ![]() , Nagalo BM, Arora M, Egan JB, Bogenberger JM, DeLeon TT, Zhou Y
, Nagalo BM, Arora M, Egan JB, Bogenberger JM, DeLeon TT, Zhou Y ![]() , Ahn DH, Borad MJ
, Ahn DH, Borad MJ
Received 3 January 2018
Accepted for publication 8 February 2018
Published 23 March 2018 Volume 2018:7 Pages 13—24
DOI https://doi.org/10.2147/OV.S161397
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chae-Ok Yun
Raquel Yokoda,1 Bolni M Nagalo,1 Mansi Arora,1 Jan B Egan,1 James M Bogenberger,1 Thomas T DeLeon,1 Yumei Zhou,1 Daniel H Ahn,1 Mitesh J Borad1–3
1Division of Hematology/Oncology, Department of Medicine, Mayo Clinic, Scottsdale, AZ, 2Department of Molecular Medicine, Center for Individualized Medicine, Mayo Clinic, Rochester, MN, 3Department of Oncology, Mayo Clinic Cancer Center, Phoenix, AZ, USA
Abstract: Upper gastrointestinal tract malignancies are among the most challenging cancers with regard to response to treatment and prognosis. Cancers of the esophagus, stomach, pancreas, liver, and biliary tree have dismal 5-year survival, and very modest improvements in this rate have been made in recent times. Oncolytic viruses are being developed to address these malignancies, with a focus on high safety profiles and low off-target toxicities. Each viral platform has evolved to enhance oncolytic potency and the clinical response to either single-agent viral therapy or combined viral treatment with radiotherapy and chemotherapy. A panel of genomic alterations, chimeric proteins, and pseudotyped capsids are the breakthroughs for vector success. This article revisits developments for each viral platform to each tumor type, in an attempt to achieve maximum tumor selectivity. From the bench to clinical trials, the scope of this review is to highlight the beginnings of translational oncolytic virotherapy research in upper gastrointestinal tract malignancies and provide a bioengineering perspective of the most promising platforms.
Keywords: oncolytic viruses, hepatopancreatobiliary, gastric cancer, pancreatic cancer, liver cancer, biliary cancer
Introduction
Gastrointestinal cancers are considered a significant challenge in public health worldwide. In the US, pancreatic adenocarcinoma and hepatocellular carcinoma (HCC) appear among the ten most deadly cancers each year.1 New drugs to combat those cancers are being developed, and arrays of oncolytic viral vectors are progressing into clinical studies. Oncolytic viral therapy in upper gastrointestinal tract (UGT) malignancies represents a promising therapeutic platform. Virotherapy can allow for combination with immunotherapies or cytotoxic chemotherapies or used as a standalone platform for gene therapy in the context of cancer ultimately being a genetic disease.
Genomic profiling of UGT malignancies has yielded compendia of mutations, regions of loss of heterogeneity, gene fusions/translocations, and copy-number alterations responsible for pathogenesis, disease recurrence, and drug resistance. This review will cover the implementation of oncolytic virotherapy (OV) in esophageal, gastric, pancreatic, liver, and biliary cancers from a bedside-to-bench perspective.
Upper gastrointestinal tract malignancies
One of the recent forays of OV in the clinic was in patients with head and neck cancers. China approved the first virus (H101) for nasopharyngeal cancers in November 2006.2 H101 is a recombinant adenovirus (Ad) very similar to ONYX15 that had been studied in the US, but efforts were unable to be sustained beyond initial clinical studies.3 The approval of H101 provided a much-needed boost to the field for ongoing investigation. More recently, a herpes simplex virus (HSV)-based vector (T-Vec) garnered US Food and Drug Administration approval in 2015 for the treatment of patients with advanced melanoma.4
Pursuant to these efforts, sustained interest in the field of OV has been fostered, aided by more comprehensive understanding of the genetic underpinnings of cancers, as well as improved understanding and success of cancer immunotherapies as a mainstay of cancer treatment and a growing appreciation of the potential for evaluating OV, combined with these existing therapeutic modalities. Given the difficult prognosis of patients with advanced UGT malignancies, OV represents a promising platform for the development of novel therapeutics.
Esophageal cancer
Esophageal cancer has an incidence of nearly 17,000 people per year in the US, and the 5-year survival is less than 19%.5 Early metastasis and diagnosis at advanced clinical stage are responsible for the poor prognosis and high mortality of the disease. One of the early attempts at use of OV in esophageal cancer used herpes simplex virus type 1 (HSV1) as the viral platform.6 The approach took advantage of the mucosal tropism of the strain equipped with GFP for visualization/monitoring. The cytotoxic effect of the virus was evaluated, first in vitro using BE3 cell lines, and thereafter in vivo with an intraperitoneal tumor model (Table 1). The study concluded that the viral platform could be used to mark esophageal cancer cells from a diagnostic approach at the time of endoscopy.
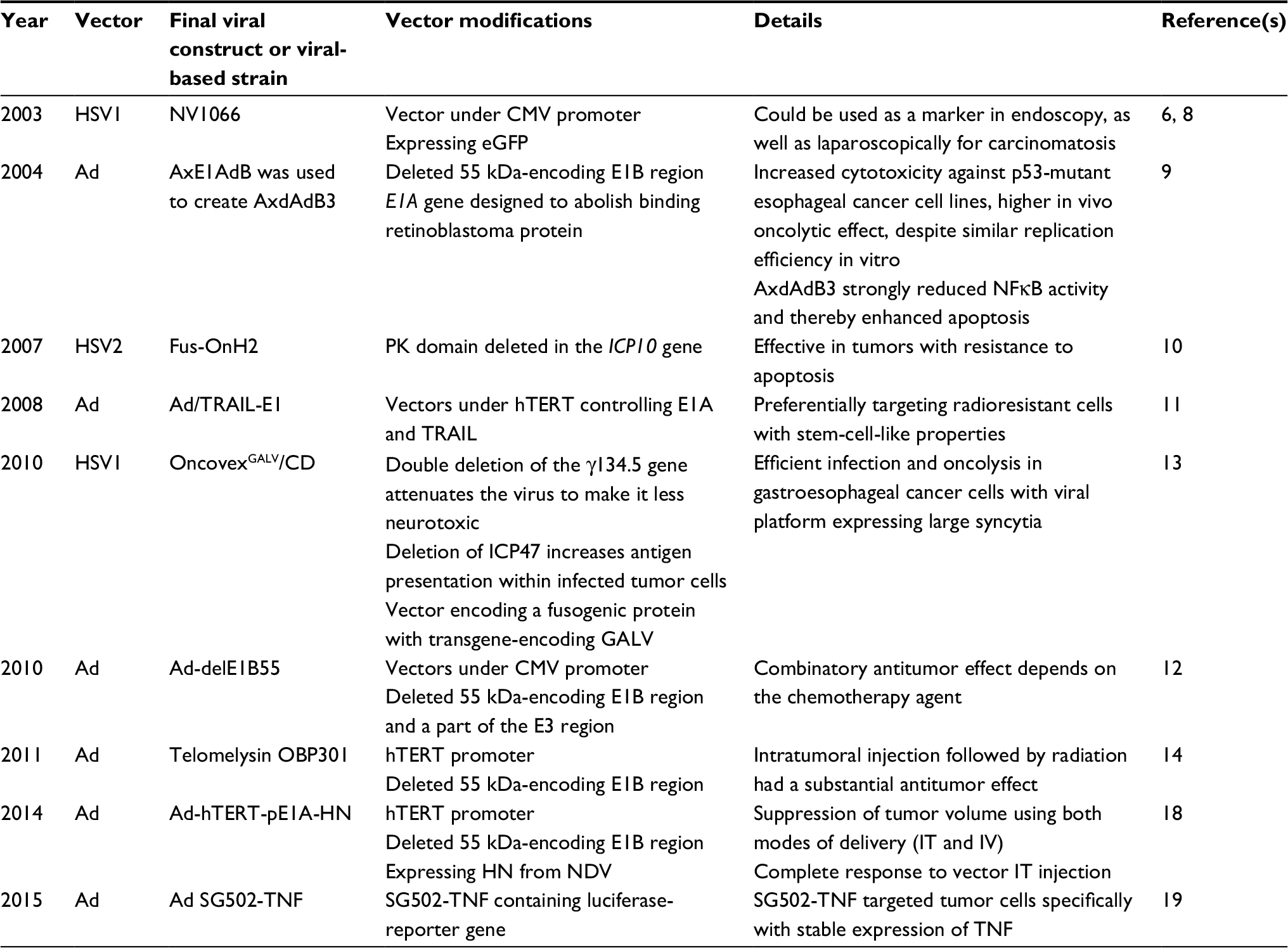 | Table 1 Oncolytic viral platforms for esophageal cancer Abbreviations: HSV1, herpes simplex virus type 1; HSV2, herpes simplex virus type 2; CMV, cytomegalovirus; Ad, adenovirus; PK, protein kinase; GALV, gibbon ape leukemia virus; HN, hemagglutinin–neuraminidase; IT, intratumoral; IV, intravenous; NDV, Newcastle disease virus; TNF, tumor necrosis factor. |
BE3 has served as a cell-line model for esophageal adenocarcinoma and Barrett’s esophagus. Interestingly, that viral platform was derived from the HSV1 strain NV1023 that was tested in squamous-cell head and neck carcinoma and was able to spread via the lymphatic system.7 The same group also tested the virus in vivo in gastric cancer models. They observed that the platform could be used to treat peritoneal carcinomatosis or demarcate it for laparoscopic intervention.8
