Back to Archived Journals » Oncolytic Virotherapy » Volume 3
Oncolytic viral therapy: targeting cancer stem cells
Authors Smith T, Roth J, Friedman GK, Gillespie GY ![]()
Received 10 August 2013
Accepted for publication 22 November 2013
Published 7 February 2014 Volume 2014:3 Pages 21—33
DOI https://doi.org/10.2147/OV.S52749
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Tyrel T Smith,1 Justin C Roth,1 Gregory K Friedman,1 G Yancey Gillespie2
1Department of Pediatrics, The University of Alabama at Birmingham, Birmingham, AL, USA; 2Department of Neurosurgery, The University of Alabama at Birmingham, Birmingham, AL, USA
Abstract: Cancer stem cells (CSCs) are defined as rare populations of tumor-initiating cancer cells that are capable of both self-renewal and differentiation. Extensive research is currently underway to develop therapeutics that target CSCs for cancer therapy, due to their critical role in tumorigenesis, as well as their resistance to chemotherapy and radiotherapy. To this end, oncolytic viruses targeting unique CSC markers, signaling pathways, or the pro-tumor CSC niche offer promising potential as CSCs-destroying agents/therapeutics. We provide a summary of existing knowledge on the biology of CSCs, including their markers and their niche thought to comprise the tumor microenvironment, and then we provide a critical analysis of the potential for targeting CSCs with oncolytic viruses, including herpes simplex virus-1, adenovirus, measles virus, reovirus, and vaccinia virus. Specifically, we review current literature regarding first-generation oncolytic viruses with their innate ability to replicate in CSCs, as well as second-generation viruses engineered to enhance the oncolytic effect and CSC-targeting through transgene expression.
Keywords: oncolytic virotherapy, cancer stem cell niche
Introduction to cancer stem cells (CSCs)
Cancer is the second-leading cause of death in the United States; and although overall cancer death rates have declined due to advances in medicine and emergence of novel therapies, one out of four deaths will be due to cancer, with an estimated 1,660,290 new cases projected for 2013.1 Despite aggressive multimodality treatments such as surgical resection, chemotherapy, and radiotherapy, deaths from aggressive cancer still persist, which are in part due to the existence of CSCs. The seminal work of John Dick’s group in 1994 provided evidence of the CSC hierarchal model by demonstrating that human acute myelogenous leukemia (AML) can be regenerated after transplantation of leukemic stem cells possessing CD34+CD38− cell surface markers in non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice.2 This small fraction of cells was more primitive than the rapidly proliferating leukemic cells and capable of initiating AML. This rare subset of cancer cells are grossly indistinguishable from other tumor cells and can elude tumor resection due to their invasive potential and propensity to establish and localize in niches peripheral to the main tumor mass. Furthermore, their “stem-like” features, such as natural quiescence and high-level expression of drug efflux transporters, render them less sensitive to chemotherapy and radiotherapy compared with normal tumor cells.3–6 These forms of therapeutic evasion provide a mechanism by which resistant CSCs are enriched and repopulate the tumor mass with resistant malignant progeny, leading to recurrence of a potentially more aggressive cancer.7–11 The irony is that while conventional therapies produce substantial benefit by destruction of the original tumor, these therapies also select for the most aggressive tumor cells. Thus, it remains crucial to identify novel ways to expand or target therapeutic toxicity to CSCs in order to prevent recurrence and increase survival and quality of life for cancer patients.
CSCs are given their name due to the “stem cell” properties they encompass, such as their ability to self-renew and divide asymmetrically while maintaining dysregulated growth and tumor-initiating potential.5 Under this definition, not all tumor-derived cells are capable of de novo tumor formation. CSCs have been identified in a number of tumor types, including brain,12 breast,13 ovarian,14 colon,15 lung,16 head and neck,17 pancreas,18 liver,19 and a variety of pediatric cancers.20 The most common way CSCs are identified and isolated is through distinct cell surface markers that are differentially present within the tumor cell population. Flow cytometry and magnetic-activated cell separation both offer means of CSC enrichment based on expression of phenotypic markers. Other ways to identify CSCs include expression of various cytoplasmic and nuclear proteins, transcription factors, and/or functional properties ascribed to CSCs such as deoxyribonucleic acid (DNA) label retention, dye efflux properties, or enzymatic metabolism. Because some of these markers are not exclusively limited to CSCs, functionality assays, such as serial transplantation in animal models, are crucial to defining CSCs. Typically, cells isolated from tumors are orthotopically xenografted into immunocompromised mice, and the ability to self-renew is demonstrated by engraftment and tumor formation in secondary recipients. Although in-vivo assays are ideal, they can take up to 6 months to complete, which is why in-vitro assays, including those based on sphere-forming potential, provide a more rapid means of assessing self-renewal and tumor propagation. Although recent reports have highlighted the potential of oncolytic virus approaches for blood cancers, we have restricted the discussion to CSCs from two common solid tumors, gliomas and breast cancer.21 We provide a summary of existing knowledge on the biology of CSCs, including their markers and their niche thought to comprise the tumor microenvironment, and then provide a critical analysis of the potential for targeting CSCs with oncolytic viruses, including herpes simplex virus-1 (HSV), adenovirus (Ad), measles virus (MV), reovirus, and vaccinia virus (VACV).
Mechanisms of CSC resistance to therapy
Many of the stem-like properties embodied by CSCs confer resistance to traditional chemotherapy and radiotherapy. Such properties include relative quiescence, which allows them to be unaffected by drugs that are cytotoxic to highly proliferative cells, such as DNA synthesis inhibitors, alkylating agents, topoisomerase inhibitors, and DNA intercalating drugs.22 Previously, hematopoietic stem cells were identified by their ability to exclude Hoechst 3342 dye, which produced a unique side population when analyzed by flow cytometry.23 Hoechst 3342 dye exclusion is mediated by a class of ATP (adenosine triphosphate)-binding cassette (ABC) transporters known as drug efflux pumps.24 CSCs possess a similar attribute to generate side population from the increased expression of ABC transporters that allow rapid efflux of Hoechst 3342 dye as well as chemotherapeutic agents.24,25 Additionally, CSCs have upregulated anti-apoptotic gene expression and constitutively active signaling pathways involved in cell survival, including PI3K/Akt and Ras pathways.26,27 Lastly, CSCs express a broad range of DNA repair proteins, which protect the cells from cytotoxic DNA damage induced by certain classes of chemotherapy agents.28 In addition to protecting CSCs from standard therapies, these properties may also serve as some of the defining markers for CSC identification and targeting.
