Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 8
Olaparib in the management of ovarian cancer
Received 29 April 2015
Accepted for publication 22 June 2015
Published 7 August 2015 Volume 2015:8 Pages 127—135
DOI https://doi.org/10.2147/PGPM.S62809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin Bluth
Kristin Bixel,1 John L Hays2
1Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, 2Department of Hematology Oncology, Ohio State University, Columbus, OH, USA
Abstract: Alterations in the homologous repair pathway are thought to occur in 30%–50% of epithelial ovarian cancers. Cells deficient in homologous recombination rely on alternative pathways for DNA repair in order to survive, thereby providing a potential target for therapy. Olaparib, a poly(ADP-ribose) polymerase (PARP) inhibitor, capitalizes on this concept and is the first drug in its class approved for patients with ovarian cancer. This review article will provide an overview of the BRCA genes and homologous recombination, the role of PARP in DNA repair and the biological rationale for the use of PARP inhibitors as cancer therapy, and ultimately will focus on the use of olaparib in the management of ovarian cancer.
Keywords: olaparib, ovarian cancer, PARP inhibitor
Introduction
Ovarian cancer is the most common cause of gynecologic cancer death and the fifth leading cause of death from cancer in women.1 It was estimated that there were 21,980 new cases of ovarian cancer and 14,270 deaths from ovarian cancer in the US in 2014.1 Aggressive surgical cytoreduction followed by platinum- and taxane-based chemotherapy remains the standard of care for patients with epithelial ovarian cancer. At this time, maintenance therapy is not recommended after first-line treatment.2 While most will initially respond to the therapy, up to 75%–80% of women with advanced ovarian cancer will experience tumor progression or recurrence. The choice of chemotherapy for recurrent ovarian cancer is guided by the treatment-free interval, which has been shown to predict response to subsequent chemotherapy.3–6 While the majority of patients will have platinum-sensitive disease at the time of initial relapse, nearly all patients with recurrent disease will develop chemoresistance.3 This highlights the need for improved treatment strategies for the management of advanced ovarian cancer.
Improved understanding of tumor biology has led to the development of targeted molecular therapies, several of which have been tested in ovarian cancer. In the genomic landscape of ovarian cancer, very few genes aside from p53 have been shown to be modulated in a significant number of patients.7 However, alterations in the homologous recombination (HR) pathway have been postulated to be associated with ~30%–50% of ovarian carcinomas.7,8 Cells that are deficient in HR rely on alternative pathways for DNA repair in order to survive thereby providing a potential target for therapy. Poly(ADP-ribose) polymerase (PARP) inhibitors capitalize on this concept. Multiple PARP inhibitors (PARPis) have or are currently being clinically investigated in ovarian cancer. Recently, the PARPi olaparib (Lynparza, formerly known as AZD2281), was granted accelerated approval by the US Food and Drug Administration (FDA) as a therapy for ovarian cancer in patients with germline BRCA mutations who have received three or more prior lines of chemotherapy. Here, we will discuss the biologic rationale for the use of PARPis and review the use of olaparib in the management of ovarian cancer.
BRCA and homologous recombination
Cellular DNA is constantly subjected to damage and it requires several coordinated repair pathways in order to maintain genomic integrity.9 At least six primary pathways of DNA repair have been identified and are used variably to address DNA break damage.10 Base excision repair (BER), nucleotide excision repair, mismatch repair, and translesional synthesis are used to identify and repair single-stranded DNA breaks.10 HR and nonhomologous end joining are repair mechanisms for double-stranded DNA breaks, the former is a high-fidelity system, and the latter is more error prone.10 Mutations in any of these DNA repair pathways can predispose cells to malignant transformation and are the hallmarks of some hereditary cancer syndromes.11,12
Germline mutations in BRCA1 and BRCA2 account for the majority of inherited breast and ovarian cancers.13 These mutations are inherited in an autosomal dominant fashion with high penetrance and are associated with a 50%–85% lifetime risk of breast cancer and a 15%–40% risk of ovarian cancer.14 In addition to germline mutations, somatic mutations of BRCA1 and BRCA2 as well as epigenetic silencing of BRCA1 may yield tumors that are predicted to behave like BRCA-deficient tumors despite their normal germline BRCA genes.15,16 The protein products of BRCA1 and BRCA2 play an essential role in the cellular response to DNA double-strand break repair through HR.17–22 Cells with nonfunctional or deficient BRCA1/2 proteins are unable to localize the DNA recombinase RAD51 to damaged DNA and therefore are unable to perform HR efficiently.22,23 Subsequently, those cells are then forced to use an alternative, error-prone DNA repair mechanisms, such as nonhomologous end joining, and are therefore subject to accumulation of DNA damage, genetic instability, and subsequent tumorigenesis or cell death secondary to excessive DNA damage.