Later, a modified Ad was developed to establish a platform that could induce cell-cycle arrest.9 Increased cytotoxicity was observed against p53-mutant esophageal cancer cells. Additionally, greater oncolytic effect with reduced NFκB and enhanced apoptosis was noted.9 Correspondingly, a newly modified herpes simplex virus type 2 (HSV2) was developed with oncolytic effect on esophageal cancer cell lines. The N-terminal of the ICP10 gene of HSV2 was modified by deleting a protein-kinase domain. This deletion increased the bystander-killing effect of the virus, enhancing apoptosis. The mutant virus was designated FusOnH2.10
A study evaluating resistant tumor cells with properties similar to the cancer stem cells was conducted using an Ad carrying an apoptotic gene, TRAIL, which would preferentially induce apoptosis in these resistant cells.11 Further, another group evaluated the combinatorial effects of chemotherapy and oncolytic therapy with an Ad.12 They used four drugs and a modified Ad5, with deletion of the 55 kDa-encoding E1B region and a part of the E3 region, generating the vector Ad-delE1B55. The four drugs tested were fluorouracil (5FU), etoposide (VP16), mitomycin C, and cisplatin (CDDP). 5FU enhanced the cell cycle at the S phase and then induced G2–M phase entry, whereas CDDP induced G1 phase arrest. Ad-delE1B55 also induced the S phase and G2/M phase such that G1 phase arrest by CDDP was inhibitory to Ad-mediated cell-cycle progression and as a result was unfavorable to Ad-mediated toxicities. On the other hand, the Ad-induced S phase and G2/M phase impeded CDDP-mediated cytotoxicity, and thus the cytotoxicity between the Ad and CDDP promoted cross-resistance.12
A third-generation HSV1 improved the oncolytic effect by incorporating a gene of fusogenic proteins with transgenes for gibbon-ape leukemia virus (GALV) encoded into the viral genome.13 Furthermore, another group built upon their work with Ad-TRAIL-E1 toward the development of the vector Ad-hTERT-E1 (Telomelysin OBP301), under the control of hTERT.14 This vector has progressed to clinical trials for esophageal cancer.
In the same year, another group evaluated the adenoviral vector H101 in esophageal carcinoma.15 Following this, an enhanced antitumor effect in esophageal cancer cell lines was observed with H101 through the overexpression of coxsackie and adenovirus receptor (CAR) using trichostatin A.16 Enhanced oncolytic activity was achieved through MAPK–ERK pathway induction of viral proliferation.16 Recently, another study has demonstrated that trichostatin A enhances the spread and replication of adenoviral vectors in several infection-resistant cancers.17
A chimeric vector, Ad-hTERTp-E1A-HN, which uses the same adenoviral backbone as H101 coupled with the hemagglutinin–neuraminidase gene from Newcastle disease virus (NDV), has been evaluated in preclinical esophageal cancer models. Hemagglutinin–neuraminidase is a 75-kDa membrane glycoprotein that has the potential to boost innate immunity. Its neuraminidase activity allows for hydrolysis of sialic acid on the receptors of host cells. It can also induce IFNα and TRAIL19 production in peripheral blood mononuclear cells, leading to enhancement of apoptotic pathways. The study of esophageal cancer observed cytotoxicity by an increase in reactive oxygen species. In vivo evaluation demonstrated complete responses and prolonged survival with intratumoral injection of the vector.18
A number of vectors have been entering clinical trials following these preclinical evaluations. A Phase I clinical trial for Telomelysin OBP301 is awaiting initiation (ClinicalTrials.gov NCT03213054). These efforts mark the transition of OV from bench to bedside in esophageal cancer. Future directions entail safety evaluation and optimal dosing of vectors, followed by combinatorial assessment with existing therapies, particularly immunotherapies.
Gastric cancer
Gastric cancer is the fifth most common cancer in the world. It has a 5-year overall survival (OS) rate of 30.6%. Surgery is the only curative treatment, but less than a quarter of cases are eligible.20 OV for gastric cancer was initially tested in vivo in mouse models of peritoneal carcinomatosis. Initially, two oncolytic HSV vectors (G207 and NV1020) were tested. Locoregional therapy was found to be efficacious. However, systemic delivery through intravenous administration failed to diminish tumor burden.21 In another study, apoptosis inhibitors were used in conjunction with viral vectors. It was found that N-acetyl cysteine improved killing of neighboring uninfected cells using the NV1066 platform.22
Later, the first tumor-specific promoters were tested for conditionally replicating Ads (CR-Ads) in gastric cancer.23 midkine and cyclooxygenases (Cox2M and Cox2L) promoters demonstrated high transcriptional activity in gastric cancer cells, and the Cox2CR-Ad showed a potent oncolytic effect. A fiber-modified vector, Ad5/3, also yielded robust infectivity in this study.23 With its serotype 3 knob, Ad5 allows the vector to bind to a receptor different from CAR. Another study used this Ad5/3 capsid in an Ad5LucRGD vector. Incorporation of peptides Arg–Gly–Asp (RGD) in the fiber knob allowed the virus to utilize αvβ-class integrins for binding and internalization. These integrins are highly expressed in gastric cancers. The vector Ad5LucRGD also had polylysine motifs – pK7 and pK21 – that bound to heparan sulfates, which are overexpressed in gastric cancers. The receptor for Ad3 is believed to be CD46, which is also highly expressed in advanced gastric cancers. From this perspective, the chimeric capsid considerably improved viral infectivity.24