Many different markers have been identified that distinguish CSCs from other non-stem tumor cells. There are currently no markers that can be used in isolation that definitively identify CSCs, but the combined use of multiple markers now offers the potential for significant CSC enrichment. The specific panel of markers varies based on the particular tumor type. These markers comprise cell-surface, nuclear, and cytoplasmic proteins, including transcription factors and enzymes. The most commonly used CSC surface marker to date is prominin-1, CD133, which is involved in regulation of MAPK and Akt signaling pathways.29 High CD133 expression in a variety of cancers is associated with poor prognosis, as it correlates with tumor aggressiveness and recurrence.30 Glioma cells acquire stem-like characteristics and upregulation of CD133 positivity in response to hypoxia, which has been linked to glioma initiation, invasion, angiogenesis, and therapeutic resistance.31–33 However, recent studies have identified both CD133-positive and CD133-negative CSC populations and purport CD133 as a marker of more or less invasive CSC subsets.34,35 Many of the markers used to identify other stem cell populations are also found in CSCs. Neuronal stem cell markers, including the nuclear protein nestin and transcription factors Sox-2 and Oct3/4, are similarly expressed on glioma CSCs.28,36 High expression levels of O6-methylguanine-DNA-methyltransferase (MGMT) have been associated with CSCs. MGMT repairs O6-alkyl lesions in DNA, rendering CSCs resistant to DNA alkylating agents such as temozolomide (TMZ).36 However, TMZ therapy can produce acquired resistance by non-MGMT mechanisms, predominantly by altering mitochondrial function.37,38
Breast cancer CSCs have repeatedly shown to be CD44+ and CD24−/low in addition to having increased aldehyde dehydrogenase (ALDH) activity.13,39 CD44 is a prominent cell adhesion marker with a high affinity for hyaluronate, and the molecular profile of cells expressing CD44 is highly associated with genes for self-renewal.40 ALDH expression is important for converting retinol into retinoic acid needed for cell proliferation and survival, but ALDH can also serve as a detoxification enzyme capable of metabolizing chemotherapeutics like cyclophosphamide for improved resistance.40,41 Furthermore, the ABC transporter permeability glycoprotein (P-gp) plays an important role in breast CSC chemoresistance and is consistently elevated in ALDH-expressing CSCs.22,42 These features of CSCs provide resistance to standard therapies; thus, innovative strategies for circumventing these resistance mechanisms should be included in multimodal approaches aimed at eradicating cancer cells as a whole, including the CSC populations responsible for recurrent tumor growth. More detailed understanding of the tumor microenvironment and CSC niche may provide additional markers or unique profiles to further enhance therapeutic targeting.
Microenvironment and the CSC niche
Specialized areas within the tumor microenvironment, specifically hypoxic zones, the perivascular space, and invading tumor front, appear to function as potential CSC niches.6 The established tumor microenvironment supports the maintenance and regulation of the CSC niche and provides an immunosuppressive environment that protects malignant cells from immune effector populations. Not only do CSCs give rise to malignant growth, but they also contribute to the recruitment and growth of non-tumorigenic cells that make up this specialized microenvironment, including the endothelial cells and suppressive immune cells.43,44 Wang et al found that chromosomal aberrations in tumors were also present in glioma-derived endothelium, suggesting the neovasculature can also be of tumor origin.43 Chemokine gradients generated in tumors can also recruit tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells that actively suppress antitumor immunity.3,44,45 CSC markers, such as epithelial cell adhesion molecule, allow CSCs to interact with specific extracellular matrix (ECM) proteins, suggesting that certain ECM profiles are more likely to contain CSCs.46,47 In addition to CSC interactions with the ECM and other extracellular factors, oxygen levels can also contribute to the CSC niche, and allow bidirectional feedback between CSCs and the tumor microenvironment.47,48
Hypoxia is a critical factor in the retention and self-renewal of CSCs in the niche, particularly in neural cancers. Hypoxic zones in tumors develop when tumor cell division outpaces angiogenesis, limiting efficient oxygen exchange. For this reason, hypoxia is frequently associated with necrotic zones in the tumor mass. In these zones specialized oxygen-sensing transcription factors called hypoxia-inducible factors become activated and induce expression of gene products, such as notch proteins, that enhance cell survival and differentiation or chemokines that recruit endothelial progenitors.49,50 Lack of extensive vasculature also protects CSCs in the hypoxic niche, as it limits the distribution of systemically administered chemotherapy. The reduced oxygen levels in solid tumors also confer radioresistance, since oxidative free radicals generated from this procedure illicit much of the toxicity.51 While hypoxia induces transcriptional changes that promote the CSC phenotype, other extracellular factors contribute to establishment of CSC niches in normoxic regions of the tumor.
The perivascular region is important for CSC development, and this zone is also the preferred niche for many non-tumor stem cells, suggesting it is a critical physiological site for maintenance of stem cells, in general. The perivascular region within the tumor contains endothelial cells that express Notch ligands and secrete nitric oxide to stimulate Notch receptors and activate pathways for CSC maintenance.52,53 Bao et al demonstrated that CD133+ glioma xenograft lines (xenolines) secrete increased levels of vascular endothelial growth factor after implantation in nude mice, which stimulated local vascular development and increased tumor incidence.8 The perivascular niche also has a distinct ECM profile favorable to CSC retention and regulation. The perivascular ECM protein laminin interacts with integrin α6 expressed on CSCs and partially drives the CSC phenotype through downstream Akt signaling.54 Matrix metalloproteinases involved in ECM plasticity are also speculated to assist in the regulation of the CSC niche, possibly through ECM architectural rearrangement, vascular development, and tumor metastasis.55
Another likely CSC niche is the invasive front of the tumor zone. The recurrence of tumors near the area of resection is an indication that many CSCs may reside beyond the primary tumor mass in the invading margin. CXCR4 is a stem cell chemokine receptor involved with the chemotactic migration of cells to stromal-derived factor (SDF)-1, which is expressed in many tissues and is concentrated in non-tumor stem cell niches such as bone marrow and lymph nodes.56 SDF-1 is concentrated at tumor invasive margins, and not surprisingly, CXCR4+ CSCs co-localize to these SDF-1-rich sites.3,4 Additionally, the presence of CXCR4 on CSCs is highly correlative to metastatic disease, particularly to organs with increased SDF-1 production.40,56 The bidirectional regulation of the tumor microenvironment provides a pool of repopulating tumor cells maintained by their progenitor CSCs, which makes the CSC niche an enticing target for cancer therapeutics.
Introduction to oncolytic virotherapy
Oncolytic virotherapy offers one such strategy to target CSCs. Oncolytic viruses take advantage of the natural propensity of cancer cells to provide an optimal state for viral replication; some viruses have been attenuated to predominantly replicate and lyse malignant cells while sparing normal tissues, while non-attenuated viruses that do not cause disease in humans can still effectively kill malignant cells in a similar fashion. Additionally, larger viruses can be armed to express immune-stimulating transgenes to recruit an influx of immune effector cells to further augment their therapeutic effect.57,58 After infection of tumor cells, oncolytic virus replication generates progeny that are released by cytolysis to infect nearby tumor cells, amplifying the therapeutic payload throughout the tumor and potentially reaching infiltrating CSCs in their niche at the tumor margin. This method of tumor killing differs from standard methods and presents a unique means of expanding therapeutic potency to drug-resistant CSCs. Furthermore, genetic manipulation of oncolytic viruses offers much more complicated, gene-based therapeutics to be utilized to specifically target cell surface receptors and activated cellular pathways unique to CSCs and to produce transgene products that can disrupt the niche and damage cells via bystander effect. This review will focus on exciting and innovative ways in which oncolytic viruses have been utilized in CSC models for brain and breast cancers.
