Back to Journals » Clinical Ophthalmology » Volume 8
Ocular manifestations of idiopathic aplastic anemia: retrospective study and literature review
Authors Mansour A ![]() , Lee J, Yahng S, Kim K, Shahin M, Hamerschlak N
, Lee J, Yahng S, Kim K, Shahin M, Hamerschlak N ![]() , Belfort R, Kurup S
, Belfort R, Kurup S
Received 9 February 2014
Accepted for publication 10 March 2014
Published 17 April 2014 Volume 2014:8 Pages 777—787
DOI https://doi.org/10.2147/OPTH.S62163
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Ahmad M Mansour,1 Jong Wook Lee,2 Seung Ah Yahng,2 Kyu Seop Kim,3 Maha Shahin,4 Nelson Hamerschlak,5 Rubens N Belfort,6 Shree K Kurup7
1Department of Ophthalmology, American University of Beirut, Rafic Hariri University Hospital, Beirut, Lebanon; 2Division of Hematology, Seoul St Mary’s Hospital, The Catholic University of Korea, Seoul, Republic of Korea; 3Department of Ophthalmology, Seoul St Mary’s Hospital, The Catholic University of Korea, Seoul, Republic of Korea; 4Department of Ophthalmology, Mansoura University, Mansoura City, Egypt; 5Oncology and Hematology Program, Instituto Israelita de Ensino e Pesquisa Albert Einstein, São Paulo, Brazil; 6Vision Institute, Hospital São Paulo, Federal University of São Paulo, Brazil; 7Department of Ophthalmology, Wake Forest University Baptist Medical Center, Winston-Salem, NC, USA
Abstract: Aplastic anemia (AA) is a rare disease with few reports on its ophthalmic manifestations. The ocular findings are described in a retrospective consecutive series of 719 AA Korean patients followed at the Hematology Clinic of The Catholic University of Korea. Out of a total of 719 patients, 269 patients had eye examinations, 156 patients had retinal evaluation, and 37 (23.7%) had retinal findings. These 37 patients had unilateral retinal hemorrhage in seven and bilateral retinal hemorrhage in 30 with mean hemoglobin of 6.6 g/dL (range 2.7–12.6 g/dL) and platelet counts of 18.8×109/L (range 4–157×109/L); central retinal vein occlusion-like picture occurred in nine patients and these had similar rheology to the rest of the subjects; optic disc edema, cotton-wool spots, macular edema, and dry eyes occurred in two, three, five, and three patients, respectively. In this Korean series of 141 subjects with AA, systemic bleeding occurred in 24.8% of subjects, retinal hemorrhage in 37% of subjects, and any bleeding site (eye or elsewhere) occurred in 47.5% of subjects with AA. A literature review (1958–2010) of 200 AA cases revealed retinal hemorrhages in 56%, subhyaloid or vitreous hemorrhage in 9%, peripheral retinal vasculopathy in 5.5%, and cotton-wool spots, Sjögren’s syndrome, or optic disc edema in 4% each. The prevalence of retinopathy among series of AA patients varied from 20% to 28.3%, which is consistent with the Korean series of 24.8%. Management of AA patients needs to involve multiple specialties, including hematologists, ophthalmologists, and infectious disease specialists.
Keywords: aplastic anemia, eye, retinopathy, retinal hemorrhage, retinal vasculopathy, thrombocytopenia
Introduction
Aplastic anemia (AA) is a bone marrow failure syndrome with an incidence of two per million in Western countries and 4–6 per million in Asia.1,2 AA commonly presents at between 15 and 25 years of age, with a second smaller peak after age 60 years. The pathophysiology involves immune-mediated destruction of hematopoietic stem cells causing pancytopenia and empty bone marrow.1 Potential factors related to bone marrow failure include viral infection, toxins, drug side effects, autoimmune diseases, and prior radiochemotherapy.1–12 Exposure to the aforementioned factors together with diverse host genetic risk factors may account for different severity of cytopenia at presentation and diverse responsiveness to treatments such as immunosuppressive therapy.1 AA needs to be differentiated from aplastic crisis (transient cessation of bone marrow activity seen in sickle cell anemia, often triggered by parvovirus B19 infection and/or folic acid deficiency).1,2 Major concerns in AA include spontaneous severe bleeding and recurrent infections. Besides gingival or nasal bleeding, patients with AA may present with ocular hemorrhages. According to previous reports, AA patients with ocular findings appeared to have various degrees of disseminated intravascular coagulopathy syndrome with serious rheological shifts and hemorrhagic diathesis.3–59 Occasionally, patients with these life-threatening hematological disorders can present initially to the ophthalmologist because this bone marrow failure syndrome can primarily manifest with ocular problems.17,34 Consequently, clinicians should be aware of the presence of the various ocular findings during physical inspection of AA patients. Herein, we analyzed single center data of patients diagnosed with idiopathic AA in Korea who were further examined for ocular manifestations seeking to relate the ocular and systemic findings. This is contrasted with a literature review and supplemented by a small collaborative case series outside Korea.