In another study, a CEA promoter was introduced into an adenoviral vector to study conditional vector replication in CEA-expressing gastric carcinoma cells, paving the path for a CEA-promoter-driven CR-Ad in gastric cancers.25 Further, a viral platform targeting the cyclin D–CDK–p16–Rb–E2F pathway was created based on an Ad by inserting a p16-expression cassette upstream of the E2F promoter, thereby controlling E1A with the E2F promoter. This AdE2F-p16 replicated preferentially in tumor cells with the desired specificity, while sparing normal cells. Luciferase assays demonstrated that the promoter conferred vector selectivity in cancer cells compared to normal cells (Table 2).26 In a contemporaneous evaluation, echovirus 1 was evaluated for peritoneal dissemination in gastric cancer models by vector dissemination through the use of α2β1 integrins.27
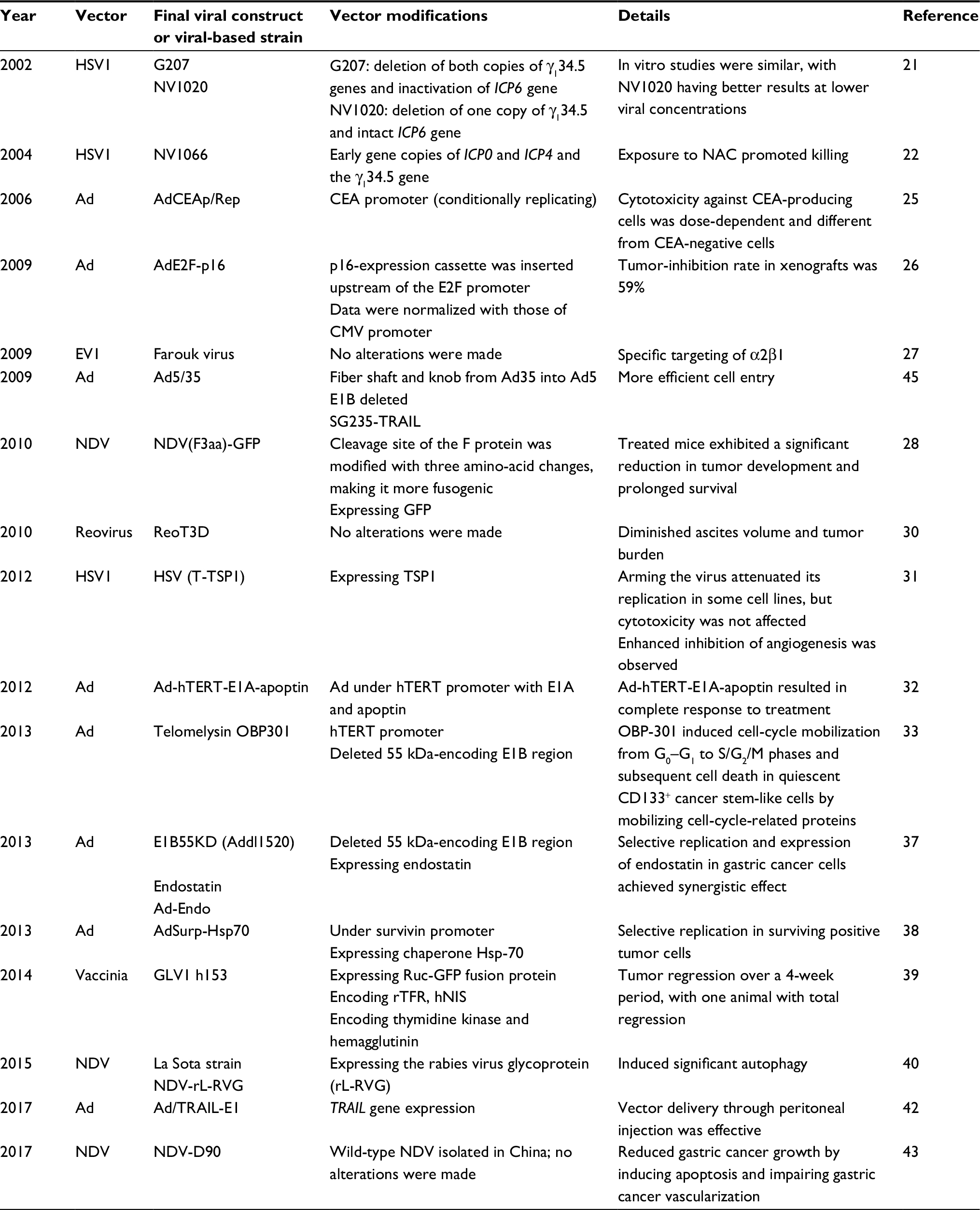 | Table 2 Oncolytic viral platforms for gastric cancer Abbreviations: HSV, herpes simplex virus; HSV1, herpes simplex virus type 1; NAC, N-acetylcysteine; CMV, cytomegalovirus; Ad, adenovirus; CEA, carcinoembryonic antigen; EV, echovirus; NDV, Newcastle disease virus; Ruc, Renilla Aequorea luciferase; rTFR, reverse-inserted human transferrin receptor; hNIS, human sodium–iodide symporter. |
A subsequent study evaluated NDV as an oncolytic in gastric cancer. A bioengineering modification improved its fusogenic proteins by deleting three amino acids through reverse genetics. The new virus, NDV(F3aa), was able to reduce the tumor burden in animal models of gastric cancer.28 Reovirus has also been studied in gastric cancer. It has a double-stranded RNA genome, and its oncolytic action depends on the presence of activated Ras-signaling pathways. This activation could be via direct Ras mutations, such as K-Ras or N-Ras, or downstream of other pathway activators, such as EGFR, Her2 (Neu/ErbB2), or SOS.29 These attributes make reovirus a desired OV candidate for targeting cancers that have Ras activity. In 2010, a study evaluating reovirus in Ras-activated gastric cancer models showed encouraging efficacy.30
Gene therapy-based approaches to modulate the tumor microenvironment have also been pursued using viral vectors in gastric cancer. An HSV armed with thrombospondin 1 was evaluated in gastric cancer.31 Another study used apoptin to induce apoptosis specifically in tumor cells. Apoptin is a p53-independent, Bcl2-insensitive apoptotic protein that can induce programmed cell death only in cancer cells. An Ad under the hTERT promoter was utilized to carry the apoptin gene (Ad-hTERT-E1A-apoptin). In vitro and in vivo evaluations yielded promising efficacy, including a number of complete tumor regressions.32 The apoptin gene is derived from the chicken anemia virus, and it does not need a functional p53 pathway nor is it hindered by Bcl2 blockage of apoptosis.
Furthermore, another group evaluated the Ad OBP301 to target quiescent stem-like cells.33 These types of cells are largely resistant to conventional cytotoxic therapies as long as they are in a dormant phase. Mobilizing these quiescent cells into the cell cycle can reenable a response to treatment. This study demonstrated that cell-cycle mobilization and S/G2/M phase trapping could be induced by adenoviral infection. This was felt to be a significant accomplishment in solid-tumor OV, given that mobilization of quiescent cells into the cell cycle had only been previously achieved in leukemia.34,35
Another study described a viral gene-therapy approach to create an Ad with endostatin.36 Endostatin is a 20 kDa C-terminal fragment of collagen XVIII known to be a potent inhibitor of angiogenesis.37 Synergistic effects of oncolysis and endostatin expression were achieved allowing for enhancement of virotherapy efficacy. In the same realm, another study evaluated an adenoviral platform with a survivin promoter and a chaperone gene – HSP70. The resultant vector (AdSurp-Hsp70) replicated selectively in survivin-positive gastric cancer cells and inhibited tumor growth in both immunodeficient and immunocompetent mice.38
Conversely, a vaccinia virus with visible markers has been used for deep-tissue imaging in gastric cancer.39 The markers used were GFP and human NIS, which can be readily imaged with 99mTc scintigraphy and 124I positron-emission tomography. These types of approaches allow for noninvasive vector monitoring. Recently, a number of studies have evaluated chimeric vectors. An example of such a vector is the NDV La Sota strain expressing the rabies virus glycoprotein. This chimeric vector was found to have increased oncolytic efficacy in gastric cancer cell lines.40
Besides vector development, delivery strategies have also been shown to have an impact on antitumor efficacy.41 An intraperitoneally delivered adenoviral vector exhibited efficacy in diminishing peritoneal carcinomatosis in a gastric cancer model.42 There has been considerable interest in evaluating wild-type virus strains known to have oncolytic properties. A wild-type NDV strain isolated in China named NDVD90 has shown promising preclinical efficacy in gastric cancer, selectively killing gastric cancer cells with no appreciable effect on normal cells.43 Vaccinia virus vectors have also garnered interest toward evaluation in gastric cancer. A clinical study evaluating a vaccinia viral oncolytic vector (GL-ONC1) was completed in Germany in gastric cancer patients with peritoneal carcinomatosis (ClinicalTrials.gov NCT01443260).44 Recently, synergistic cytotoxicity has been reported with concomitant OV and chemotherapy.45
Pancreatic cancer
Pancreatic cancer is one the most challenging malignancies, with a 5-year OS of only 8.2%.46 Unfortunately, this rate has not changed over the last 20 years, despite some recent modest advances in systemic therapy with FOLFIRINOX and gemcitabine–nab-paclitaxel. As such, novel therapeutic approaches are in imminent need and oncolytic viruses represent a promising treatment approach in this regard. A number of clinical studies evaluating oncolytic viruses are ongoing (Table 3). Translational research is the main focus of these vectors being evaluated in pancreatic cancer.