While mutations in DNA repair pathways predispose cells to malignant transformation, they also impart vulnerabilities that may increase susceptibility to certain cancer therapies.23,24 It has been reported that HR-deficient cells are highly sensitive to platinum chemotherapeutic agents as they are less likely to repair the DNA damage caused by platinum adducts.25,26 This translates clinically as women with BRCA-associated ovarian carcinoma have been shown to have a better response to platinum chemotherapy.26,27 Despite this, the majority still experience recurrence and ultimately succumb to their disease, which led to the question: is there a way to target the HR deficiency in BRCA mutation carriers or BRCA-like tumors? Theoretically, inhibition of complimentary DNA repair pathways may be selectively cytotoxic to cells deficient in HR.
PARP inhibition
The PARP family consists of a number of proteins encoded by different genes that contain a conserved catalytic domain.28 A true PARP can transfer the first ADP-ribose moiety from nicotinamide adenine dinucleotide (NAD+) to an accepter protein and can sequentially add multiple ADP-ribose units to the preceding ones to form poly(ADP-ribose) chains (PAR chains).28 While there are several members of this family, PARP1 and PARP2 appear to play a significant role in DNA damage repair. PARP1, the most well studied member of the family, is an abundant nuclear protein that detects and binds DNA nicks or breaks through its N-terminal zinc finger motifs.29 After DNA binding, the activation of the catalytic domain hydrolyzes oxidized NAD+ to produce linear and branched PAR chains on itself and other proteins.29–31 Activation of PARP1 has been implicated in several distinct DNA repair pathways including BER.30,32–34 The addition of PAR moieties to PARP1 and surrounding proteins serves multiple functions including the recruitment and activation of several DNA repair factors.30,35 Additionally, the formation of PAR chains diminishes the affinity of PARP1 for DNA allowing PARP1 to be removed from damaged DNA allowing for subsequent repair.30 Finally, in conditions that cause excessive DNA damage, PARP1 hyperactivation produces PAR chains, at the expense of cellular NAD+ and ATP, which become depleted leading to cell death by necrosis or parthanatos.30,36,37 Additional studies have shown that PARP2 can be activated by DNA damage but is thought to be responsible for only a small portion of the PAR synthesis stimulated by DNA strand breaks.30,38
The development of PARPis for use in cancer therapy has taken two different strategic approaches.30 The anticancer activity of many chemotherapeutic agents relies on the cytotoxic consequences of DNA damage.39 Thus, the role of PARP in DNA damage repair has led to interest in the use of PARPis as chemo-sensitizers in cancer therapy.30 The second approach capitalizes on the concept of synthetic lethality.40 Synthetic lethality refers to a situation where a defect in one gene or protein is compatible with cell viability but results in cell death when combined with another gene or protein defect.23 Cells deficient in HR have been shown to be sensitive to the inhibition of BER by PARPis, with resulting chromosome instability, cell cycle arrest, and subsequent apoptosis.20
Olaparib, in addition to the other small molecule PARPis, appears to have multiple modes of action (Figure 1). Current small molecule inhibitors compete with NAD+ binding, impairing the ability of PARP to produce PAR chains.30,41 Olaparib is an inhibitor of PARP1, PARP2, and PARP3.42 Inhibition of enzymatic activity results in the inability to recruit the appropriate DNA repair factors to the site of DNA damage, resulting in the accumulation of single strand breaks and ultimately the formation of double strand breaks secondary to the stalling and collapsing of replication forks.43,44 If the synthetic lethality of PARPi in HR-deficient cells is based solely on catalytic inhibition, one would expect that PARP deletion would have a similar effect. Horton et al, however, demonstrated that Parp1wt mouse fibroblasts are more sensitive to a DNA damaging agent when also treated with a PARPi than Parp1−/− mouse fibroblasts treated with the same DNA damaging agent, suggesting its function goes beyond enzymatic inhibition.45 Murai et al found that olaparib-induced chromatin binding/trapping of PARP1 and PARP-2 in the presence of a DNA damaging agent that could be reversed with the removal of the drug. Stablilization of the PARP–DNA complex resulted in significant cytotoxicity beyond that caused by unrepaired SSBs caused by PARP inactivation.41 The concentrations required to readily detect PARP–DNA complexes (<10 μmol/L) are well below the peak concentration of olaparib (24 μmol/L) in clinical trials.41,46 With a better understanding of the multiple possible mechanisms on the action of olaparib, it is possible to more rationally design clinical trials with cytotoxic agents or other targeted agents.