Ocular findings in Korean series
The methodology consisted of a retrospective case review of idiopathic AA cases seen in the Department of Hematology (by two of us: JWL, SAY), The Catholic University of Korea from 1981 to 2011. Idiopathic AA is a diagnosis by exclusion, after ruling out secondary bone marrow aplasia from various entities (neoplasms, chemotherapy, and others). AA was diagnosed based on bone marrow findings (hematopoietic cells <30% of residual cells) and blood count with at least two of the following: hemoglobin <10 g/dL, platelet count <50×109/L, and neutrophil count <1.5×109/L. Correlation was done using Student’s t-test or nonparametric tests for small sample sizes with a P-value <0.05 being considered significant. The P-value was computed from a 2×2 contingency table using two-tailed Fisher’s tests. Of the 719 AA patients, 269 patients had eye examinations and 37 patients of the 156 patients who underwent funduscopy by one of us (KSK) had retinal abnormalities (retinal hemorrhages: 23.7%; Figures 1–4). There was no protocol to perform ocular examination during the study period. We tried to correlate the presence of retinal hemorrhage with age, lowest hemoglobin, lowest platelet, concomitant low hemoglobin and low platelets, and other variables (Table 1). The lowest hematocrit or hemoglobin refers to the lowest value on record in case of negative retinal finding or to the value of the blood parameter at the first detection of retinal hemorrhage. In these 37 patients, median initial visual acuity was 6/9.5 in the right eye and 6/10 in the left eye (range: counting finger 30 cm –6/6). Median final visual acuity was 6/6 in either eye on last follow-up (range: 6/300–6/6). Follow-up varied from 2–146 months with a mean of 64 months (median 59 months). These 37 patients were studied in detail (Table 1): seven had unilateral retinal hemorrhages and 30 had bilateral retinal hemorrhages; the mean age was 34.5 years (range: 16–69) with 25 men and 12 women; the mean lowest hemoglobin level was 6.6 g/dL (range: 2.7–12.6 g/dL); and the mean lowest platelet count was 18.8×109/L (range: 4×109/L–157×109/L); central retinal vein occlusion-like picture occurred in nine patients and these had similar rheology to the remaining 28 subjects (hemoglobin level 6.8 g/dL versus [vs] 6.5 g/dL, P=0.8; platelet count 32×109/L vs 14,300×109/L, P=0.3); optic disc edema, cotton wool spots, macular edema, and dry eyes occurred in two, three, five, and three patients, respectively; seven patients died and these had low hemoglobin levels (hemoglobin level 5.4 g/dL vs 6.8 g/dL, P=0.1), significantly low platelet count (platelet count 8.3×109/L vs 21.2×109/L, P=0.03), and significantly low hemoglobin level times platelet count (470,400 g/mL2 vs 1,850,700 g/mL2, P=0.04). In this group of subjects with retinal hemorrhage, the etiology of AA was idiopathic (34 cases), autoimmune (one case), virus-induced (one case), and syndromic (one case of paroxysmal nocturnal hemoglobinmuria [PNH]). Systemic diseases included diabetes mellitus (four cases), hypertension (two cases), rheumatic disease (one case), autoimmune disease (one case), hepatitis B (two cases), IgA nephropathy (one case), goiter (one case), and PNH (one case). The mean heart rate was 85.5 bpm (50–124). Systemic bleeding occurred in five patients (13.5%), including gingival hemorrhage in four patients. Superinfection occurred in four patients (10.8%). A single patient with central retinal vein occlusion-like picture underwent surgical intervention (intravitreal injection of tissue plasminogen activator combined with perfluoropropane gas injection). Therapies included blood immunotherapy based on antithymocyte globulin (ATG) regimen in combination with cyclosporine in 33 cases.