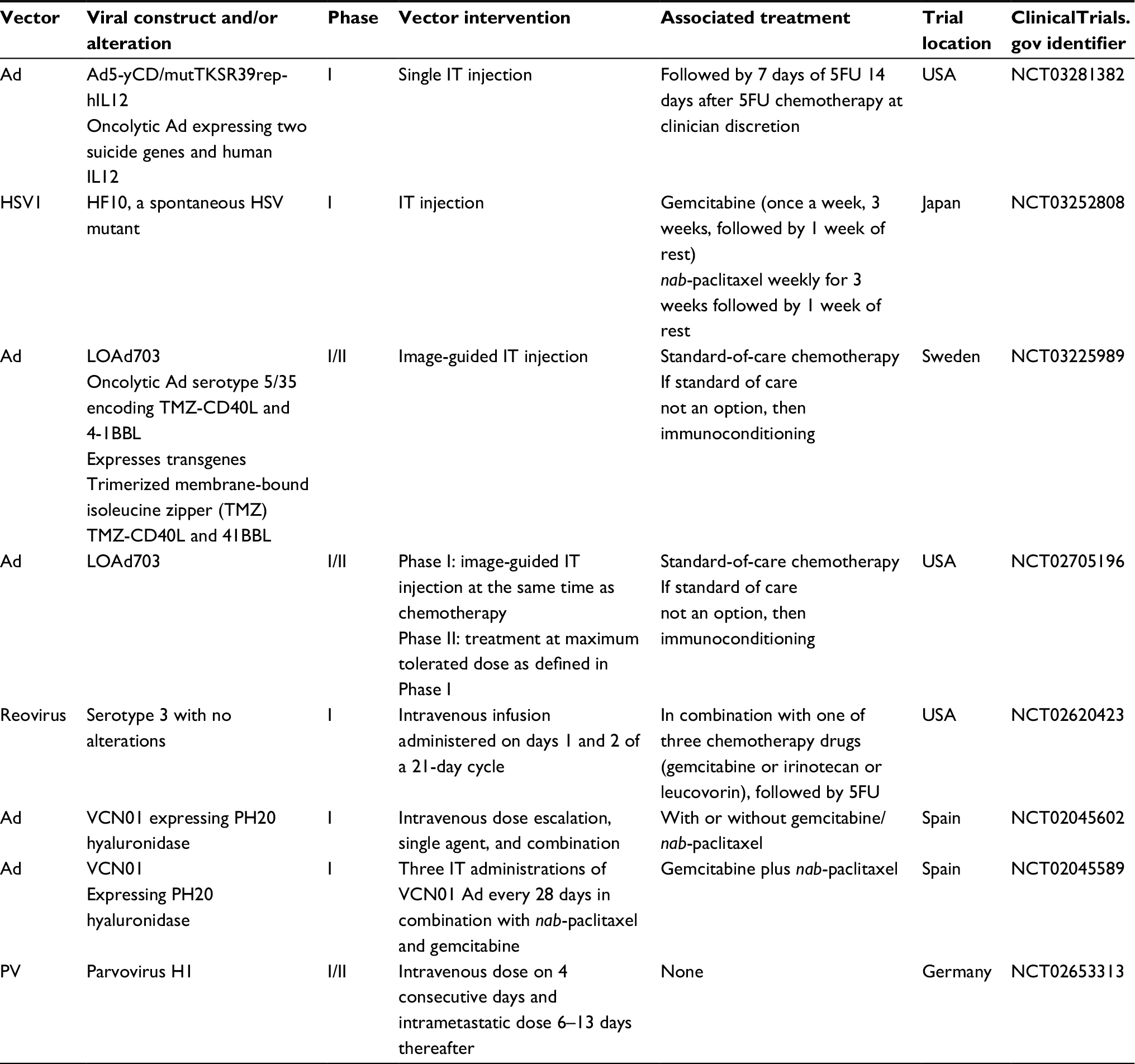 | Table 3 Current clinical trials using oncolytic viruses for pancreatic cancer Abbreviations: Ad, adenovirus; IT, intratumoral; HSV, herpes simplex virus; HSV1, herpes simplex virus type 1; FU, fluorouracil. |
Initially, such vectors as ONYX015 (Ad with E1B 55 kDa deletion) developed to treat other malignancies were evaluated in pancreatic tumor cancer cells. Findings were similar to other tumors, with the vector replicating in TP53-deficient cells.47 Since then, the field has quickly evolved to address specific challenges related to pancreatic tumors. From this perspective, research efforts have focused on overcoming 5FU resistance and using a gene-therapy approach: this effort has yielded the Ad AxE1AdB-UPRT. This Ad vector is capable of expressing UPRT, which can overcome 5FU resistance.47 Given that host antiviral immunity can be an impediment to efficacy, evaluation was undertaken in immunocompetent models. Other studies have utilized immunosuppression with cyclophosphamide accompanying vector delivery in immunocompetent models, with encouraging results.48
In 2007, an Ad carrying suicide genes was successfully tested in preclinical models of pancreatic cancer. Ad5-yCD/mutTKSR39rep-ADP is an Ad5 vector carrying Ad cytosine deaminase and HSV thymidine kinase (TKSR39). This vector augmented the effectiveness of pancreatic radiotherapy without resulting in excessive toxicity.49 Pursuant to this, other adenoviral vectors carrying TK genes in combination with ganciclovir were tested using an intraductal delivery approach, resulting in improved survival in vivo.50
The La Sota strain of NDV has been tested in pancreatic cancer models, with favorable efficacy. Pancreatic tumor-cell lines were 700 times more sensitive to cytotoxicity from the NDV La Sota strain in in vivo pancreatic cancer models than normal pancreatic cells.51 Likewise, reovirus has been evaluated in pancreatic cancer. Given that it replicates only in cells that express an active Ras pathway and the nearly ubiquitous nature of Ras activation in pancreatic cancer, this virus became particularly interesting in this disease.52 In one study evaluating reovirus in pancreatic cancer, the vector proliferated to yield a considerable amount of viral particles and ubiquitinated proteins in the endoplasmic reticulum, causing apoptosis mediated by endoplasmic reticulum stress in vitro and in vivo.52
Human and avian influenza A viruses (IAVs) have also been evaluated in pancreatic cancer models.53 The appeal of these platforms was based on the natural tropism of IAV for pancreatic cells. The study confirmed the expression of α2,3- and α2,6-linked polysaccharide receptors, respectively, for avian and human IAV in pancreatic ductal adenocarcinoma (PDAC) cell lines. Correspondingly, IAVs did not infect normal pancreatic cells. Tropism and selectivity were observed, with promising antitumor efficacy both in vitro and in vivo.53
Tumor selectivity was a critical impetus for a study in pancreatic cancer that involved targeting MMP with an Ad bioengineered with a TAT-like peptide and linked to a blocking domain by an MMP-cleavable sequence and achieving MMP-dependent transduction. This viral platform, AdTATMMP, was shown to reduce local and distant metastases, with no significant toxicity in in vivo pancreatic cancer models.54
From the perspective of tumor selectivity, one research group identified a pancreatic cancer-targeting ligand (SYENFSA) that would boost the specificity of an Ad under a survivin promoter, achieving higher transduction efficiency. Ad Sur-SYE resulted in potent oncolysis in PDAC55 and superior efficiency when compared to the non-targeting virus AdSur for neuroendocrine pancreatic tumors when administrated intratumorally.56
In 2015, the oncolytic Ad VCN01 was also tested in vitro and in vivo for PDAC. VCN01 is characterized by the expression of hyaluronidase and the RGD shaft-retargeting ligand. As such, VCN01 employs a strategy whereby the tumor microenvironment can be modulated using a viral vector. Encouraging safety and efficacy have been noted with VCN01 in preclinical studies, paving its way toward clinical development.57
Second-generation HSV1 vectors have been evaluated in pancreatic cancer. Using recombinant HSV1, a novel viral platform was created with ICP6-defective expression and HSV1 γ134.5 gene expression, regulated by the cellular B-Myb promoter and giving rise to the viral vector Myb34.5.58 Tumor progression was inhibited with intratumoral injection of this virus alone, and tumor regression was achieved when virotherapy was combined with standard chemotherapeutic agents. In light of the promising preclinical data generated in these and other evaluations, a number of oncolytic vectors have progressed into clinical studies (Table 3).