 | Figure 1 Dual mechanism of action of PARPi. |
Olaparib in the management of ovarian cancer
Monotherapy
Given the compelling preclinical data demonstrating selective targeting of BRCA deficient cells by PARPis, clinical trials using olaparib were initiated. In a Phase I single-agent dose escalation study, the maximum tolerated dose was 400 mg by mouth (PO) twice daily (capsule formulation). This study demonstrated a 47% overall response rate (ORR) and a 63% clinical benefit rate (CBR) in patients with BRCA-associated breast, ovarian, or prostate cancers.46 Additionally, an expansion cohort of ovarian cancer patients with germline BRCA mutations were treated with olaparib monotherapy at a dose of 200 mg bid. Forty percent of patients demonstrated a response as defined by the Response Evaluation Criteria in Solid Tumors (RECIST) or a decline in CA125 (a tumor marker that is commonly elevated in advanced epithelial ovarian cancer and is frequently followed during treatment and posttreatment surveillance). Notably, those with platinum-sensitive disease had a greater response to olaparib as compared to patients with platinum-resistant or platinum-refractory disease.47
Monotherapy with olaparib for the treatment of recurrent ovarian cancer has now been studied in several Phase II trials (Table 1). Audeh et al provided evidence of a dose–response relationship with olaparib in patients with germline BRCA mutations and recurrent ovarian cancer. This Phase II study included two nonrandomized sequentially enrolled cohorts who received either 400 mg or 100 mg of olaparib bid. Patients receiving olaparib 400 mg bid had a response rate of 33% in comparison to 13% in those receiving only 100 mg bid.48 Although this data is compelling, it does have to be interpreted with caution as this study was nonrandomized, and patients in the low-dose cohort had poorer prognostic factors. Clinical benefit was also demonstrated in two additional nonrandomized Phase II trials. Gelmon et al evaluated the use of olaparib 400 mg bid in women with triple negative breast cancer, high-grade serous, or poorly differentiated ovarian carcinoma with or without germline BRCA mutations. The ORR for women with ovarian cancer was 29%, though notably higher (41%) for patients who carried a BRCA mutation.49
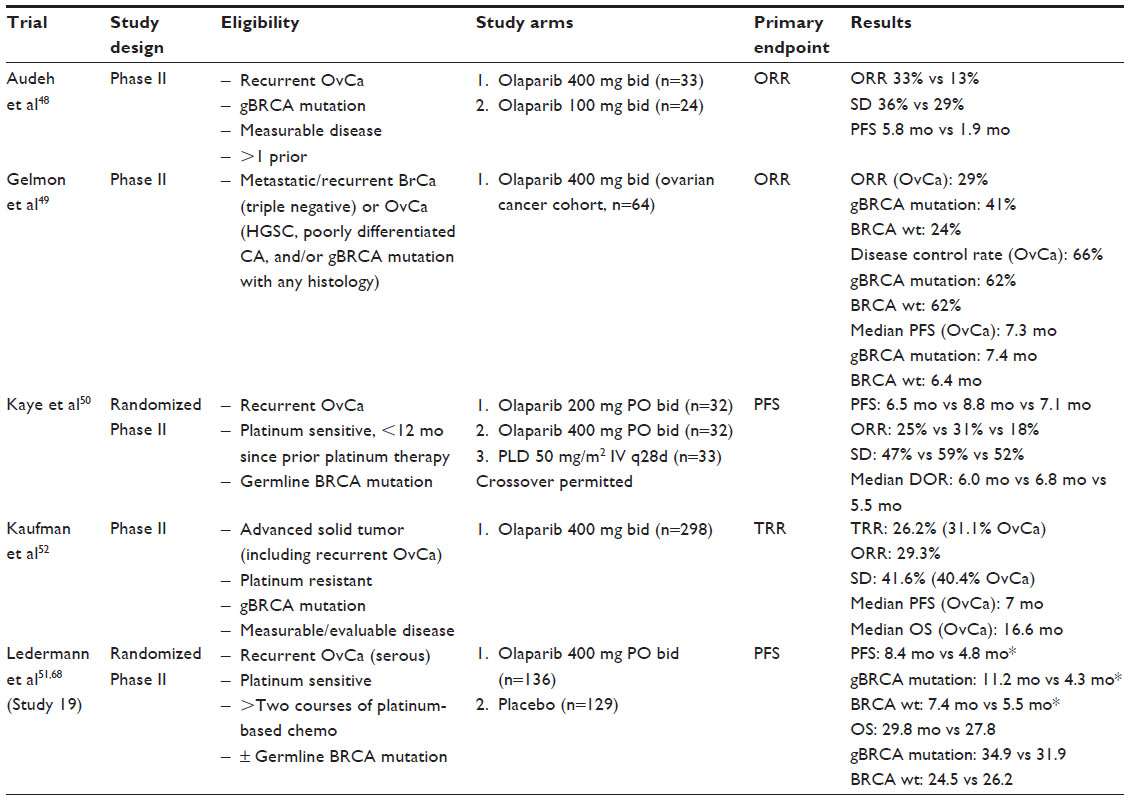 | Table 1 Olaparib monotherapy for the treatment of recurrent ovarian cancer |