 | Figure 1 A 27-year-old patient with central retinal vein occlusion-like picture in both eyes. A: diffuse nerve fiber layer retinal hemorrhages with exudative macular edema. B: 40 days later with same findings. C: after 3 weeks, retinal hemorrhages have decreased in size. D: 6 months after last photograph (8 months after initial photograph), many retinal hemorrhages around macula have resolved. |
 | Figure 2 A: a 38-year-old patient with layered subhyaloid hemorrhage sparing the fovea in the right eye. B: 2 weeks later, the fovea is fully covered by preretinal hemorrhage. C: 1 month after last photograph, the preretinal hemorrhage has partially resorbed. D: 1 month thereafter, the residual organized blood clot has given a pseudo-hypopyon appearance. |
 | Figure 3 A: a 14-year-old patient with bilateral-layered subinternal limiting membrane involving the fovea in the left eye with prominent striae of the internal limiting membrane. B: 2 weeks later, the fovea is half covered by preretinal hemorrhage with epiretinal membrane formation. C: 3 weeks after last photograph, the preretinal hemorrhage has resorbed. D: 40 days thereafter, the epiretinal membrane is less prominent. |
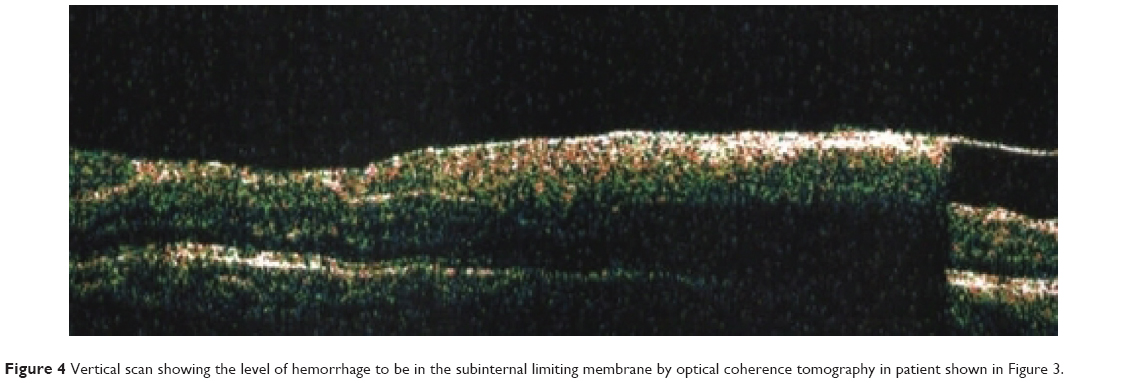 | Figure 4 Vertical scan showing the level of hemorrhage to be in the subinternal limiting membrane by optical coherence tomography in patient shown in Figure 3. |
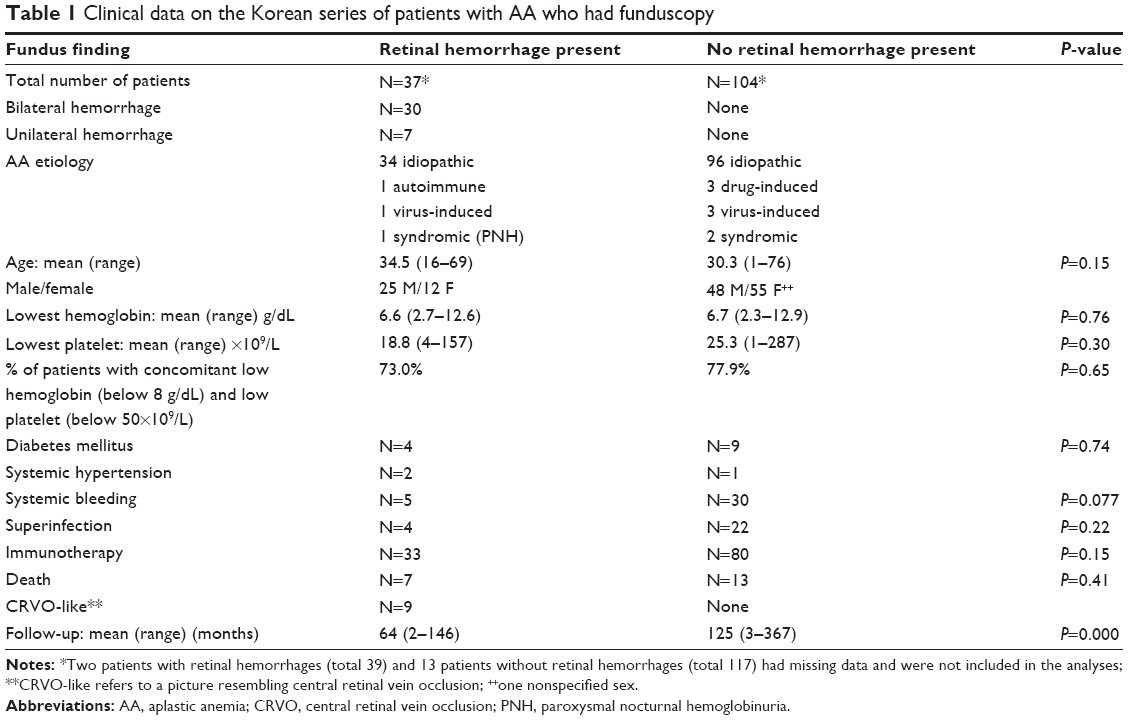 | Table 1 Clinical data on the Korean series of patients with AA who had funduscopy |
The variables in 104 consecutive AA patients who had “normal” funduscopy (no retinal hemorrhages) were analyzed (Table 1). Unrelated findings included: diabetic retinopathy, glaucoma, cytomegalovirus retinitis, or exotropia (three cases each), preeclampsia, or epiretinal membranes (two cases each), central serous retinopathy, epiretinal membranes, uveitis, hypertensive retinopathy, or retinal macroaneurysms (one case each). Follow-up varied from 3–367 months with a mean of 125 months (median 123 months). The mean age was 30.3 years (range 1–76 years). The mean lowest hemoglobin level was 6.7 g/dL (range: 2.3–12.9 g/dL) (similar to the group of 37 patients with retinal hemorrhage) and the mean lowest platelet count was 25.3×109/L (range 1–287×109/L). Thirteen deaths occurred in the series. In these subjects without retinal hemorrhage, the etiology of AA was idiopathic (96 cases), drug-induced (three cases), virus-induced (three cases), and genetic or syndromic (two cases). Systemic diseases included diabetes mellitus (nine cases), hypertension (one case), rheumatic disease (three cases), chronic renal failure (two cases), angina, congestive heart failure, subacute thyroiditis, paroxysmal supraventricular tachycardia, hepatitis B, and hepatitis C in one case each. Systemic bleeding occurred in 30 patients (28.8%) with gingival hemorrhage in eight patients. Superinfection occurred in 22 patients (21.2%), including one case of sepsis. Therapies included blood transfusions (13 cases) and immunotherapy based on ATG (80 cases) (Table 1).