HSV
HSV is a double-stranded, enveloped DNA virus that has been extensively studied for its ability to lyse tumor cells. The virus is well researched, and the genome is extensively characterized: it does not integrate into the chromosome, it is naturally neurotropic, and antivirals such as intravenous acyclovir can be administered as a safety mechanism to shut off viral replication. Several conditionally replicative herpes viruses have been genetically engineered for the treatment of cancer.59 A common element among HSV used for brain tumor therapy is the deletion of the γ34.5 genes.60 The translated product of γ34.5 is infected cell protein (ICP) 34.5, a neurovirulence protein the herpes virus uses to circumvent shutdown of host protein synthesis.61 HSV transcription in host cells produces double-stranded RNA intermediates that initiate the protein kinase RNA-activated (PKR) response.61 PKR phosphorylates eukaryotic initiation factor 2α (eIF2α), which shuts off translation in the cell, including that of viral proteins.61 The late gene product, ICP34.5, complexes with a phosphatase that dephosphorylates eIF2α in order to prevent translational arrest.61 Deletion of the γ34.5 genes prevents viral replication in healthy cells due to the inability to circumvent the PKR response. Many gliomas have constitutively active MEK that attenuates PKR activity and renders γ34.5-deleted HSV (oncolytic HSV [oHSV]) permissive to replication and cell lysis.62–64 Furthermore, transgenes under constitutive or tissue-specific promoters can be inserted into the deleted γ34.5 regions to express products that may aid in an antitumor bystander effect.65 Several Phase I trials have been conducted with oHSV, G207, and HSV1716, for patients with recurrent glioblastoma multiforme (GBM), revealing no serious adverse events related to the virus and further demonstrating clinical responses resulting from several long-term survivors.66–68
The oHSV, G207, which has been used in clinical high-grade glioma trials, has both copies of γ34.5 deleted with an additional attenuation by the insertion of LacZ in place of the viral gene UL39 encoding the large subunit of ribonucleotide reductase.66,67 Friedman et al first showed that G207 killed CSCs marked by CD133 in human GBM xenolines similarly to non-stem tumor cells.69 Interestingly, they suggest all glioma cells, including CD133+ CSCs, are susceptible to γ34.5-deleted oHSV oncolysis when there is adequate cell receptor (CD111) expression which allows for viral entry.69 If this holds true, the potential impact of oHSV treatment may be predicted from receptor expression levels after tumor biopsy or resection. To determine whether G47Δ, a derivative of G207 that lacks the α47 coding region, placing the US11 gene under an immediate-early promoter, enhanced killing of glioma CSCs, Wakimoto et al compared G207 and G47Δ replication and cell killing in four glioma CSC lines derived from surgical specimens.70 Loss of the α47 gene leads to increased MHC (major histocompatibility complex) class I expression in infected cells because the gene product is responsible for inhibiting the transporter associated with antigen presentation (TAP), and the US11 gene encodes an RNA-binding protein that may protect viral RNA when present at earlier expression.71 All four of the glioma CSC lines exhibited functional characteristics of CSCs, including tumor-initiating potential with as few as 50 cells implanted, CD133 and nestin expression, asymmetric division reproducing cellular heterogeneity, and neurosphere formation during in-vitro culturing.70 Results revealed enhanced glioma CSC killing and viral replication in vitro with G47Δ, indicating early transcription of the US11 gene rescued the attenuated replication of oHSV lacking ICP34.5.70 Furthermore, G47Δ was shown to suppress CSC self-renewal in vitro, confirming CSC susceptibility to oncolytic virotherapy.70
The majority of high-grade malignant gliomas possess low oxygen levels (ranging from 2.5%–0.1%), which can lead to a necrotic tumor core.72 In order to evaluate oHSV with respect to the hypoxic CSC niche, Friedman et al examined the ability of 34.5-deleted virus to infect, replicate, and kill three GBM xenolines in 1% hypoxia and normoxia.73 They showed that hypoxia dramatically increased the CSC fraction, as measured by CD133 expression. The CD133+ CSC fraction was as permissive as CD133− tumor cells to virus replication regardless of oxygen tension. However, infection, replication, and cytotoxicity were diminished in all three xenolines under hypoxia. Considering the critical role of hypoxia in the microenvironment of GBMs, these findings may partially explain the moderate responses seen in oHSV clinical trials and will likely have important implications for future clinical applications of oHSV therapies.
In similar studies, Sgubin et al measured G47Δ viral replication and glioma CSC toxicity under hypoxic conditions, both in vitro and in vivo.72 In these studies, CSCs isolated from primary human GBM specimens that were cultured under hypoxic conditions demonstrated elevated self-renewal and CD133 expression, compared with normoxic conditions, suggesting hypoxic conditions support the stem-like status and increase tumorigenic potential of CSCs.72 In agreement with the findings by Friedman et al, the viral yields were consistently lower in CSCs cultured in hypoxic conditions, but G47Δ was capable of infecting and replicating in CSCs.72 This resulted in decreased numbers of CD133+ cells, suggesting oncolytic viruses could be utilized to counteract CSC enrichment in hypoxic tumors.72 Furthermore, when CSCs were implanted in SCID mice and challenged with G47Δ, hypoxic regions were shown to be disrupted as visualized by hypoxyprobe immunofluorescence when compared with saline-treated controls.72 Overall, CSCs may be infected by oHSV in hypoxia; however, replication and cytotoxicity of some oHSV are not as robust as in normoxic conditions.
To better understand the effects of oHSV in conjunction with current standard of care chemotherapy, Kanai et al investigated the combined use of TMZ with G47Δ in a human glioma CSC model implanted in nude mice.74 Glioma CSC lines were first screened for resistance to TMZ, and resistant lines were found to have elevated MGMT activity, as expected.74 Combination therapy of TMZ and G47Δ in vitro was reported to significantly reduce the viability in CSCs compared with either treatment alone; however, the synergistic effect was only observed in MGMT-negative cells.74 Adding an MGMT inhibitor, O6-benzylguanine (BG), restored the synergistic killing observed in the MGMT-positive CSCs.74 To elucidate the synergistic role of oHSV, DNA damage response proteins were evaluated by immunocytochemistry. These data revealed that oHSV sequesters phosphorylated ataxia telangiectasia mutated (ATM) to replication compartments after infection.74 Thus, the absence of MGMT allowed TMZ-induced strand breaks, and virus-based sequestration of ATM prevented DNA repair via homologous recombination. Mouse studies were then used to evaluate long-term survival after intracranial implants of MGMT-negative CSCs were challenged with TMZ and G47Δ. Eventually, all mice succumbed after implantation with MGMT-positive CSCs; however, administration of BG re-sensitized tumors to combination treatment, extending median survival from 36 to 56 days.74 In this model, it appears that oHSV in CSCs prior to TMZ and BG exposure is required for the synergistic effects. In a similar effort to evaluate whether synergy is achieved with other types of DNA damaging agents, Cheema et al examined the effect of low-dose etoposide and G47Δ in the previously characterized primary xenoline BT74 chosen for its MGMT-positive, etoposide-resistant phenotype.75 In-vivo results provided evidence for apoptosis-induced tumor killing from activated caspase 3 with improved survival in cohorts treated with both agents.75 Although synergistic killing was observed for CSCs in the presence of chemotherapeutics, synergistic killing is only achieved in non-drug-resistant cells or through the combined use of inhibitors that target the drug-resistance mechanism.