Liver and biliary cancers
Liver and intrahepatic bile duct cancer affect around 40,000 people per year in the US, with nearly 29,000 deaths each year and a dismal 5-year OS of only 17.6%.59 Toxicity to neighboring hepatocytes is a significant concern, and vectors have been developed to increase tumor specificity and decrease off-target toxicity. In this section, we review both DNA and RNA viruses used as oncolytic vectors for liver and biliary cancers and acknowledge current oncolytic clinical trials addressing those tumors in Table 4.
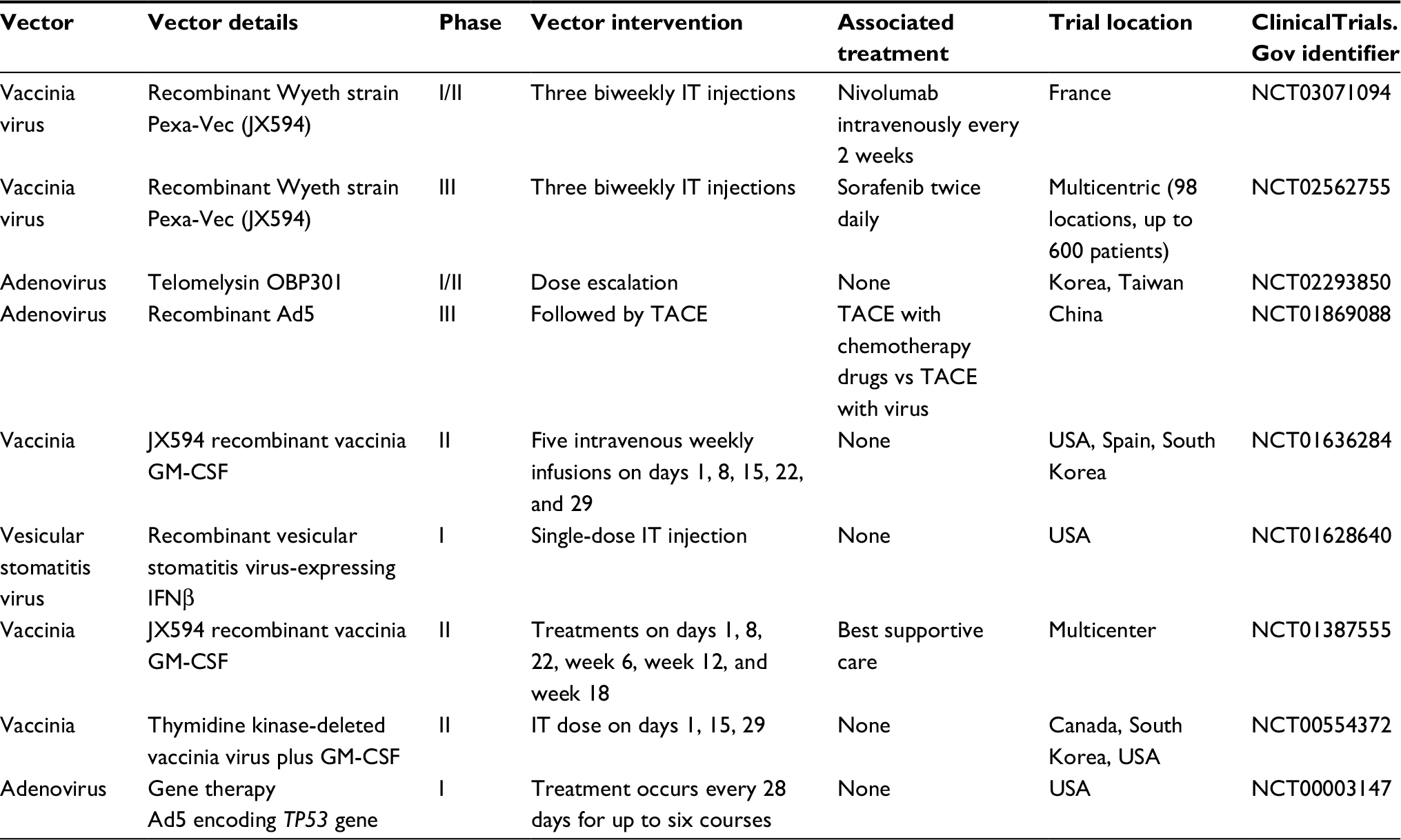 | Table 4 Clinical trials with oncolytic viruses for hepatobiliary tumors Abbreviations: Pexa-Vec, pexastimogene devacirepvec; IT, intratumoral; Ad, adenovirus; TACE, transarterial chemoembolization. |
DNA viruses: adenovirus, herpes viruses, and vaccinia
The first HCC-specific Ad (CV890) was reported using transcriptional regulatory elements of AFP as replication drivers for Ad5. The vector has E1A and E1B genes under the control of a bicistronic AFP-E1A-IRES-E1B cassette.60 In vivo results demonstrated tumor-volume reduction below baseline 4 weeks after a single viral dose. When combined with doxorubicin, there was synergistic antitumor efficacy, with complete remission after a single instance of combination therapy.60 Using the ONYX015 adenoviral vector platform, a murine endostatin-carrying vector was evaluated in HCC. A favorable safety profile and antitumor efficacy were both exhibited.61
A dual-promoter approach has been evaluated as a strategy to enhance tumor specificity and delivery of gene therapy in HCC. A conditionally replicative gene–viral vector system (CNHK500-p53) employed insertion of a TP53 gene-expressing cassette into the viral genome coupled with an hTERT promoter to drive the E1A adenoviral gene and a hypoxia-response-element promoter to drive the adenoviral E1B gene, with the objective of producing p53 protein in hypoxic drive, telomerase-positive HCC cells.62 In vitro studies demonstrated that p53 expression enhanced virus oncolytic effect.62 Besides TP53, another tumor suppressor gene, TSLC1, was evaluated using an Ad platform (SD55-TSLC1).63 In order to increase oncolytic efficacy, another strategy has been to add lethal genes from mitochondria to be expressed by the vector in tumor cells. In 2007, a vector encoding the Smac protein was developed (ZD55-Smac). ZD55-Smac was a superior oncolytic vector but cytotoxicity towards normal cells was a concern.64
A number of enhancements to increase safety have been made beyond the initial vector approaches. Tumor-specific promoters have aided with vector selectivity toward tumors in HCC. AFP and transthyretin65 promoters have been deployed in adenoviral vectors. Similarly, an AFP-regulated ribonucleotide reductase was instituted in a HSV vector, resulting in increased oncolytic effect.66 A number of promoters have been evaluated in HCC in HSV vectors, which include ANGPTL3, CYP2C8, vitronectin, ADH6, APOB, FBC, ITIH3, and ITIH1.67 Recently, the promoter for a Golgi protein, GOLPH2 (also called GP73), was successfully instituted as an HCC-specific Ad promoter.68
Hybrid promoters are also being used. One such example is HRE-AFP, used in a construct with the bee-venom toxic peptide melittin. Melittin can induce HCC apoptosis, and the vector QG511-HA-melittin exerted an inhibitory effect in HCC and was described as triple-targeting mechanism addressing AFP-positive cells, cells in a hypoxic environment, and cells with p53 deficiency.69 Furthermore, the use of microRNA (Let7) to control replication was introduced to diminish toxicity.70 A study utilizing the micro-RNA miR34a for vector specificity allowed for delivery of IL24.71 Recently, an Ad-expressing lncRNA that can competitively bind oncogenic miRNAs has been shown to achieve promising antitumor efficacy.72
A wide range of genes that can elicit oncolytic effects has been evaluated to date. SOCS3 can downregulate cyclin D1 and antiapoptotic proteins and enhance antitumor effects.73 Similarly, SOCS1, a negative regulator of STAT3, can inhibit STAT3 phosphorylation and downregulate survivin, cyclin D1, Bcl-xL and C-myc.74 Another strategy to enhance potency relies on oxygen-dependent degradation domain-regulated vectors that attract NK92 cells into hypoxic tumor microenvironments in animal models.75 Similarly, a construct using manganese superoxide dismutase (AD55-Mn-SOD) has been shown to suppress HCC growth effectively in xenografted nude mice.76
Despite Ad and cisplatin being described as having competitive modes of action, one study has found that cisplatin can be used in combination with Ad. Growth inhibition of HCC by XAF1 in an adenoviral platform (ZD55-XAF1) was pursued in these efforts.77 XAF1 is a counterregulator of the IAP protein family. With this vector, reduced cisplatin doses were possible and tumor-cell apoptosis enhanced via the activation of the caspase 9–PARP pathway.77
Another DNA virus, the vaccinia-based construct JX594, has been shown to be a promising candidate for viroimmunotherapy. Immunostimulation was augmented by GM-CSF transgene expression and evaluated in three patients with hepatitis B-related HCC. In this study, virotherapy was found to induce antivascular cytokines and was associated with tumor vascular shutdown. Interestingly, it also appeared to suppress hepatitis B replication.78 Correspondingly, another vaccinia strain, GLV1h68, was evaluated in vitro and exhibited cytotoxicity in both sorafenib-sensitive and -resistant HCC cell lines.79 Preliminary consideration for an immunomediated mechanism of action has been proposed, given observation of tumor response in distant, noninjected tumors in an ongoing clinical study with JX594.80