The two groups (with and without retinal hemorrhage) did not differ in terms of rheology and immunotherapy (ATG intake). Superinfections were more commonly encountered in the group without retinal hemorrhage (Table 1). In the total of 141 patients with funduscopy (and complete clinical data), 35 (24.8%) had systemic bleeding, 37 (26.2%) had retinal hemorrhage, and a total of 67 (47.5%) had any bleeding in the retina or elsewhere. Systemic bleeding occurred in 30 patients with combined anemia and thrombocytopenia and five patients not belonging to this group, but this was not significant compared to the group who did not experience systemic bleeding (P=0.17). Patients with either retinal hemorrhage or systemic bleeding did not differ from patients without any bleeding regarding platelet and hemoglobin status (P=0.55). Also, bleeding can also occur in the context of PNH (rare syndrome of aplastic anemia, hemolysis, and thrombosis) and a single case was found in the current study (a 27-year-old man with bilateral retinal hemorrhages, 6/6 vision, no sign of retinal venous stasis, perianal abscess, hemoglobin of 6.6 g/dL, and platelet count of 11×109/L).
Ocular findings in the literature
A literature review (Table 2)3–59 is also presented from 1958 to 2010 of cases with AA not receiving any surgical therapy (such as bone marrow transplant). We searched Medline and Scopus from 1958 to 2010 for cases written in English, French, or Spanish with aplastic anemia (excluding secondary to chemotherapy or leukemia or metastatic carcinoma) that received medical therapy (excluding bone marrow transplant) and had one of the following ocular findings: dry eye, conjunctival hemorrhage, lid hematoma, orbital hemorrhage, retinal hemorrhage, central retinal vein occlusion, vitreous hemorrhage, subhyaloid hemorrhage, cotton-wool spots, and optic neuropathy. We also present in a table form the clinical findings collected in eight unreported cases from the four collaborating medical centers outside Korea (Table 3) (after Institutional Review Board approval). A literature review of ocular findings in 200 patients with idiopathic AA that did not receive surgical treatment (bone marrow transplant) emphasizes the most common reported manifestations: retinal hemorrhages in 56%, subhyaloid or vitreous hemorrhage in 9%, peripheral retinal vasculopathy in 5.5%, cotton-wool spots, Sjögren’s syndrome, and optic disc edema in 4% each. The prevalence of retinopathy among reported series (not case reports) of AA patients varied from 20% to 28.3% (these figures are consistent with the finding of 23.7% of retinal hemorrhages in the Korean series). The current series involves one subtype of anemia (idiopathic AA prior to surgical intervention), while the literature is a mixture of anemias of various etiologies (malaria, hookworm, nutritional, cirrhosis, leukemia, cancer, postpartum hemorrhage, duodenal ulcer rupture, tuberculosis, cyanotic heart disease, sickle cell anemia, thalassemia, lupus, and chronic renal disease)60–62 and the confounding pathology severely limits the conclusions of these studies.
 | Table 2 Ocular findings in 200 patients with idiopathic aplastic anemia in the Western literature |
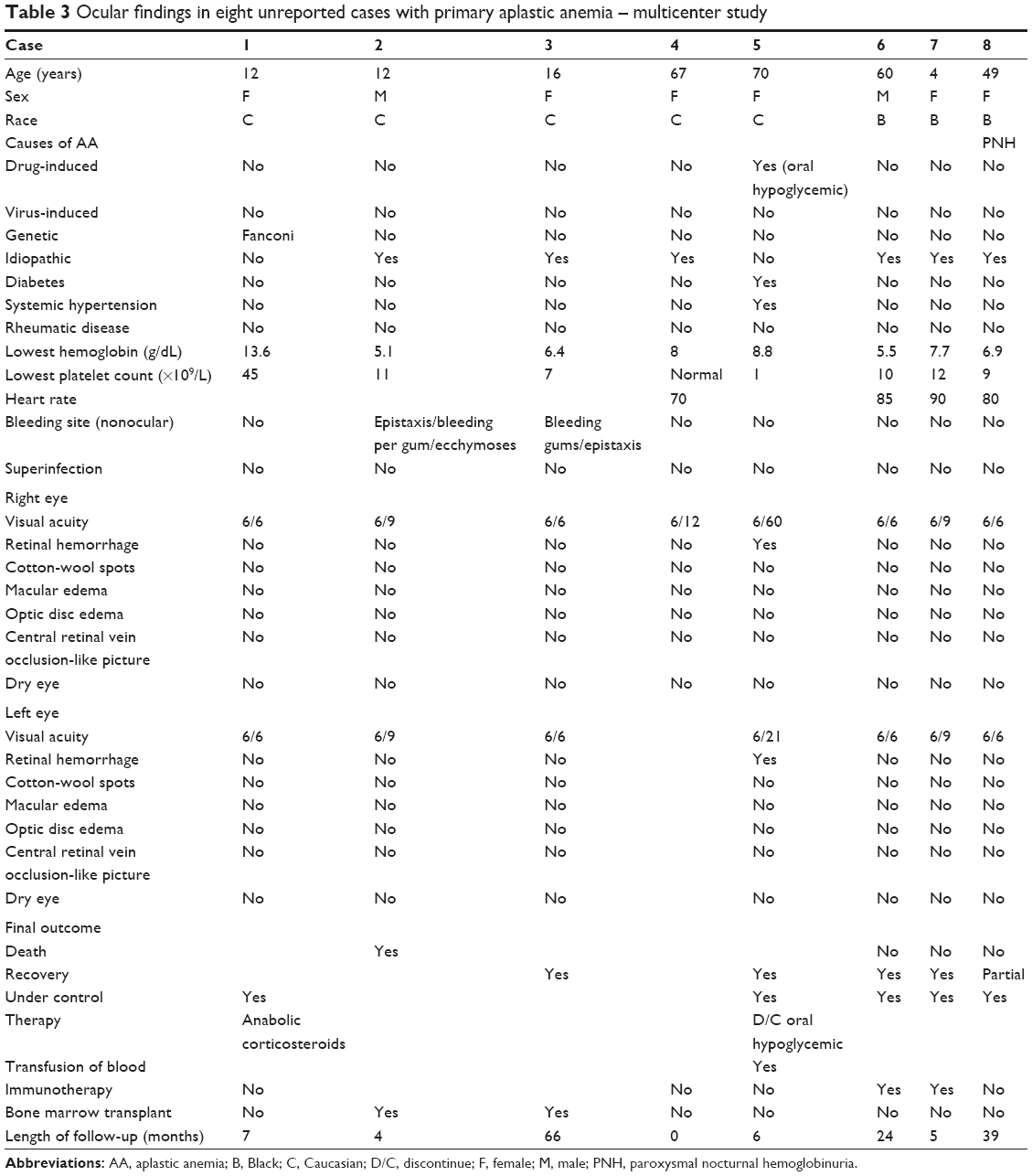 | Table 3 Ocular findings in eight unreported cases with primary aplastic anemia – multicenter study |