CSCs and the tumor microenvironment maintain bidirectional communication to ensure tumor support and immune evasion. One such mechanism for immune evasion is the recruitment and expansion of TAMs and regulatory T cells (Tregs). GBMs are associated with extensive vasculature, which allows the recruitment of TAMs that can limit the spread of oHSV infection by phagocytosis. Oncolytic viruses armed with interleukin (IL)-12 have the ability to skew the immune profile from a tumor-supportive T-helper (Th)-2 response to an antitumor Th-1 response.76 Zhang et al demonstrated the significance of armed oHSV in the treatment of the MGG4 glioma CSC xenoline after implantation in nude mice.77 The unarmed G47Δ virus provided little protection compared with saline-treated controls, whereas G47Δ armed with IL-12 and angiostatin significantly prolonged survival.77 This study suggests that CSC-mediated neovascularization and recruitment of tumor-supportive TAMs can be abrogated with oHSV armed with IL-12/angiostatin. Arming virus with IL-12 and angiostatin will likely increase oHSV efficacy by limiting TAM-associated viral clearance while simultaneously limiting nutrient supplies to malignant cells.77 Since these in-vivo studies were performed in athymic nude mice, the antitumor immunological responses could not be addressed.
To evaluate oHSV therapy in an immunocompetent GBM CSC model, Cheema et al generated mouse CSCs from lentiviral transduction of activated H-Ras and Akt in GFAP (glial fibrillary acidic protein) positive cells from C57BL/6 mice.78 Functionality tests showed the resulting 005 glioma CSC line mimicked human GBM and maintained immune evasion mechanisms, including the recruitment and expansion of Tregs.78 G47Δ armed with IL-12 was utilized in these studies and provided greater therapeutic efficacy compared with the unarmed version of the virus.78 Flow cytometric analyses revealed the armed virus also generated significantly increased interferon-γ and interferon gamma-inducible protein 10 (IP-10) production, which are important for Th-1 responses and angiostatic effects, respectively.78 Although the percentages of T-cell infiltration were not significant, the IL-12-armed virus achieved a significant reduction of Tregs.78 Additionally, depletion of T-cells in the model abrogated the enhanced survival from G47Δ-mIL-12. Therefore, arming viruses with IL-12 has the potential to indirectly target CSCs by disrupting angiogenesis in the perivascular CSC niche. It also interrupts recruitment of TAMs and Tregs that support the immunosuppressive microenvironment. Disrupting the immunosuppressive microenvironment improves T-cell infiltration and mounts a robust immune response against malignant cells, including CSCs.
Since chemotherapy and oHSV toxicity are mediated through separate mechanisms, Zhuang et al evaluated whether doxorubicin could be used to control tumor growth, followed by oHSV treatment as a means to target the resistant CSCs remaining.79 CSCs were isolated from established syngeneic murine 4T1 tumors, based on ALDH expression.79 In ex vivo studies, increased ALDH expression provided resistance to repeated doxorubicin treatment, but the cells remained sensitive to oHSV-mediated toxicity.79 In animal studies, the combined treatment enhanced survival, compared with single-agent controls. Increased CD8+ T-cells were noted in the dual-treated cohort, but limited tumor-specific T-cell response was detected.79 This suggests the therapeutic gains may indeed have been mediated by oHSV-based depletion of the doxorubicin-resistant cells.
Ad
A number of studies have explored the use of oncolytic Ads for CSC targeting and therapy. Ads are double-stranded, non-enveloped viruses that are capable of infecting both dividing and non-dividing cells.80 Their replication is dependent on their ability to promote cell cycle entry into the G1 phase through the viral immediate-early protein E1A binding to retinoblastoma (Rb) protein, which releases active transcriptional factor E2F.80 Two strategies are used to achieve tumor-specific or conditionally replicating Ad (CRAd). Type I CRAds contain deletions in the Rb-binding site of E1A (Δ24), which abrogates viral replication in normal, non-cycling cells that contain functional Rb.80 Type II CRAds place E1A under transcriptional regulation by tumor-specific promoters.80 The viral capsids can also be genetically engineered to target tumor-associated entry receptors rather than entry through the coxsackie-Ad receptor, which is often limited on tumor cells. The Ad, H101 completed a Phase III trial for the treatment of squamous cell carcinoma of the head and neck and esophagus, indicating a 78.8% response rate with virus and combination therapy of cisplatin plus 5-fluorouracil compared with 39.6% response rate with chemotherapy alone.81
To evaluate the oncolytic potency of CRAds for breast cancer, Eriksson et al derived CSCs from metastatic disease in patient pleural effusions. The CSCs expressed CD44+CD24−/low, Oct4, and Sox-2, and were capable of Hoechst 3342 dye exclusion. Among a panel of CRAds, the highest in-vitro efficacy was achieved with Ad5/3-Δ24 and Ad5.pK7-Δ24 derivatives, which are modified for entry through the Ad serotype 3 receptor and heparin sulfate proteoglycans, respectively.82 CD44+CD24−/low CSCs injected into the fat pads of SCID mice were reduced to 11% of the cell population after tumor development, which could be contributed to multi-lineage differentiation of CSCs and subsequent proliferation to form solid tumors.82 Treatment with oncolytic CRAd demonstrated an antitumor effect after three weekly intratumoral injections for 5 weeks.82 Although CSC infection and cell killing were observed in vitro, limited tumor clearance observed in the mouse studies may have been due to asymmetric division that gave rise to both cells permissive to CRAd replication and non-permissive CSCs capable of repopulating the tumors.
To evaluate different tumor-specific promoters for CSC oncolysis, Bauerschmitz et al screened CRAds containing a variety of tumor-specific promoters and capsid configurations for infectivity and replication in CSCs.83 The Ad5/3-multidrug resistance (MDR)-Δ24 and Ad5/3-cyclooxygenase (Cox)-2L-Δ24 CRAds demonstrated the highest oncolytic activity administered at the lowest doses.83 Both of these viruses were created with a 5/3 capsid modification with E1AΔ24 under two different tumor-associated promoters: MDR and Cox2L.83 MDR genes encode energy-dependent pumps like P-gp that decrease the intracellular accumulation of chemotherapeutics, and cyclooxygenase-2 is involved in tumorigenesis and cell survival by stimulating cell growth, invasiveness, and angiogenesis, all similar functions attributed to CSCs.7,9 Again, asymmetrical cell division occurred during tumor development after implanting 100% CD44+CD24−/low CSCs in the fat pads of nude mice. CSC killing from promoter-controlled oncolytic viruses in vivo was suggested after FACS (fluorescence-activated cell sorting) analysis of treated tumors demonstrated a decreased proportion of CSCs compared with mock-treated tumors.83 These data provide support for utilization of CSC-specific promoters for preferential infection and potential eradication of CSCs. However, limiting virus replication to such a small subset of cells may also limit the degree of virus amplified for spread and infection throughout tumor. Indeed, the capsid modifications expand the tropism of the virus, suggesting the much more abundant non-CSC population would rapidly sequester the viral progeny generated.