A randomized Phase II trial compared olaparib with pegylated liposomal doxorubicin (PLD) for the treatment of recurrent ovarian cancer in women with a BRCA mutation. The authors reported an ORR of 25%, 31%, and 18% for olaparib 200 mg bid, olaparib 400 mg bid, and PLD 50 mg/m2 intravenously (IV) every 4 weeks, respectively. Although no statistically significant differences in progression-free survival (PFS, the primary outcome chosen) or overall survival (OS) were observed, it should be noted that the efficacy of PLD in this study was greater than expected and crossover was permitted.50 At the very least, these findings are compelling in that an oral biologic therapy yielded a similar response rate and PFS to an intravenous cytotoxic chemotherapeutic agent in the management of recurrent ovarian cancer. In Study 19 reported by Ledermann et al, women with platinum sensitive, recurrent high-grade serous ovarian cancer were randomized to olaparib 400 mg PO bid or placebo after completion of platinum-based chemotherapy. Olaparib was associated with a 3.6-month improvement in PFS in the entire study population (8.4 months vs 4.8 months, hazard ratio [HR] 0.35, P<0.0001). When stratified by BRCA mutation status, patients with a germline BRCA mutation treated with olaparib had a median PFS of 11.2 months compared to 4.3 months for patients who received placebo (P<0.0001). Patients without a BRCA mutation had a significant improvement in PFS, though to a lesser degree (PFS 7.4 months vs 5.5 months, HR 0.54, P=0.0075).51 Kaufman et al evaluated olaparib 400 mg bid in a cohort of patients with germline BRCA mutations and advanced solid tumors including 193 patients with platinum-resistant ovarian cancer. In this subset of patients, a tumor response rate of 31.1% was observed with an additional 40% of patients achieving stable disease for at least 8 weeks.52 The results of this study were pivotal to the FDA approval of olaparib as monotherapy in patients with BRCA mutated advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy.53
To date, there are no published randomized Phase III trials evaluating the use of olaparib in ovarian cancer though three are underway. The current studies are limited to women with germline BRCA mutations and each will serve to answer an important question regarding the use of olaparib in the management of ovarian cancer. Olaparib maintenance therapy after first line platinum-based chemotherapy in newly diagnosed ovarian cancer will be investigated in SOLO-1 (NCT01844986). SOLO-2 (NCT01874353) will evaluate olaparib maintenance after platinum-based chemotherapy in the recurrent setting. Finally, SOLO-3 (NCT02282020) will compare olaparib monotherapy to physician’s choice single-agent chemotherapy for the treatment of recurrent ovarian cancer.