Concomitant severe anemia and thrombocytopenia in a mixture of hematologic entities was found to be important risk factors for developing retinopathy.60–62 Merin and Freund61 found a 22% prevalence of retinopathy among anemic patients, similar to the 20% prevalence found by Aisen et al and 28.3% prevalence published by Carraro et al.60 Rubenstein et al found a 44% prevalence of retinopathy in 67 patients with concomitant anemia and thrombocytopenia. In the series by Carraro et al60 involving 33 subjects, retinopathy was observed in 28.3% of the patients as a whole, with the presence of fundus lesions being closely associated with severe anemia (hemoglobin <8 g/dL) and severe thrombocytopenia (platelet count <50×109/L). Among the patients with concomitant anemia and thrombocytopenia, the incidence of retinopathy was 38%.60 A prospective study was conducted to evaluate the prevalence and severity of ocular lesions in dogs with anemia (packed cell volume ≤20%) and/or thrombocytopenia (platelet count <150×109/L).62 The dogs were divided into four groups: 1) anemic (n=17); 2) thrombocytopenic (n=36); 3) anemic and thrombocytopenic (n=24); and 4) healthy controls (n=26). The prevalence of ophthalmic lesions in these four groups was 12%, 42%, 42%, and 0%, respectively. Anemia was not associated with the presence of ocular lesions or their severity. Thrombocytopenia was significantly associated with the presence of ocular lesions and with their severity.62
Prevalence of ocular findings in the literature
The prevalence of ocular findings is unknown because of the rarity of the disease. The only study to address this point was published by Mansour et al3 who reviewed the ocular findings in 18 patients with AA as follows: cotton-wool spots (38%), nerve fiber layer or preretinal hemorrhages (67%), vitreous hemorrhages (13%), a picture resembling central retinal vein occlusion (13%; Figure 1), and optic disc edema (6%).
Pathophysiology of ocular findings in AA (Figure 5)
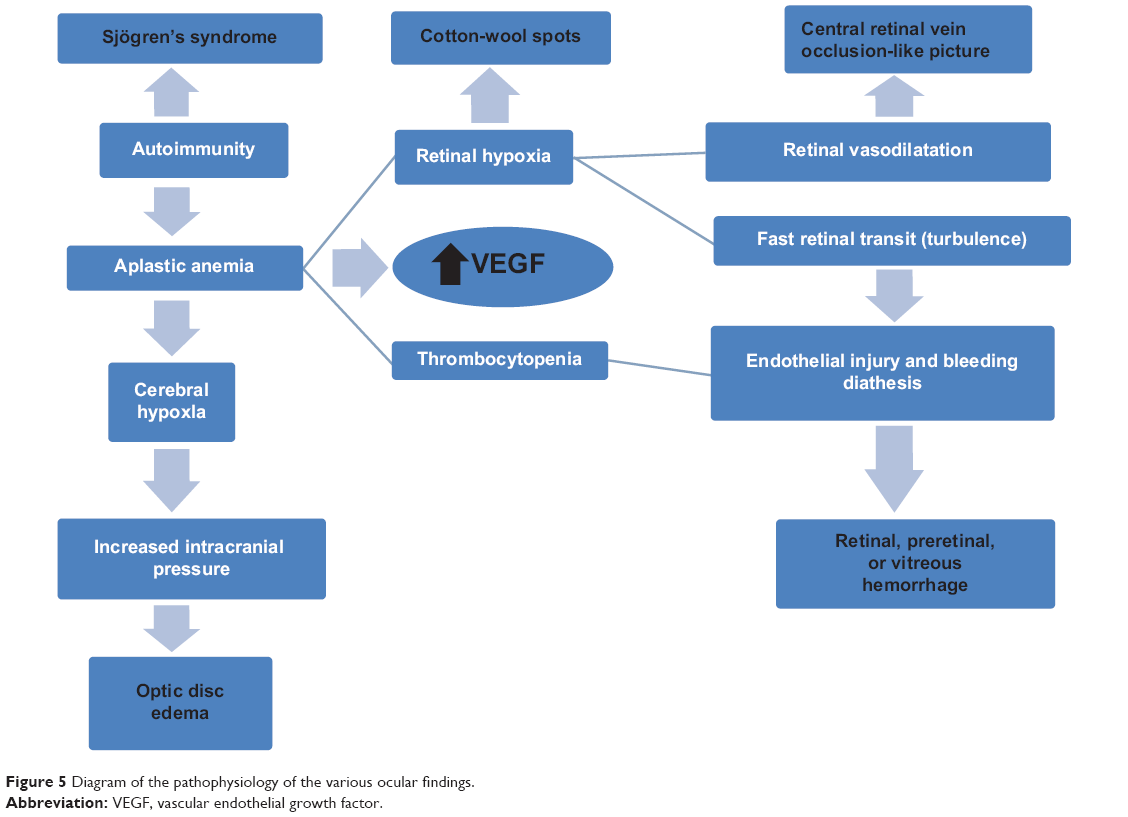 | Figure 5 Diagram of the pathophysiology of the various ocular findings. |
The main physiologic role of red blood cells is to deliver oxygen to the tissues. Physiologic adjustments to compensate for the lack of oxygen delivery in AA include: 1) increased cardiac output; 2) shunting of blood to vital organs; and 3) increased 2,3-diphosphoglycerate in the red blood cells, which causes reduced oxygen affinity, shifting the oxygen dissociation curve to the right and thereby enhancing oxygen release to the tissues. The clinical effects of anemia depend on its duration and severity. When anemia is acute, the body does not have enough time to make the necessary physiologic adjustments, and the symptoms are more likely to be pronounced and dramatic like cotton-wool spots or nonarteritic anterior ischemic optic neuropathy. In contrast, when anemia develops gradually, the body is able to adjust, ameliorating the symptoms relative to the degree of the anemia. In order to compensate for the low oxygen carrying capacity, there is elongation and dilatation of the retinal venous system with turbulent and enhanced flow. This places the endothelium under stress: ischemia, dilatation, and turbulence, leading to bleeding diathesis inside the eye. Valsalva maneuvers and ocular rubbing enhance intraocular bleeding.3