RNA viruses: vesicular stomatitis virus, measles virus, Newcastle disease virus, and retroviruses
Vesicular stomatitis virus (VSV) is a negative-strand RNA virus that been tested in vitro and in vivo in immunocompetent mice, with evidence of tumor shrinkage and minimal toxicity.81 Subsequently, other recombinant forms of VSV, such as rVSV-β-galactosidase, which expresses β-galactosidase to facilitate X-galactosidase staining of tumors, were tested in HCC. rVSV-β-gal was enabled with tracking capabilities to evaluate multifocal viral spread after arterial delivery of the vector.82
Further interest in VSV with chimeric genes to enhance its antitumor effect led to the development of a fusogenic VSV expressing the fusion protein of NDV (rVSV-NDV/F). This new vector was superior to wild-type VSV, with enhanced oncolysis due to syncytia formation and resulted in prolonged survival of rats in vivo.83 Continued VSV genome engineering yielded improved vector safety using an MΔ51 deletion in the viral genome. Pursuant to this, the expression of M3, a broad-spectrum and high-affinity chemokine-binding protein from murine γHSV68, enhanced oncolytic potency, resulting in the vector rVSV(MΔ51)-M3.84 VSV-hIFNβ was also built around the cornerstone of achieving enhanced safety by using human IFNβ to protect neighboring normal cells from oncolytic damage.85 This vector is currently undergoing clinical trials.
Other RNA viruses have also been tested in HCC. In 2006, the measles virus Edmonston strain (MV-Edm) entered the scene, due to the establishment of overexpression of CD46 receptors in HCC. CD46 is one of the entry receptors used by MV allowing for cellular entry and syncytia formation. Engineered MV-Edm vectors expressing CEA or human NIS were evaluated in HCC, and demonstrated promising oncolytic potential.86 MV has also been bioengineered with the suicide gene super-cytosine deaminase, which promotes apoptosis-like death that is not dependent on an intact apoptotic pathway.87
NDV is another RNA virus that has been tested in HCC models. An NDV vector harboring an L289A mutation within the F gene resulted in enhanced fusion and cytotoxicity of HCC cells in vitro compared to an rNDV/F3aa control virus.88 Experiments with RNA retroviruses have demonstrated oncolytic effect in HCC. Vectors derived from the murine leukemia virus-based replicating retrovirus vector (RRV) and GALV have been evaluated in HCC. GALV RRV-mediated suicide gene therapy efficiently suppressed HCC tumor growth in vivo, and no detectable RRV signals were observed outside the tumor.89
Cholangiocarcinoma receptors have exhibited a wider level of heterogeneity with regard to receptors for viral entry. One study addressed this issue by modifying the vector with ligands that would better enable integrin-dependent infection. An RGD-fiber modification was used to improve the infectivity and subsequent efficacy of adenoviral vectors in biliary cancers.90 Biliary cancers remain somewhat understudied with regard to OV. Additional studies are needed in this regard to better inform OV approaches in biliary cancers.
Future directions
Despite the continuous evolution of vectors, particularly at the genomic level, and the increasing tumor specificity achieved, it is clear that the overall efficacy of OV therapy needs to be improved. One aspect of significant concern is the delivery method, which needs to be designed in a way to enhance viral bioavailability and viral dissemination throughout the tumor. An excellent delivery tool would provide a shield to immunoneutralization and would also increase virus bystander-killing potency by enhancing tumor-cell infection. Another approach to improve oncolytic efficacy is to enhance the replication capacity of tumor-selective viral strains to achieve better tumor penetration. The challenges observed to date show that the more bioengineering modifications a virus is subjected to, the less potent it becomes in relation to its wild type. There is no genomic manipulation thus far that has been able to increase potency. As such, after safety and tumor specificity have been developed, it is about time for future work to show an increase in killing power of safe vectors.
Conclusion
Oncolytic vectors are very diverse, and their development has evolved to establish tumor selectivity and safety. For cancers with dismal OS rates, virotherapy represents a promising therapeutic platform for further evaluation. A broader array of clinical studies evaluating OV is anticipated in the years to come. In particular, combinations of virotherapies with immunotherapies, such as immunocheckpoint inhibitors and standard cytotoxic chemotherapies, will be of particular interest. More tailored, precision-medicine-informed approaches will likely also constitute an area of future exploration. A number of challenges are yet to be addressed to allow for the realization of the full potential of OV. These include drug delivery of vectors to tumors with sequestration in normal tissues/organs, ability to overcome/avoid host immunoresponse to allow for repeated dosing, and ability to enable systemic administration of vectors to permit broader adaptation of these therapies. The framework to address these challenges and explore the myriad available opportunities has been laid, and as such, a brave new era awaits this burgeoning field.
Disclosure
The authors report no conflicts of interest in this work.