Combination therapy
Though olaparib is currently only FDA approved as monotherapy in a select patient population with recurrent ovarian cancer, it has been studied in combination with other targeted therapeutic and cytotoxic agents (Table 2). The combination of olaparib and cediranib (an oral ATP-competitive tyrosine kinase inhibitor of VEGFR 1, 2, and 3) in women with recurrent ovarian cancer yielded an ORR of 44% and a CBR of 61% in the Phase I study published by Liu et al.54 Subsequently, a randomized Phase II trial of olaparib 400 mg PO bid versus combination therapy with olaparib 200 mg PO bid and cediranib 30 mg PO daily was conducted in women with platinum-sensitive recurrent ovarian cancer. Combination therapy was associated with a significantly improved PFS (9.0 months vs 17.7 months, HR 0.42, P=0.005) and ORR (47.8% vs 79.6%, odds ratio 4.24, P=0.002). There was a trend toward improved OS at 2 years, but this data is not mature. Interestingly, a post hoc exploratory analysis suggested that patients with BRCA wild type or unknown status appeared to benefit the most from combination therapy.55 Results from two Phase I studies evaluating olaparib in combination with other targeted therapies (AKT inhibitor, AZD5363; PI3K inhibitor, BKM120) were presented at the annual American Association for Cancer Research meeting in April, 2015.56,57 While preliminary results indicate that these combinations are tolerable with promising response rates, we await the final publication.
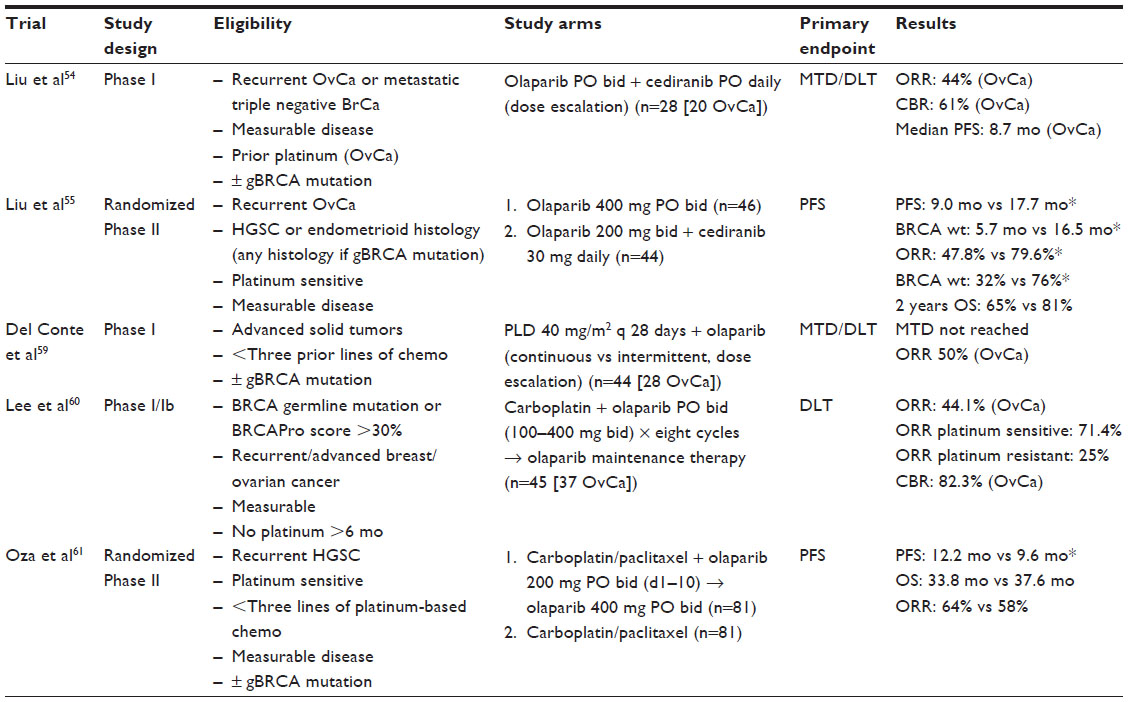 | Table 2 Olaparib in combination therapy for the treatment of recurrent ovarian cancer |
Preclinical data suggest that olaparib may potentiate the efficacy of cytotoxic chemotherapy. There have now been several trials assessing olaparib in combination with cytotoxic chemotherapy in patients with advanced ovarian cancer (in addition to other solid tumors). PLD has shown efficacy in the treatment of recurrent ovarian cancer and is approved for patients failing platinum and taxane chemotherapies.58 A Phase I study combining PLD with olaparib yielded ORR of 50% for patients with recurrent ovarian cancer who had received no more than three prior lines of chemotherapy.59 Lee et al evaluated the use of olaparib in combination with carboplatin followed by olaparib maintenance therapy in BRCA1 or BRCA2 mutation associated breast or ovarian cancer. In this Phase I/Ib, combination therapy resulted in an ORR 44.1% and a CBR 82.3% in patients with ovarian cancer.60 Oza et al compared olaparib in combination with carboplatin and paclitaxel followed by olaparib maintenance therapy with carboplatin and paclitaxel alone for the treatment of recurrent platinum-sensitive ovarian cancer. This study demonstrated a 2.6-month improvement in PFS with the addition of olaparib, however, no difference in OS was found. The magnitude of benefit in PFS was greatest for women with germline BRCA mutations though still no OS benefit was observed.61 Results from a Phase Ib trial (NCT01650376) evaluating olaparib in combination with weekly paclitaxel and carboplatin were presented at the 2014 American Society of Clinical Oncology (ASCO) annual meeting. Intermittently dosed olaparib was tolerable in combination with paclitaxel and carboplatin with promising clinical response.62 While olaparib in combination with PLD, carboplatin, and/or paclitaxel has shown promise, combination with cisplatin and gemcitabine,63 dacarbazine,64 and topotecan65 have been limited by severe toxicity without clear clinical benefit. Further investigation will be required to better understand the combinations which may be tolerable and beneficial in the management of ovarian cancer.