The occurrence of macular edema in AA may be related to ischemia, venous impedance (central retinal vein occlusion-like picture), and increased vascular endothelial growth factor in AA.63 The occurrence of opportunistic infections relates to the immunocompromised status of subjects with AA, with reports of nocardial choroidal infiltration,54 cytomegalovirus retinitis,55 and orbital infiltration from adjacent sinus aspergillosis.56–59
Disc edema is multifactorial in AA. Besides cerebral vascular thrombosis, raised intracranial pressure may result from cerebral hypoxia and cerebral edema, both secondary to low hemoglobin levels and inadequate oxygen-carrying capacity associated with decreased iron-dependent enzymes. It is also possible that low hemoglobin levels may result in increased cerebral blood volume, thereby producing intracranial hypertension.
Paradoxical coagulopathy in AA
Severe anemia is occasionally accompanied by a hypercoagulable state and has been associated with venous and arterial cerebral thrombosis in numerous reports. There is a decrease in blood viscosity in AA, and hence it is expected that this state is protective against hypercoagulability. However, there is paradoxical hypercoagulability in some severe AA cases in the context of associated infections, acute bleeding, intravascular hemolysis,65 or use of cyclosporine,66 often resulting in a disseminated intravascular coagulopathy state.67,68 This paradox is similar to the paradox of bleeding tendency in some patients with thrombocythemia.69 Hence, the mechanism of coagulation activation in some cases of AA is likely to be multifactorial as described in the Russian literature.67,68 The increased cardiac output, increased turbulence of flow, and stretch of the blood vessel lumen in AA lead to vascular endothelial activation, increased proinflammatory cytokines, increased thrombin and fibrin generation, increased tissue factor activity, and increased platelet activation.70 Some of these AA patients appeared to have disseminated intravascular coagulation syndrome with serious rheological shifts and hemorrhagic diathesis.68
Additional causes for disseminated intravascular coagulopathy include the use of ATG (the standard medical therapy for AA),72 as well as severe infections or sepsis often seen in AA. Also, PNH can be associated with disseminated intravascular coagulopathy (DIC).73 Around fifty percent of patients with AA have expanded populations of PNH cells, as detected by flow cytometry, hence the theoretical increased risk for thrombosis and DIC in AA.1
Therapeutic avenues in ocular complications of AA
Acute control of severe bilateral visual loss necessitates red blood cell and or platelet transfusion with all the known risks. Visual recovery occurs after blood transfusion.3,5,41–43 Gupta et al43 presented a 20-year-old pregnant woman at 20 weeks of gestation who had sudden visual loss to the level of hand motion in the right eye. The right eye had subhyaloid hemorrhage and disc edema.43 She received multiple blood transfusions. Upon delivery, the ocular findings were found to have regressed and there was visual recovery to the level of 6/15.43 In the long-term management of AA patients, immunosuppressive therapy17 with anti-thymocyte globulins (ATG) and cyclosporine or stem cell transplantation has enhanced the survival of patients with AA. Immunosuppressive therapies are most widely used because of a lack of histocompatible sibling donors and immediate cost of surgery.