References
US Cancer Statistics Working Group. United States Cancer Statistics: 1999–2014 Incidence and Mortality Web-Based Report. Atlanta: Department of Health and Human Services; 2017. | ||
Liang M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr Cancer Drug Targets. 2018;18(2):171–176. | ||
Heise C, Sampson-Johannes A, Williams A, McCormick F, von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3(6):639–645. | ||
Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. | ||
Surveillance, Epidemiology, and End Results Program. Cancer stat facts: esophageal cancer. 2016. Available from: https://seer.cancer.gov/statfacts/html/esoph.html. Accessed February 21, 2018. | ||
Stiles BM, Bhargava A, Adusumilli PS, et al. The replication-competent oncolytic herpes simplex mutant virus NV1066 is effective in the treatment of esophageal cancer. Surgery. 2003;134(2):357–364. | ||
Wong RJ, Joe JK, Kim SH, Shah JP, Horsburgh B, Fong Y. Oncolytic herpesvirus effectively treats murine squamous cell carcinoma and spreads by natural lymphatics to treat sites of lymphatic metastases. Hum Gene Ther. 2002;13(10):1213–1223. | ||
Stanziale SF, Stiles BM, Bhargava A, Kerns SA, Kalakonda N, Fong Y. Oncolytic herpes simplex virus-1 mutant expressing green fluorescent protein can detect and treat peritoneal cancer. Hum Gene Ther. 2004;15(6):609–618. | ||
Yamada K, Moriyama H, Yasuda H, et al. Modification of the Rb-binding domain of replication-competent adenoviral vector enhances cytotoxicity against human esophageal cancers via NF-κB activity. Hum Gene Ther. 2007;18(5):389–400. | ||
Fu X, Tao L, Zhang X. An HSV-2-based oncolytic virus deleted in the PK domain of the ICP10 gene is a potent inducer of apoptotic death in tumor cells. Gene Ther. 2007;14(16):1218–1225. | ||
Zhang X, Komaki R, Wang L, Fang B, Chang JY. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin Cancer Res. 2008;14(9):2813–2823. | ||
Ma G, Kawamura K, Li Q, et al. Combinatory cytotoxic effects produced by E1B-55kDa-deleted adenoviruses and chemotherapeutic agents are dependent on the agents in esophageal carcinoma. Cancer Gene Ther. 2010;17(11):803–813. | ||
Wong J, Kelly K, Mittra A, et al. A third-generation herpesvirus is effective against gastroesophageal cancer. J Surg Res. 2010;163(2):214–220. | ||
Fujiwara T. A novel molecular therapy using bioengineered adenovirus for human gastrointestinal cancer. Acta Med Okayama. 2011;65(3):151–162. | ||
Zheng H, Li MS, Zhao GQ, Dong ZM. [Effect of CEA gene regulation on the anti-tumor activity of oncolytic adenovirus H101 to esophageal carcinoma]. Zhonghua Zhong Liu Za Zhi. 2011;33(11):822–826. Chinese. | ||
Ma J, Zhao J, Lu J, et al. Coxsackievirus and adenovirus receptor promotes antitumor activity of oncolytic adenovirus H101 in esophageal cancer. Int J Mol Med. 2012;30(6):1403–1409. | ||
Ma J, Li N, Zhao J, et al. Histone deacetylase inhibitor trichostatin A enhances the antitumor effect of the oncolytic adenovirus H101 on esophageal squamous cell carcinoma in vitro and in vivo. Oncol Lett. 2017;13(6):4868–4874. | ||
He D, Sun L, Li C, et al. Anti-tumor effects of an oncolytic adenovirus expressing hemagglutinin-neuraminidase of Newcastle disease virus in vitro and in vivo. Viruses. 2014;6(2):856–874. | ||
Jiang YQ, Zhang Z, Cai HR, Zhou H. Killing effect of TNF-mediated by conditionally replicating adenovirus on esophageal cancer and lung cancer cell lines. Int J Clin Exp Pathol. 2015;8(11):13785–13794. | ||
Surveillance, Epidemiology, and End Results Program. Cancer stat facts: stomach cancer. 2016. Available from: https://seer.cancer.gov/statfacts/html/stomach.html. Accessed December 15, 2017. | ||
Bennett JJ, Delman KA, Burt BM, et al. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene Ther. 2002;9(11):935–945. | ||
Stanziale SF, Petrowsky H, Adusumilli PS, Ben-Porat L, Gonen M, Fong Y. Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activity. Clin Cancer Res. 2004;10(9):3225–3232. | ||
Ono HA, Davydova JG, Adachi Y, et al. Promoter-controlled infectivity-enhanced conditionally replicative adenoviral vectors for the treatment of gastric cancer. J Gastroenterol. 2005;40(1):31–42. | ||
Kangasniemi L, Kiviluoto T, Kanerva A, et al. Infectivity-enhanced adenoviruses deliver efficacy in clinical samples and orthotopic models of disseminated gastric cancer. Clin Cancer Res. 2006;12(10):3137–3144. | ||
Araki Y, Fujiwara H, Inada S, Atsuji K, Yamagishi H. [An antitumor effect of oncolytic adenovirus capable of selectively replicating in CEA-expressing cancer cells and its enhancement by 5-FU]. Gan Kagaku Ryoho. 2006;33(12):1754–1755. Japanese. | ||
Ma J, He X, Wang W, et al. E2F promoter-regulated oncolytic adenovirus with p16 gene induces cell apoptosis and exerts antitumor effect on gastric cancer. Dig Dis Sci. 2009;54(7):1425–1431. | ||
Haley ES, Au GG, Carlton BR, Barry RD, Shafren DR. Regional administration of oncolytic echovirus 1 as a novel therapy for the peritoneal dissemination of gastric cancer. J Mol Med (Berl). 2009;87(4):385–399. | ||
Song KY, Wong J, Gonzalez L, Sheng G, Zamarin D, Fong Y. Antitumor efficacy of viral therapy using genetically engineered Newcastle disease virus (NDV[F3aa]-GFP) for peritoneally disseminated gastric cancer. J Mol Med (Berl). 2010;88(6):589–596. | ||
Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17(12):3351–3362. | ||
Kawaguchi K, Etoh T, Suzuki K, et al. Efficacy of oncolytic reovirus against human gastric cancer with peritoneal metastasis in experimental animal model. Int J Oncol. 2010;37(6):1433–1438. | ||
Tsuji T, Nakamori M, Iwahashi M, et al. An armed oncolytic herpes simplex virus expressing thrombospondin-1 has an enhanced in vivo antitumor effect against human gastric cancer. Int J Cancer. 2013;132(2):485–494. | ||
Liu L, Wu W, Zhu G, et al. Therapeutic efficacy of an hTERT promoter-driven oncolytic adenovirus that expresses apoptin in gastric carcinoma. Int J Mol Med. 2012;30(4):747–754. | ||
Yano S, Tazawa H, Hashimoto Y, et al. A genetically engineered oncolytic adenovirus decoys and lethally traps quiescent cancer stem-like cells in S/G2/M phases. Clin Cancer Res. 2013;19(23):6495–6505. | ||
Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–1808. | ||
Sutterluty H, Chatelain E, Marti A, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1(4):207–214. | ||
Li LX, Zhang YL, Zhou L, et al. Antitumor efficacy of a recombinant adenovirus encoding endostatin combined with an E1B55KD-deficient adenovirus in gastric cancer cells. J Transl Med. 2013;11:257. | ||
O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–285. | ||
Wang W, Ji W, Hu H, et al. Survivin promoter-regulated oncolytic adenovirus with Hsp70 gene exerts effective antitumor efficacy in gastric cancer immunotherapy. Oncotarget. 2014;5(1):150–160. | ||
Jun KH, Gholami S, Song TJ, et al. A novel oncolytic viral therapy and imaging technique for gastric cancer using a genetically engineered vaccinia virus carrying the human sodium iodide symporter. J Exp Clin Cancer Res. 2014;33:2. | ||
Bu XF, Wang MB, Zhang ZJ, Zhao YH, Li M, Yan YL. Autophagy is involved in recombinant Newcastle disease virus (rL-RVG)-induced cell death of stomach adenocarcinoma cells in vitro. Int J Oncol. 2015;47(2):679–689. | ||
Yokoda R, Nagalo BM, Vernon B, et al. Oncolytic virus delivery: from nano-pharmacodynamics to enhanced oncolytic effect. Oncolytic Virother. 2017;6:39–49. | ||
Zhou W, Dai S, Zhu H, et al. Telomerase-specific oncolytic adenovirus expressing TRAIL suppresses peritoneal dissemination of gastric cancer. Gene Ther. 2017;24(4):199–207. | ||
Sui H, Wang K, Xie R, et al. NDV-D90 suppresses growth of gastric cancer and cancer-related vascularization. Oncotarget. 2017;8(21):34516–34524. | ||
Lauer U, Zimmermann M, Beil J, et al. Final results of the first phase I study of intraperitoneal administration of GL-ONC1, a marker gene modified vaccinia virus, in patients with peritoneal carcinomatosis. Z Gastroenterol. 2015;53:KG204. | ||
Chen L, Chen D, Gong M, et al. Concomitant use of Ad5/35 chimeric oncolytic adenovirus with TRAIL gene and taxol produces synergistic cytotoxicity in gastric cancer cells. Cancer Lett. 2009;284(2):141–148. | ||