Safety and tolerability of olaparib
In the Phase I dose escalation study of olaparib capsules, reversible dose-limiting toxicity was seen in one out of eight patients receiving 400 mg bid (grade 3 mood alteration and fatigue) and two out of five patients receiving 600 mg bid (grade 4 thrombocytopenia, grade 3 somnolence). Therefore, it was determined that the maximum tolerated dose was 400 mg bid.46 The majority of studies to date have used the capsule formulation including the aforementioned study. For patient convenience and compliance a tablet formulation was developed. Studies have been conducted to compare the relative bioavailability and efficacy of the tablet formulation to the capsule formulation. The dose normalized maximum plasma concentration was found to be higher in the tablet than the capsule formulation.66 Mateo et al validated the dose of olaparib at 300 mg bid as the optimal dosing schedule and the recommended tablet dose in the maintenance setting.67 This is the dose being used in the ongoing Phase III trials.
Generally, olaparib is well tolerated. The most commonly reported adverse events in women taking olaparib include nausea (59%–78%), fatigue (41%–65%), vomiting (34%–50%), and anemia (12%–32%), the majority of which are low grade.50–52 In the largest randomized Phase II study with olaparib monotherapy, 40% of patients receiving olaparib and 22% of patients receiving placebo experienced a grade 3 or higher toxicity. More patients in the olaparib group had dose interruptions (36% vs 16%) or reductions (42% vs 22%) with nausea, vomiting, and fatigue being the most common causes. Discontinuation rates were low overall with seven (5%) patients in the olaparib group and two (1.5%) in the placebo group discontinuing study treatment due to adverse events.51
Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are rare adverse events that have been reported following treatment with olaparib. Overall, MDS/AML was reported in <1% of all patients treated with olaparib according to the package insert.42 Kaufman et al reported two cases of AML and one case of MDS in patients with ovarian cancer who received olaparib monotherapy in their single-arm trial.52 In Study 19, no cases of MDS/AML were reported at the time of publication with a median follow-up of 37.3 months.51 Kaye et al and Audeh et al each reported one case of MDS/AML out of 64 and 57 patients treated with olaparib, respectively.48,50 All patients diagnosed with MDS/AML in these trials had been heavily pretreated with cytotoxic chemotherapy, and thus causality is difficult to determine.
Patient focused perspectives (quality of life)
In addition to response rates, disease recurrence or progression, and survival, one must consider the impact of a treatment on a patient’s quality of life (QOL). Olaparib is an attractive anticancer therapy as it is a relatively well tolerated oral medication dosed twice daily as compared with the majority of standard cytotoxic therapies, which require intravenous infusion at least monthly. Data regarding the impact of olaparib on health-related QOL are limited at this time to secondary endpoints in the randomized Phase II trials. The Functional Assessment of Cancer Therapy-Ovarian (FACT-O) Questionnaire, the FACT-National Comprehensive Cancer Network Ovarian Symptom Index (FOSI) and the Trial Outcome Index (TOI) have been used to measure health-related QOL. Study 19 found no significant differences in disease-related symptoms or rates of improvement in health-related QOL in women receiving olaparib versus placebo. Additionally, there was no significant difference in the time to worsening of these endpoints.68 Kaye et al found no significant differences in health-related QOL between the olaparib treatment groups and the PLD group. However, a significantly higher improvement rate was noted for patients receiving olaparib 400 mg compared with PLD for the total FACT-O score (odds ratio 7.23, P=0.039).50 Further investigation is needed to completely understand the impact of olaparib on patient QOL. It will be important to include QOL assessment in future trials in which olaparib may be used as monotherapy or in combination with cytotoxic chemotherapy and/or other targeted therapies. Additionally, in studies addressing maintenance therapy with olaparib, QOL should be a primary endpoint along with progression free and OS.