Although small preretinal hemorrhages improve spontaneously, the presence of a large amount of blood may cause permanent macular changes before it resolves. Posterior hyaloidotomy17 enabled rapid resolution of premacular subhyaloid hemorrhage, thereby restoring vision (especially in bilateral involvement) and preventing the need for vitreoretinal surgery. Pars plana vitrectomy can be performed in eyes of patients for non-clearing vitreous hemorrhage after receiving immunosuppressive therapy, including cyclosporine and anti-thymocyte globulin, and platelet and blood transfusions. Pars plana vitrectomy resulted in functional and anatomic success in the majority of eyes operated on by Agarwal et al.33 Coordination of medical and surgical care with the hematology service is strongly advisable to stabilize hematologic parameters prior to undertaking a vitreoretinal procedure. The role of antivascular endothelial growth factor agents is promising due to the markedly increased blood vascular endothelial growth factor71 in AA and the likely increase in intraocular vascular endothelial growth factor with the setting of retinal ischemia. Finally, we need to mention the prophylactic role of avoiding Valsalva and ocular trauma, although mild aerobic exercises in mild AA cases can be tolerated.64
Conclusion
The current retrospective review of a Korean series and collective case reports has its limitations. Moreover, review of the literature is also limited by a selection bias towards severe cases, often with unusual outcome. Prospective studies are not feasible due to the extreme rarity of the disease under study. Hence, we need to analyze the data at hand with caution. In conclusion, retinal hemorrhage is the most common ocular finding as it occurred in 23.7% of AA patients in one series. Central retinal vein occlusion-like picture is the second most frequent finding, occurring in 5.8% of AA patients; its occurrence does not seem related to a more severe rheology. The occurrence of ocular abnormalities in AA patients is associated with the severity and concomitance of anemia and thrombocytopenia. Small preretinal hemorrhages respond to blood transfusions while large hemorrhages require posterior hyaloidotomy or vitrectomy to prevent permanent macular damage. AA patients should be advised to refrain from Valsalva maneuvers, ocular rubbing, and vigorous exercise to prevent ocular morbidity. AA is a life-threatening and a potentially blinding disease if not treated promptly and ophthalmologists may be the first physicians to diagnosis this rare condition. We also emphasize the importance of coordination of the medical and surgical care of AA with the hematology service and the need for periodic retinal exam.
Disclosure
Presented at the 2013 American Academy of Ophthalmology, New Orleans, LA, USA, 2013 Association for Vision and Ophthalmology, Seattle, WA, USA, 2012 World Congress of Ophthalmology, Abu Dhabi, UAE. The authors report no conflicts of interest in this work.
References
Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–2519. | ||
Issaragrisil S, Kaufman DW, Anderson T, et al. The epidemiology of aplastic anemia in Thailand. Blood. 2006;107:1299–1307. | ||
Mansour AM, Salti HI, Han DP, et al. Ocular findings in aplastic anemia. Ophthalmologica. 2000;214:399–402. | ||
Lilley ER, Bruggers CS, Pollock SC. Papilledema in a patient with aplastic anemia. Arch Ophthalmol. 1990;108:1674–1675. | ||
Mansour AM. Central retinal vein occlusion-like picture inaplastic anemia. Am J Ophthalmol. 1985;100:478–479. | ||
Jenkyn LR, Budd RC, Fein SH, Cornell CJ. Insecticide/herbicide exposure, aplastic anaemia, and pseudotumor cerebri. Lancet. 1979;2(8138):368. | ||
Aisen ML, Bacon BR, Goodman AM, Chester EM. Retinal abnormalities associated with anemia. Arch Ophthalmol. 1983;101:1049–1052. | ||
Wang KS. Aplastic anemia in Korea: a clinical study of 309 cases. In: Hibino S, Takahu S, Shahidi N, editors. Aplastic anemia. Baltimore, MA: University Park Press; 1978:225–242. | ||
Holt JM, Gordon-Smith EC. Retinal abnormalities in diseases of the blood. Br J Ophthalmol. 1969;53:145–160. | ||
Cosnett JE, Macleod IN. Retinal hemorrhages in severe anemia. Br Med J. 1959;2:1002–1004. | ||
Lowenthal MN, Jones IG, Desai M. Aplastic anemia and optic fundus hemorrhages due to traditional herbal remedies. J Trop Med Hyg. 1978;81:177–179. | ||