MV
A case report revealing spontaneous regression of Burkitt’s lymphoma after wild-type measles infection provided some of the first evidence of the oncolytic potential of viruses.84 MV is an antisense, non-integrating RNA virus that induces syncytia formation from viral protein binding and fusion of receptor proteins in neighboring cells. Syncytia formation generates giant multinucleated cells that inevitably undergo apoptosis.85 Oncolytic MV (oMV) seizes the malignant cell’s biosynthetic pathway for the transcription of eight viral genes: nucleocapsid (N), phospho (P), matrix (M), fusion (F), hemagglutinin (H), large (L) proteins, as well as the accessory proteins, C and V.85 Receptor binding occurs through recognition of H with CD46, a ubiquitous complement regulatory protein that protects nucleated cells from complement-mediated lysis and is upregulated on tumor cells. Entry is mediated through downstream interaction with F for membrane fusion.85 The attenuated Edmonton vaccine strain (oMV-Edm) was first isolated from throat washings of a measles patient and was further attenuated from serial passaging in tissue culture. The oMV-Edm strain offers potent antitumor properties with a superb safety profile, as it has been administered to the general public as a vaccine over the years with no reported incidents of severe pathogenicity.85 Tumor selectivity is achieved through increased expression of the entry receptor on tumor cells, as well as the attenuated innate antiviral responses found in malignant cells.85 A Phase I trial of oMV expressing carcinoembryonic antigen for real-time monitoring of viral gene expression in recurrent ovarian cancer resulted in a dose-dependent response to the virus that was well tolerated in patients.86
Allen et al investigated the ability of oMV-Edm to infect and kill glioma CSCs. Among ten different glioma CSC lines attained from surgical specimens, all tested positive for the oMV entry receptor, CD46, and oMV infection revealed syncytia in cells with the hallmark CSC marker, CD133.87 Replication was confirmed in vivo after injection of oMV compared with ultraviolet-inactivated virus in orthotopic CSC implants in nude mice, and prolonged survival was achieved.87 It is important to note that virus injected in CSC-derived tumors can induce syncytia with subsequent spreading to CD133+ glioma CSCs; however, FACS analysis displayed variability in expression levels of CD46 among the glioma CSC xenolines compared with isotype controls.87 This variability in entry receptor expression could explain the difference in survival percentages between the different glioma CSC lines implanted. Enhanced infectivity or syncytia formation of CSCs by oMV must be employed to ensure total tumor eradication.
To preferentially target glioma CSCs, Bach et al genetically fused the oMV H protein to a single-chain antibody (scFV) specific to CD133.88 The CD133-retargeted oMV was selective for CD133 even within close proximity to CD133-negative cells. Although the virus was retargeted to CD133+ cells, oMV spared the CD133+ somatic cells such as hematopoietic stem cells, which is likely due to the mutations in the viral accessory V protein that enhances oMV susceptibility to β-interferon responses.88,89
Friedrich et al revealed an improved way of retargeting oMV using designed ankyrin repeat proteins (DARPins) that exclusively bind the HER2 (human epidermal growth factor receptor 2)/neu tumor receptor.90 Initially, the group compared the oncolytic efficacy of DARPin-targeted, scFV-targeted, and non-targeted oMV to HER2/neu-positive SK-OV-3 subcutaneous xenografts in SCID mice. The DARPin-targeted oMV treated tumors grew slower than tumors treated with scFV-targeted virus; however, the non-targeted virus showed comparable results for delaying tumor progression.90 Importantly, the DARPin-targeted oMV attenuated off-target toxicity.90 The size and lateral position of the binding domains of DARPins offer a means by which bispecific targeting of two separate tumor-associated antigens can be achieved with one infectious viral unit and syncytium.90 The group next chose to engineer a DARPin targeted to HER2/neu and the epithelial cell adhesion molecule CSC marker.90 This was done in an effort to enhance tumor remission by limiting antigen loss due to mutation, transient expression, or general heterogeneity in recurrent tumors.90 Although limitations can arise when conducting experiments solely in vitro, the bispecific DARPin-targeted oMV displayed infection solely in the CHO cell lines stably expressing the intended entry receptors, and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays showed comparable cell killing of SK-OV-3 between the bispecific DARPin virus and monospecific HER2/neu-targeted oMV. Retargeting oMV to a single marker might provide a means of tumor escape; therefore, targeting additional markers, especially receptors involved in CSC niche maintenance, may offer additional protection.
Reovirus
Reovirus (respiratory enteric orphan) is considered an orphan virus due to its ubiquitous nature and absence of severe pathophysiology. In most cases, reovirus infection in the respiratory or gastrointestinal tract is asymptomatic and considered benign; however, in cancer cells, reovirus infection displays severe cytopathic effects and oncolytic potential. Reovirus is a double-stranded RNA virus comprised of 10 segments encoding a single protein with the exception of the bicistronic S1 segment.91 The main contributing factor to tumor-specific viral replication and oncolysis is the presence of oncogenic Ras signaling pathways that attenuate the translational arrest associated with antiviral PKR response.92 Thus, in Ras-activated tumors, reovirus utilizes the cellular translational machinery for viral replication, as demonstrated in the Phase I trial using reolysin to treat recurrent gliomas, where overall survival was 25% at 1 year.93
Taking advantage of natural Ras mutations found in many cancers, Marcato et al utilized reovirus in a CSC breast tumor mouse model and observed tumor remission.27 This group isolated CSCs from core breast tumor samples passaged in SCID mice based on expression of breast CSC markers CD44+CD24− and ALDH-positivity.27 Instead of observing enrichment in breast CSCs commonly observed after chemotherapy treatment, reovirus infection resulted in a decrease in CSC percentages, which coincided with apoptotic cell death.27 Western blot analysis demonstrated Ras activity levels in ALDH+ and CD44+CD24− cells comparable to non-stem tumor cells, which was consistent with reovirus sensitivity.27 A limitation to this study was the inability to compare Ras activation in different breast cancer surgical specimens and their associated CSCs, as reovirus permissiveness is regulated by activation status of Ras signaling pathways.94
VACV
VACV has played a vital role in eradicating smallpox; now the virus is undergoing studies that may lend itself useful in the fight against cancer. VACV is a double-stranded, enveloped DNA virus in the poxvirus family.95 VACV is a non-integrating virus, as the entire replication cycle takes place in the cytoplasm.95 VACV elicits a robust immune response, as evidenced by its use in vaccinations throughout history. Currently, there are several antiviral agents available to limit viral-associated toxicity if complications were to arise from treatment.95 VACV possesses a natural tropism to tumors after systemic administration, which could be due to the combined systemic stability of the virus and the leaky vasculature associated with tumors.95 The oncolytic potential of VACV is verified by efficient tumor infection with rapid replication and cell death, as well as the cytotoxic immune cell response generated in reaction to viral pathogen-associated molecular patterns.95 In a Phase I trial for hepatocellular carcinoma, vaccinia JX-594 expressing granulocyte macrophage-colony stimulating factor demonstrated clinical responses in four out of five patients after intravenous administration of virus.96
Wang et al demonstrated that VACV replicates more efficiently in breast CSCs with increased ALDH activity.97 CSC presence was confirmed in the breast tumor line GI-101A through ALDH activity and resistance to chemotherapy and radiation.97 Further characterization revealed high tumorigenicity with CD44+CD24+ subsets compared with the standard breast CSC markers CD44+CD24−/low.97 When cells were challenged with recombinant VACV strain GLV-1h68 in vitro, replication was statistically greater in ALDH-positive cells compared with the non-CSC ALDH-negative cells.97 Additionally, VACV demonstrated greater replication in the CD24+ subset of ALDH+ cells compared with CD24−/low.97 In a breast cancer model, mice received tumor implants in left and right fat pads with CD44+CD24−/low and CD44+CD24+ cells, respectively, and were then challenged with retro-orbital delivery of VACV.97 Better tumor elimination was observed in the CD24+ subset; however, both left and right breast tumors were generally inhibited after treatment.97 These data suggest VACV can be systemically administered to treat breast cancer and has enhanced replication and oncolysis of CSCs that have a greater tumorigenic profile.