Conclusion and future perspectives
PARPis are one of the most promising new classes of targeted agents for use in ovarian cancer. Olaparib is an oral PARPi that has undergone the most extensive clinical investigation thus far and is currently the only FDA-approved PARPi for the treatment of ovarian cancer. While the FDA approved indication for use is limited to women with recurrent ovarian cancer with a germline BRCA mutation and three prior lines of chemotherapy, ongoing and future studies will focus on expanding the role of PARPi in the management of women with ovarian cancer. Ang et al recently demonstrated that treatment with effective doses of olaparib did not compromise the benefit of future chemotherapy regimens (including platinum-based regimens) for women with recurrent ovarian cancer which provides support to the use of olaparib earlier in ovarian cancer management.69 Current Phase III studies are investigating the use of olaparib for 1) maintenance therapy after platinum-based chemotherapy for women with newly diagnosed ovarian cancer, 2) maintenance therapy after platinum-based chemotherapy for women with recurrent ovarian cancer, and 3) monotherapy for recurrent ovarian cancer in women with BRCA mutations. An additional area of study includes combination therapy with cytotoxic chemotherapeutics and/or other targeted agents.
While the use of olaparib is currently restricted to patients with germline BRCA mutations, there are other genetic or epigenetic abnormalities of HR pathway genes (amplification of EMSY, deletion of PTEN, mutation of ATM or ATR, and mutation of the Fanconi anemia genes) that may contribute to sensitivity to PARPi.7 Identification of these patients remains an important challenge. Additionally, the use of other targeted agents (such as angiogenesis inhibitors) may have the potential to sensitize HR proficient tumors to PARPi. Finding and validating biomarkers that can accurately predict HR deficiency and/or response to PARP inhibition will help to tailor therapy to those most appropriate.
Ovarian cancer remains the most lethal of the gynecologic malignancies despite relatively high response to initial therapy. In order to advance the management of this disease, we must identify agents that improve the rates of durable remission and/or have activity against chemoresistant disease. Olaparib has demonstrated the potential to fill both of these roles and is likely to play a significant part in ovarian cancer treatment in the near future.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. | |
NCCN Clinical Practice Guidelines in Oncology: Ovarian Cancer. Version 1. 2015. Available from: http://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf. Accessed April 28, 2015. | |
Luvero D, Milani A, Ledermann JA. Treatment options in recurrent ovarian cancer: latest evidence and clinical potential. Ther Adv Med Oncol. 2014;6(5):229–239. | |
Dizon DS, Dupont J, Anderson S, et al. Treatment of recurrent ovarian cancer: a retrospective analysis of women treated with single-agent carboplatin originally treated with carboplatin and paclitaxel. The Memorial Sloan-Kettering Cancer Center experience. Gynecol Oncol. 2003;91(3):584–590. | |
Markman M, Rothman R, Hakes T, et al. Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J Clin Oncol. 1991;9(3):389–393. | |
Gore ME, Fryatt I, Wiltshaw E, Dawson T. Treatment of relapsed carcinoma of the ovary with cisplatin or carboplatin following initial treatment with these compounds. Gynecol Oncol. 1990;36(2):207–211. | |
Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | |
Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. | |
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. | |
Plummer R. Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res. 2010;16(18):4527–4531. | |
Bernstein C, Bernstein H, Payne CM, Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res. 2002;511(2):145–178. | |
Risinger MA, Groden J. Crosslinks and crosstalk: human cancer syndromes and DNA repair defects. Cancer Cell. 2004;6(6):539–545. | |
Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The breast cancer linkage consortium. Am J Hum Genet. 1998; 62(3):676–689. | |
Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25(11):1329–1333. | |
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. | |
Walsh CS. Two decades beyond BRCA1/2: homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol Oncol. 2015;137(2):343–350. | |
Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4(4):511–518. | |
Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263–272. | |
O’Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 2010;31(6):961–967. | |
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | |
Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408(6811):429–432. | |
Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014;343(6178):1470–1475. | |
Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. | |
Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. | |
Tutt AN, Lord CJ, McCabe N, et al. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb Symp Quant Biol. 2005;70:139–148. | |
Tan DS, Rothermundt C, Thomas K, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008;26(34):5530–5536. | |
Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. 2003;97(9):2187–2195. | |
Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26(8):882–893. | |
Gradwohl G, Ménissier de Murcia JM, Molinete M, et al. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc Natl Acad Sci U S A. 1990;87(8):2990–2994. | |
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301. | |
Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J Biol Chem. 1984;259(8):4770–4776. | |
Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8–24. | |
Haince JF, Kozlov S, Dawson VL, et al. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem. 2007;282(22):16441–16453. | |
Haince JF, McDonald D, Rodrigue A, et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283(2):1197–1208. | |
Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. | |
David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci. 2009;14:1116–1128. | |
Wang Y, Kim NS, Haince JF, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal. 2011;4(167):ra20. | |
Shieh WM, Amé JC, Wilson MV, et al. Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J Biol Chem. 1998;273(46):30069–30072. | |
Ljungman M. Targeting the DNA damage response in cancer. Chem Rev. 2009;109(7):2929–2950. | |
Schultz N, Lopez E, Saleh-Gohari N, Helleday T. Poly(ADP-ribose) polymerase (PARP1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003;31(17):4959–4964. | |
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72(21):5588–5599. | |
LYNPARZA [package insert]. Wilmington, DE: AstraZeneca; 2014. | |
Liu JF, Konstantinopoulos PA, Matulonis UA. PARP inhibitors in ovarian cancer: current status and future promise. Gynecol Oncol. 2014; 133(2):362–369. | |
Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. Ablation of PARP1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene. 2004;23(21):3872–3882. | |
Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J Biol Chem. 2005; 280(16):15773–15785. | |
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | |
Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. | |
Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. | |
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | |
Kaye SB, Lubinski J, Matulonis U, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30(4):372–379. | |
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–861. | |
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. | |
FDA approves Lynparza to treat advanced ovarian cancer. Available from: http://www.FDA.gov. Accessed April 28, 2015. | |
Liu JF, Tolaney SM, Birrer M, et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur J Cancer. 2013;49(14):2972–2978. | |
Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15(11):1207–1214. | |
Michalarea V, Lorente D, Lopez J, et al. Accelerated phase I trial of two schedules of the combination of the PARP inhibitor olaparib and AKT inhibitor AZD5363 using a novel intrapatient dose escalation design in advanced cancer patients [abstract CT323]. Paper presented at: 106th Annual Meeting of the American Association for Cancer Research; April 18–22, 2015; Philadelphia, PA. | |
Matulonis UA, Wulf G, Barry W, et al. Phase I of oral BKM120 and olaparib for high-grade serous ovarian cancer or triple-negative breast cancer. Final results of the BKM120 plus olaparib cohort [abstract CT324]. Paper presented at: 106th Annual Meeting of the American Association for Cancer Research; April 18–22, 2015; Philadelphia, PA. | |
Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19(14):3312–3322. | |
Del Conte G, Sessa C, von Moos R, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer. 2014;111(4):651–659. | |
Lee JM, Hays JL, Annunziata CM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106(6):dju089. | |
Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(1):87–97. | |
Saul ER, Desiree I, Heather S, Cara W, James M. Phase Ib/II with expansion of patients at the MTD study of olaparib plus weekly (metronomic) carboplatin and paclitaxel in relapsed ovarian cancer patients (Suppl; abstr 5527). Paper presented at: 2014 ASCO Annual Meeting, Chicago, IL. | |
Rajan A, Carter CA, Kelly RJ, et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res. 2012;18(8):2344–2351. | |
Khan OA, Gore M, Lorigan P, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104(5):750–755. | |
Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30(4):1493–1500. | |
Molife LR, Forster MD, Krebs M, et al. A phase I study to determine the comparative bioavailability of two different oral formulations of the PARP inhibitor, olaparib (AZD2281), in patients with advanced solid tumors (Suppl; abstr 2599). Paper presented at: 2010 ASCO annual meeting 2010. | |
Mateo J, Friedlander M, Sessa C, et al. Administration of continuous/intermittent olaparib in ovarian cancer patients with a germline BRCA1/2 mutation to determine an optimal dosing schedule for the tablet formulation. Eur J Cancer. 2013;49:s161. | |
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012; 366(15):1382–1392. | |
Ang JE, Gourley C, Powell CB, et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin Cancer Res. 2013;19(19):5485–5493. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.