Surveillance, Epidemiology, and End Results Program. Cancer stat facts: pancreatic cancer. 2016. Available from: https://seer.cancer.gov/statfacts/html/pancreas.html. Accessed December 15, 2017. | ||
Sunamura M, Hamada H, Motoi F, et al. Oncolytic virotherapy as a novel strategy for pancreatic cancer. Pancreas. 2004;28(3):326–329. | ||
Hasegawa N, Abei M, Yokoyama KK, et al. Cyclophosphamide enhances antitumor efficacy of oncolytic adenovirus expressing uracil phosphoribosyltransferase (UPRT) in immunocompetent Syrian hamsters. Int J Cancer. 2013;133(6):1479–1488. | ||
Freytag SO, Barton KN, Brown SL, et al. Replication-competent adenovirus-mediated suicide gene therapy with radiation in a preclinical model of pancreatic cancer. Mol Ther. 2007;15(9):1600–1606. | ||
Jose A, Sobrevals L, Camacho-Sanchez JM, et al. Intraductal delivery of adenoviruses targets pancreatic tumors in transgenic Ela-myc mice and orthotopic xenografts. Oncotarget. 2013;4(1):94–105. | ||
Walter RJ, Attar BM, Rafiq A, Tejaswi S, Delimata M. Newcastle disease virus LaSota strain kills human pancreatic cancer cells in vitro with high selectivity. JOP. 2012;13(1):45–53. | ||
Carew JS, Espitia CM, Zhao W, et al. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013;4:e728. | ||
Kasloff SB, Pizzuto MS, Silic-Benussi M, Pavone S, Ciminale V, Capua I. Oncolytic activity of avian influenza virus in human pancreatic ductal adenocarcinoma cell lines. J Virol. 2014;88(16):9321–9334. | ||
Jose A, Rovira-Rigau M, Luna J, et al. A genetic fiber modification to achieve matrix-metalloprotease-activated infectivity of oncolytic adenovirus. J Control Release. 2014;192:148–156. | ||
Yamamoto Y, Hiraoka N, Goto N, et al. A targeting ligand enhances infectivity and cytotoxicity of an oncolytic adenovirus in human pancreatic cancer tissues. J Control Release. 2014;192:284–293. | ||
Yamamoto Y, Nagasato M, Rin Y, et al. Strong antitumor efficacy of a pancreatic tumor-targeting oncolytic adenovirus for neuroendocrine tumors. Cancer Med. 2017;6(10):2385–2397. | ||
Rodriguez-Garcia A, Gimenez-Alejandre M, Rojas JJ, et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res. 2015;21(6):1406–1418. | ||
Gayral M, Lulka H, Hanoun N, et al. Targeted oncolytic herpes simplex virus type 1 eradicates experimental pancreatic tumors. Hum Gene Ther. 2015;26(2):104–113. | ||
Surveillance, Epidemiology, and End Results Program. Cancer stat facts: liver and intrahepatic bile duct cancer. 2016. Available from: https://seer.cancer.gov/statfacts/html/livibd.html. Accessed December 15, 2017. | ||
Li Y, Yu DC, Chen Y, et al. A hepatocellular carcinoma-specific adenovirus variant, CV890, eliminates distant human liver tumors in combination with doxorubicin. Cancer Res. 2001;61(17):6428–6436. | ||
Li G, Sham J, Yang J, et al. Potent antitumor efficacy of an E1B 55kDa-deficient adenovirus carrying murine endostatin in hepatocellular carcinoma. Int J Cancer. 2005;113(4):640–648. | ||
Zhao HC, Zhang Q, Yang Y, et al. P53-expressing conditionally replicative adenovirus CNHK500-p53 against hepatocellular carcinoma in vitro. World J Gastroenterol. 2007;13(5):683–691. | ||
He G, Lei W, Wang S, et al. Overexpression of tumor suppressor TSLC1 by a survivin-regulated oncolytic adenovirus significantly inhibits hepatocellular carcinoma growth. J Cancer Res Clin Oncol. 2012;138(4):657–670. | ||
Pan QW, Zhong SY, Liu BS, et al. Enhanced sensitivity of hepatocellular carcinoma cells to chemotherapy with a Smac-armed oncolytic adenovirus. Acta Pharmacol Sin. 2007;28(12):1996–2004. | ||
Hsieh JL, Lee CH, Teo ML, et al. Transthyretin-driven oncolytic adenovirus suppresses tumor growth in orthotopic and ascites models of hepatocellular carcinoma. Cancer Sci. 2009;100(3):537–545. | ||
Pin RH, Reinblatt M, Fong Y. Utilizing α-fetoprotein expression to enhance oncolytic viral therapy in hepatocellular carcinoma. Ann Surg. 2004;240(4):659–666. | ||
Foka P, Pourchet A, Hernandez-Alcoceba R, et al. Novel tumour-specific promoters for transcriptional targeting of hepatocellular carcinoma by herpes simplex virus vectors. J Gene Med. 2010;12(12):956–967. | ||
Wang Y, Liu T, Huang P, et al. A novel Golgi protein (GOLPH2)-regulated oncolytic adenovirus exhibits potent antitumor efficacy in hepatocellular carcinoma. Oncotarget. 2015;6(15):13564–13578. | ||
Qian CY, Wang KL, Fang FF, et al. Triple-controlled oncolytic adenovirus expressing melittin to exert inhibitory efficacy on hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8(9):10403–10411. | ||
Jin H, Lv S, Yang J, et al. Use of microRNA Let-7 to control the replication specificity of oncolytic adenovirus in hepatocellular carcinoma cells. PLoS One. 2011;6(7):e21307. | ||
Lou W, Chen Q, Ma L, et al. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J Mol Med (Berl). 2013;91(6):715–725. | ||
Li X, Su Y, Sun B, et al. An artificially designed interfering lncRNA expressed by oncolytic adenovirus competitively consumes oncoMiRs to exert antitumor efficacy in hepatocellular carcinoma. Mol Cancer Ther. 2016;15(7):1436–1451. | ||
Wei RC, Cao X, Gui JH, et al. Augmenting the antitumor effect of TRAIL by SOCS3 with double-regulated replicating oncolytic adenovirus in hepatocellular carcinoma. Hum Gene Ther. 2011;22(9):1109–1119. | ||
Liu L, Li W, Wei X, et al. Potent antitumor activity of oncolytic adenovirus-mediated SOCS1 for hepatocellular carcinoma. Gene Ther. 2013;20(1):84–92. | ||
Li J, Liu H, Li L, et al. The combination of an oxygen-dependent degradation domain-regulated adenovirus expressing the chemokine RANTES/CCL5 and NK-92 cells exerts enhanced antitumor activity in hepatocellular carcinoma. Oncol Rep. 2013;29(3):895–902. | ||
Huang F, Ma B, Wang Y, et al. Targeting gene-virus-mediated manganese superoxide dismutase effectively suppresses tumor growth in hepatocellular carcinoma in vitro and in vivo. Cancer Biother Radiopharm. 2014;29(10):403–411. | ||
Ma B, Wang Y, Zhou X, et al. Synergistic suppression effect on tumor growth of hepatocellular carcinoma by combining oncolytic adenovirus carrying XAF1 with cisplatin. J Cancer Res Clin Oncol. 2015;141(3):419–429. | ||
Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16(9):1637–1642. | ||
Ady JW, Heffner J, Mojica K, et al. Oncolytic immunotherapy using recombinant vaccinia virus GLV-1h68 kills sorafenib-resistant hepatocellular carcinoma efficiently. Surgery. 2014;156(2):263–269. | ||
Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–336. | ||
Ebert O, Shinozaki K, Huang TG, Savontaus MJ, Garcia-Sastre A, Woo SL. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 2003;63(13):3605–3611. | ||
Shinozaki K, Ebert O, Kournioti C, Tai YS, Woo SL. Oncolysis of multifocal hepatocellular carcinoma in the rat liver by hepatic artery infusion of vesicular stomatitis virus. Mol Ther. 2004;9(3):368–376. | ||
Ebert O, Shinozaki K, Kournioti C, Park MS, Garcia-Sastre A, Woo SL. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res. 2004;64(9):3265–3270. | ||
Wu L, Huang TG, Meseck M, et al. rVSV(MΔ51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum Gene Ther. 2008;19(6):635–647. | ||
Jenks N, Myers R, Greiner SM, et al. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-β in rodents and nonhuman primates. Hum Gene Ther. 2010;21(4):451–462. | ||
Blechacz B, Splinter PL, Greiner S, et al. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology. 2006;44(6):1465–1477. | ||
Lampe J, Bossow S, Weiland T, et al. An armed oncolytic measles vaccine virus eliminates human hepatoma cells independently of apoptosis. Gene Ther. 2013;20(11):1033–1041. | ||
Altomonte J, Marozin S, Schmid RM, Ebert O. Engineered Newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol Ther. 2010;18(2):275–284. | ||
Lu YC, Chen YJ, Yu YR, et al. Replicating retroviral vectors for oncolytic virotherapy of experimental hepatocellular carcinoma. Oncol Rep. 2012;28(1):21–26. | ||
Wakayama M, Abei M, Kawashima R, et al. E1A, E1B double-restricted adenovirus with RGD-fiber modification exhibits enhanced oncolysis for CAR-deficient biliary cancers. Clin Cancer Res. 2007;13(10):3043–3050. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.