Critical analysis and concluding remarks
Recent advances in cancer therapy have led to higher median survival rates and increased quality of life; however, tumor relapse after multimodality treatments remains a key barrier to long-term survival. Current research has implicated CSCs in chemo- and radio-resistance and tumor progression. Therefore, in addition to standard therapy, including treatments specifically designed to target or expand toxicity to CSCs is essential to eradicate malignant cancers. As research in oncolytic virotherapy advances, scientists have realized the potential for viruses to kill CSCs by exploiting the ability of viruses to kill tumor cells in mechanisms that differ from current chemotherapies (Table 1). Furthermore, the virus type and transgene products expressed via armed viruses may be utilized to destroy CSCs directly or disrupt their niche associated with tumor recurrence and immune evasion.
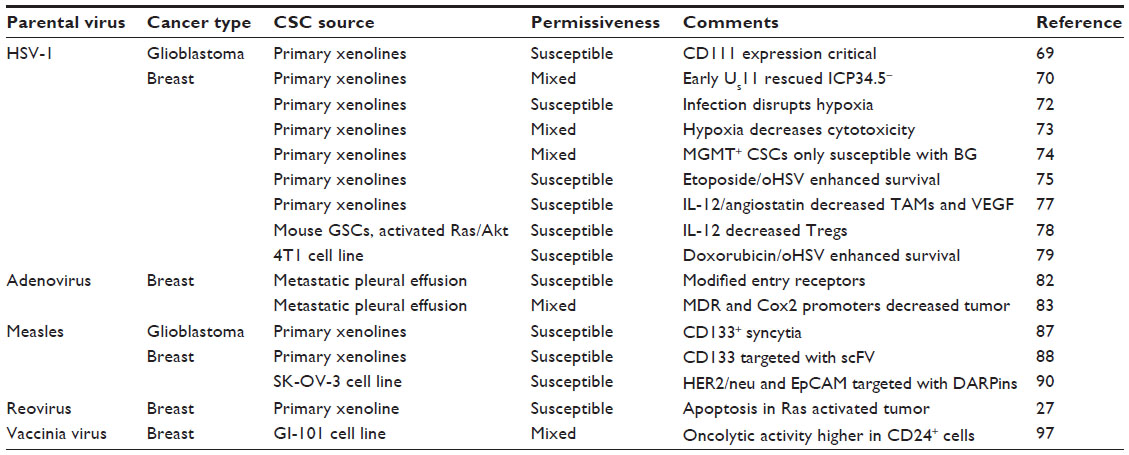 | Table 1 Summary of oncolytic virotherapy studies utilizing cancer stem cell models |
Another term ascribed to CSCs is “cancer initiating cells” due to their ability to generate tumors after inoculation of limiting cell numbers. Only a few CSCs are needed to generate a high-grade tumor, making their eradication essential for combating recurrence. Additionally, metastasis has been linked to CSCs in several accounts; therefore, targeting CSCs early in therapy may prevent cancer spread to secondary organs. Greater efficacy for oncolytic virus-mediated elimination of CSCs may be achieved through broadening the permissiveness of viral replication to include CSCs, rather than restricting viral entry to CSC-specific markers. This may achieve increased viral loads for better distribution and potential for reaching the rare CSC population. Modeling tumorigenesis by implantation of CSCs isolated based on specific tumor markers may overlook the natural heterogeneity of malignant cancers. Most of the existing models are also limited by the use of immune deficient mice, as the immune response, particularly in the setting of oncolytic viruses, is likely to play a critical role in both virus amplification and tumor regression. Next-generation viruses are being developed to enhance direct targeting of CSCs and indirect targeting of the niche through the localized production of small molecules.
The tumor microenvironment represents an ideal indirect target for manipulation of CSC activity and function. The microenvironment offers a source of blood supply and nutrients for the growing tumor and facilitates bidirectional communication with CSCs. This highly immunosuppressive microenvironment allows tumor immune evasion, and high frequency of TAMs is associated with increased viral clearance.98,99 Flipping the “immunological switch” to a Th-1-directed antitumor response may offer an advantage by inhibiting tumor vasculature formation and initiating tumor clearance by cytotoxic T-cells or natural killer cells. While immune stimulation may enhance viral clearance, antiviral immune responses will be directed at infected tumor cells and may promote antitumor immunity.100,101
Physical and physiological barriers within the tumor microenvironment can also limit viral spread to CSCs. Necrotic masses at tumor cores do not support viral replication; therefore, strategic injection sites must be considered to maximize infection after intratumoral administration. Administration of virus into prominent CSC niche locations may increase the probability of CSC infection and subsequent elimination. In this regard, systemic delivery of virus within tumor vasculature may offer better penetration and targeting of CSCs in the perivascular niche. Abnormal vessels and lack of lymphatics in tumors generates enhanced permeability and retention (EPR) effects responsible for accumulation of drugs and nanoparticles in tumor tissues.102 A similar situation may arise after intravenous injection of virus, in which case the EPR effect may serve a valuable function. However, the increased antibody concentrations and immune cell infiltration associated with the EPR effect may also limit viral spread. Most oncolytic viruses have demonstrated selectivity for tumor cell-derived endothelial cells, which suggests that intravascular administration of oncolytic viruses may also serve to destroy tumor support cells derived from CSCs.103 Among invasive tumors, CSCs are often concentrated in satellite sites (micro-metastases) that are surrounded by healthy tissues. In this case, viral entry or sequestration in healthy tissues limits the effective load available to spread and infect nearby metastases. Carrier cell delivery may provide a means by which the virus can spread among invasive tumor satellites, particularly if the carrier cells express matrix metalloproteinases, in which case the degraded tumor ECM may also improve viral dissemination. Lastly, hypoxia has been described to negatively affect first-generation oHSV replication and may have a similar affect on other viruses.73 In order to target hypoxic fractions of solid tumors, Post et al created a CRAd with a hypoxia-dependent promoter driving virus replication, creating a setting where hypoxia becomes the driving force for efficacy instead of limiting viral spread.104 Since CSCs are rare, targeting the cells encompassing or supporting the CSC niche may be a more effective means of CSC elimination.
Among the viruses discussed, each is associated with specific benefits and limitations, which are based on virus size, tropism, or prevalence of preexisting immunity. Larger viruses like herpes and Ad are more amenable to insertion of transgenes, which allows the virus to be armed with gene-based therapeutics that synergize with the oncolytic activity or augment the overall anticancer effect. Tropism plays an integral role in oncolytic therapies. Viruses that display a wide range of tropism, such as vaccinia, can be used for the treatment of multiple cancer types. However, viruses that utilize ubiquitously expressed receptors are also likely to be more prone to uptake and sequestration in non-tumor tissues. Recombinant Ads have been made to overcome the absence of their primary receptor (coxsackie-Ad receptor) on cancer cells by viral fiber modification and serotype switching.105 Similarly, the measles entry ligand has been modified to overcome the preferential tropism to cells expressing CD46. In other cases, the virus’s natural tropism may provide advantage for the treatment of tissue-specific malignancies, such as neurotropic herpes for the treatment of gliomas. Some viruses may only be effective when tumors possess certain oncogenic profiles, such as reovirus dependence on an activated Ras pathway for efficient replication in tumors.92 Finally, preexisting immunity to certain viruses, such as vaccinia, Ad, and herpes, may limit the efficacy of viral therapy. In this regard, neutralizing antibodies and phagocytic cells have the potential to eradicate the virus before it can destroy the tumor. Alternatively, the potent immune responses generated from viral infections may also stimulate anticancer immunity by disrupting immunosuppressive mechanisms active in the tumor microenvironment. In the future, virus selection for cancer therapy may become another component of personalized medicine based on a patient’s preexisting immunity and tumor type.
Oncolytic viruses have demonstrated an outstanding potential in targeting CSCs and preventing tumor relapse. Next-generation viruses can be utilized to target entry receptors specific to tumors and CSCs, conditionally replicate in malignant cells, and disrupt the CSC niche from localized production of immune-stimulating cytokines or small molecules toxic to CSCs and their progeny. Furthermore, viruses may be successfully used as an adjuvant with traditional chemotherapeutics and radiotherapy or with other novel targeted agents, including other viruses with different mechanisms of killing. As researchers continue to dissect out the specific mechanisms of CSC maintenance, tumorigenicity, and resistance, new viruses can be developed to further limit CSC function and thereby improve outcomes.
Acknowledgments
This work was supported in part by a USPHS NIH grant from the NCI (P01 CA071933) and by grants from private funders, St. Baldrick’s Foundation, Rally Foundation and Hyundai Hope on Wheels Foundation.
Disclosure
GYG is a stockholder, co-founder, and consultant for Catherex, Inc., which has licensed an oHSV (M032) from the University of Alabama at Birmingham. GYG holds partial intellectual property rights to M032 that expresses human interleukin-12. The authors report no other conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. | |
Wang S-C, Hong J-H, Hsueh C, Chiang C-S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab Invest. 2012;92(1):151–162. | |
Gelmini S, Mangoni M, Serio M, Romagnani P, Lazzeri E. The critical role of SDF-1/CXCR4 axis in cancer and cancer stem cells metastasis. J Endocrinol Invest. 2008;31(9):809–819. | |
Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12(2):133–143. | |
Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Stem Cell. 2011;8(5):482–485. | |
Labialle S, Gayet L, Marthinet E, Rigal D, Baggetto LG. Transcriptional regulators of the human multidrug resistance 1 gene: recent views. Biochemical Pharmacology. 2002;64(5–6):943–948. | |
Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66(16):7843–7848. | |
Rizzo MT. Cyclooxygenase-2 in oncogenesis. Clinica Chimica Acta. 2011;412(9–10):671–687. | |
Eramo A, Ricci-Vitiani L, Zeuner A, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13(7):1238–1241. | |
Chuthapisith S, Eremin J, El-Sheemey M, Eremin O. Breast cancer chemoresistance: emerging importance of cancer stem cells. Surg Oncol. 2010;19(1):27–32. | |
Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. | |
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | |
Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Natl Acad Sci U S A. 2011;108(16):6468–6473. | |
Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–115. | |
Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15(3):504–514. | |
Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104(3):973–978. | |
Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. | |
Friedman GK, Yancey Gillespie G. Cancer stem cells and pediatric solid tumors. Cancers. 2011;3(4):298–318. | |
Friedman GK, Cassady KA, Beierle EA, Markert JM, Gillespie GY. Targeting pediatric cancer stem cells with oncolytic virotherapy. Pediatr Res. 2012;71(4 Pt 2):500–510. | |
Rahman MM, Madlambayan GJ, Cogle CR, McFadden G. Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with Myxoma virus. Cytokine Growth Factor Rev. 2010;21(2–3):169–175. | |
Raguz S, Yagüe E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer. 2008;99(3):387–391. | |
Goodell MA, Rosenzweig M, Kim H, et al. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3(12):1337–1345. | |
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. | |
Wan F, Zhang S, Xie R, et al. The utility and limitations of neurosphere assay, CD133 immunophenotyping and side population assay in glioma stem cell research. Brain Pathol. 2010;20(5):877–889. | |
Kanai R, Wakimoto H, Martuza RL, Rabkin SD. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17(11):3686–3696. | |
Marcato P, Dean CA, Giacomantonio CA, Lee PWK. Oncolytic reovirus effectively targets breast cancer stem cells. Mol Ther. 2009;17(6):972–979. | |
Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. | |
Takenobu H, Shimozato O, Nakamura T, et al. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene. 2010;30(1):97–105. | |
Zeppernick F, Ahmadi R, Campos B, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14(1):123–129. | |
Platet N, Liu SY, Atifi ME, et al. Influence of oxygen tension on CD133 phenotype in human glioma cell cultures. Cancer Lett. 2007;258(2):286–290. | |
Griguer CE, Oliva CR, Gobin E, et al. CD133 is a marker of bioenergetic stress in human glioma. PLoS One. 2008;3(11):e3655. | |
Jensen RL. Brain tumor hypoxia: tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J Neurooncol. 2009;92(3):317–335. | |
Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67(9):4010–4015. | |
Wang J, Sakariassen PO, Tsinkalovsky O, et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer. 2008;122(4):761–768. | |
Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. | |
Oliva CR, Moellering DR, Gillespie GY, Griguer CE. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS One. 2011;6(9):e24665. | |
Griguer CE, Cantor AB, Fathallah-Shaykh HM, et al. Prognostic Relevance of Cytochrome c Oxidase in Primary Glioblastoma Multiforme. PLoS One. 2013;8(4):e61035. | |
Chuthapisith S, Eremin J, El-Sheemey M, Eremin O. Breast cancer chemoresistance: emerging importance of cancer stem cells. Surg Oncol. 2010;19(1):27–32. | |
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. | |
Sreerama L, Sladek NE. Cellular levels of class 1 and class 3 aldehyde dehydrogenases and certain other drug-metabolizing enzymes in human breast malignancies. Clin Cancer Res. 1997;3(11):1901–1914. | |
Bao L, Haque A, Jackson K, et al. Increased expression of P-glycoprotein is associated with doxorubicin chemoresistance in the metastatic 4T1 breast cancer model. Am J Pathol. 2011;178(2):838–852. | |
Wang R, Chadalavada K, Wilshire J, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468(7325):829–833. | |
Beier CP, Kumar P, Meyer K, et al. The cancer stem cell subtype determines immune infiltration of glioblastoma. Stem Cells Dev. 2012;21(15):2753–2761. | |
Raychaudhuri B, Rayman P, Ireland J, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13(6):591–599. | |
Gires O, Klein CA, Baeuerle PA. On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer. 2009;9(2):143; author reply 143. | |
Sneddon JB, Werb Z. Location, location, location: the cancer stem cell niche. Cell Stem Cell. 2007;1(6):607–611. | |
Yang Z-J, Wechsler-Reya RJ. Hit ‘em where they live: targeting the cancer stem cell niche. Cancer Cell. 2007;11(1):3–5. | |
Zhang S, Luo X, Wan F, Lei T. The roles of hypoxia-inducible factors in regulating neural stem cells migration to glioma stem cells and determinating their fates. Neurochem Res. 2012;37(12):2659–2666. | |
Gustafsson MV, Zheng X, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9(5):617–628. | |
Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. 2007;25(26):4066–4074. | |
Fan X, Khaki L, Zhu TS, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28(1):5–16. | |
Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates Notch signaling and promotes stem-like character in PDGF-induced glioma cells. Stem Cell. 2010;6(2):141–152. | |
Lathia JD, Gallagher J, Heddleston JM, et al. Integrin alpha 6 regulates glioblastoma stem cells. Stem Cell. 2010;6(5):421–432. | |
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. | |
Kucia M, Reca R, Miekus K, et al. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells. 2005;23(7):879–894. | |
Gaston DC, Whitley RJ, Parker JN. Engineered herpes simplex virus vectors for antitumor therapy and vaccine delivery. Future Virol. 2011;6(3):321–340. | |
Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9(1):64–71. | |
Liu T-C, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Prac Oncol. 2007;4(2):101–117. | |
Parker JN, Bauer DF, Cody JJ, Markert JM. Oncolytic viral therapy of malignant glioma. Neurotherapeutics. 2009;6(3):558–569. | |
He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1997;94(3):843–848. | |
Farassati F, Lee PWK. Ras signalling pathway: a gateway for HSV-1 infection. Scientific World Journal. 2003;3:533–535. | |
Pan W, Bodempudi V, Esfandyari T, Farassati F. Utilizing ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS One. 2009;4(8):e6514. | |
Smith KD, Mezhir JJ, Bickenbach K, et al. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)134.5 mutants of herpes simplex virus 1. J Virol. 2006;80(3):1110–1120. | |
Parker J, Pfister L, Quenelle D, et al. Genetically engineered herpes simplex viruses that express IL-12 or GM-CSF as vaccine candidates. Vaccine. 2006;24(10):1644–1652. | |
Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867–874. | |
Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17(1):199–207. | |
Harrow S, Papanastassiou V, Harland J, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004;11(22):1648–1658. | |
Friedman GK, Langford CP, Coleman JM, et al. Engineered herpes simplex viruses efficiently infect and kill CD133+ human glioma xenograft cells that express CD111. J Neurooncol. 2009;95(2):199–209. | |
Wakimoto H, Kesari S, Farrell CJ, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69(8):3472–3481. | |
Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A. 2001;98(11):6396–6401. | |
Sgubin D, Wakimoto H, Kanai R, Rabkin SD, Martuza RL. Oncolytic herpes simplex virus counteracts the hypoxia-induced modulation of glioblastoma stem-like cells. Stem Cells Transl Med. 2012;1(4):322–332. | |
Friedman GK, Haas MC, Kelly VM, Markert JM, Gillespie GY, Cassady KA. Hypoxia moderates γ(1)34.5-deleted herpes simplex virus oncolytic activity in human glioma xenoline primary cultures. Transl Oncol. 2012;5(3):200–207. | |
Kanai R, Rabkin SD, Yip S, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104(1):42–55. | |
Cheema TA, Kanai R, Kim GW, et al. Enhanced antitumor efficacy of low-dose Etoposide with oncolytic herpes simplex virus in human glioblastoma stem cell xenografts. Clin Cancer Res. 2011;17(23):7383–7393. | |
Kilinc MO, Aulakh KS, Nair RE, et al. Reversing tumor immune suppression with intratumoral IL-12: activation of tumor-associated T effector/memory cells, induction of T suppressor apoptosis, and infiltration of CD8+ T effectors. J Immunol. 2006;177(10):6962–6973. | |
Zhang W, Fulci G, Wakimoto H, et al. Combination of oncolytic herpes simplex viruses armed with angiostatin and IL-12 enhances antitumor efficacy in human glioblastoma models. Neoplasia. 2013;15(6):591–599. | |
Cheema TA, Wakimoto H, Fecci PE, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110(29):12006–12011. | |
Zhuang X, Zhang W, Chen Y, et al. Doxorubicin-enriched, ALDH(br) mouse breast cancer stem cells are treatable to oncolytic herpes simplex virus type 1. BMC Cancer. 2012;12:549. | |
Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy. Nat Biotechnol. 2000;18(7):723–727. | |
Xia Z-J, Chang J-H, Zhang L, et al. [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus]. Ai Zheng. 2004;23(12):1666–1670. Chinese. | |
Eriksson M, Guse K, Bauerschmitz G, et al. Oncolytic adenoviruses kill breast cancer initiating CD44+CD24−/low cells. Mol Ther. 2007;15(12):2088–2093. | |
Bauerschmitz GJ, Ranki T, Kangasniemi L, et al. Tissue-specific promoters active in CD44+CD24−/low breast cancer cells. Cancer Res. 2008;68(14):5533–5539. | |
Bluming AZ, Ziegler JL. Regression of Burkitt’s lymphoma in association with measles infection. Lancet. 1971;2(7715):105–106. | |
Msaouel P, Iankov ID, Dispenzieri A, Galanis E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr Pharm Biotechnol. 2012;13(9):1732–1741. | |
Galanis E, Hartmann LC, Cliby WA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70(3):875–882. | |
Allen C, Opyrchal M, Aderca I, et al. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Ther. 2013;20(4):444–449. | |
Bach P, Abel T, Hoffmann C, et al. Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus. Cancer Res. 2013;73(2):865–874. | |
Devaux P, Hodge G, McChesney MB, Cattaneo R. Attenuation of V- or C-defective measles viruses: infection control by the inflammatory and interferon responses of rhesus monkeys. J Virol. 2008;82(11):5359–5367. | |
Friedrich K, Hanauer JR, Prüfer S, et al. DARPin-targeting of measles virus: unique bispecificity, effective oncolysis, and enhanced safety. Mol Ther. 2013;21(4):849–859. | |
Kim M, Chung Y-H, Johnston RN. Reovirus and tumor oncolysis. J Microbiol. 2007;45(3):187–192. | |
Norman KL, Hirasawa K, Yang A-D, Shields MA, Lee PWK. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci U S A. 2004;101(30):11099–11104. | |
Forsyth P, Roldan G, George D, et al. A Phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16(3):627–632. | |
Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17(12):3351–3362. | |
Guse K, Cerullo V, Hemminki A. Oncolytic vaccinia virus for the treatment of cancer. Expert Opin Biol Ther. 2011;11(5):595–608. | |
Park B-H, Hwang T, Liu T-C, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533–542. | |
Wang H, Chen NG, Minev BR, Szalay AA. Oncolytic vaccinia virus GLV-1h68 strain shows enhanced replication in human breast cancer stem-like cells in comparison to breast cancer cells. J Transl Med. 2012;10:167. | |
Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res. 2006;66(4):2314–2319. | |
Fulci G, Dmitrieva N, Gianni D, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67(19):9398–9406. | |
Hirohashi Y, Torigoe T, Inoda S, et al. Immune response against tumor antigens expressed on human cancer stem-like cells/tumor-initiating cells. Future Oncol. 2010;6(5):717–732. | |
Hirohashi Y, Torigoe T, Inoda S, Morita R, Kochin V, Sato N. Cytotoxic T lymphocytes: sniping cancer stem cells. Oncoimmunology. 2012;1(1):123–125. | |
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | |
Wojton J, Kaur B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010;21(2–3):127–134. | |
Post DE, Van Meir EG. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene. 2003;22(14):2065–2072. | |
Cripe TP, Dunphy EJ, Holub AD, et al. Fiber knob modifications overcome low, heterogeneous expression of the coxsackievirus-adenovirus receptor that limits adenovirus gene transfer and oncolysis for human rhabdomyosarcoma cells. Cancer Res. 2001;61(7):2953–2960. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.