Marshall RA. A review of lesions in the optic fundus in various diseases of the blood. Blood. 1959;14:882–891. | ||
Mohler DN, Leavell BS. Aplastic anemia: an analysis of 50 cases. Ann Intern Med. 1958;49:326–362. | ||
Rubenstein RA, Yanoff M, Albert DM. Thrombocytopenia, anemia, and retinal hemorrhage. Am J Ophthalmol. 1968;65:435–439. | ||
Damian C, Irimia A, Preda M, et al. [Retinal lesions in hereditary aplastic anemia]. Oftalmologia. 2007;51:62–64. Romanian. | ||
Grové JD, Meyer D. Aplastic anemia presenting as spontaneous orbital hemorrhage. Orbit. 2008;27:391–393. | ||
Ghosh S, Biswas PN, Mukhopadhyay S, Bhaduri G. Posthepatitis aplastic anaemia presenting only with bilateral vision loss. J Indian Med Assoc. 2007;105:524, 526, 543. | ||
Reddy MA, Bibby K. Vitreous hemorrhage in Fanconi’s anaemia. J R Soc Med. 1998;91:540–541. | ||
Chan WM, Liu DT, Tham CC, et al. Bilateral subhyaloid hemorrhage in aplastic anaemia. Br J Haematol. 2003;123:757. | ||
Furuse N, Hayasaka S, Yamamoto Y, Setogawa T. Retinal microaneurysms in a patient with drug-induced aplastic anemia. Ophthalmologica. 1987;195:188–191. | ||
Chai SM, Mathur R, Ong SG. Retinal vasculopathy in Fanconi anemia. Ophthalmic Surg Lasers Imaging. 2009;40:498–500. | ||
Sudhir RR, Rao SK, Shanmugam MP, Padmanabhan P. Bilateral macular hemorrhage caused by azathioprine-induced aplastic anemia in a corneal graft recipient. Cornea. 2002;21:712–714. | ||
Pogorelov P, Gusek-Schneider GC, Mardin CY. [Acute bilateral vision loss in Fanconi anemia]. Ophthalmologe. 2007;104:329–331. German. | ||
Jain V, Shome D, Maiti A, Natarajan S. An unusual ocular manifestation in fanconianaemia: anterior ischaemic syndrome. Eye. 2007;21:1449–1450. | ||
Yahia SB, Touffahi SA, Zeghidi H, et al. Ocular neovascularization in a patient with Fanconi anemia. Can J Ophthalmol. 2006;41:778–779. | ||
Bahar I, Weinberger D, Kramer M, Axer-Siegel R. Retinal vasculopathy in Fanconi anemia: a case report. Retina. 2004;25:799–800. | ||
Wong VG, Bodey GP. Hemorrhagic retinoschisis due to aplastic anemia. Arch Ophthalmol. 1968;80:433–435. | ||
Suzuki J, Goto H, Usui M, Sakai J. Serous retinal detachment in a patient with aplastic anemia associated with parvovirus B19 infection. Graefes Arch Clin Exp Ophthalmol. 2007;245:324–326. | ||
Revesz T, Fletcher S, al-Gazali LI, DeBuse P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? J Med Genet. 1992;29:673–675. | ||
Niedermayer I, Reiche W, Graf N, et al. Cerebroretinal vasculopathy and leukoencephalopathy mimicking a brain tumor. Report of two early-onset cases with Fanconi’s anemia-like phenotypes suggesting an autosomal-recessive inheritance pattern. Clin Neuropathol. 2000;19:285–295. | ||
Tsilou ET, Giri N, Weinstein S, et al. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology. 2010;117:615–622. | ||
Gayatri NA, Hughes MI, Lloyd IC, Wynn RF. Association of the congenital bone marrow failure syndromes with retinopathy, intracerebral calcification and progressive neurological impairment. Eur J Paediatr Neurol. 2002;6:125–128. | ||
Agarwal M, Yeh S, Faia LJ, et al. Posterior segment ophthalmic complications of aplastic anemia. Ophthalmic Surg Lasers Imaging. 2010;41:e1–e6. | ||
Wechsler DZ, Tay TS, McKay DL. Life-threatening haematological disorders presenting with opthalmic manifestations. Clin Experiment Ophthalmol. 2004;32:547–550. | ||
Teixeira LF, Shields CL, Marr B, et al. Bilateral retinal vasculopathy in a patient with dyskeratosiscongenita. Arch Ophthalmol. 2008;126:134–135. | ||
Roth K, Lange CE. [Fundus changes in Zinsser-Engman-Cole syndrome]. Klin Monatsbl Augenheilkd. 1975;166:695–698. German. | ||
Kajtar P, Mehes K. Bilateral Coats retinopathy associated with aplastic anaemia and mild dyskeratotic signs. Am J Med Genet. 1994;49:374–377. | ||
Johnson CA, Hatfield M, Pulido JS. Retinal vasculopathy in a family with autosomal dominant dyskeratosiscongenita. Ophthalmic Genet. 2009;30:181–184. | ||
Shome D, Jain V, Jayadev C, Natarajan S. Scleral necrosis in a patient with aplastic anaemia. Eye. 2007;21:1017. | ||
Riyaz A, Riyaz N, Jayakrishnan MP, et al. Revesz syndrome. Indian J Pediatr. 2007;74:862–863. | ||
Jeng MR, Rieman M, Bhakta M, et al. Pseudotumor cerebri in two adolescents with acquired aplastic anemia. J Pediatr Hematol Oncol. 2002;24:765–768. | ||
Nazir SA, Siatkowski RM. Pseudotumor cerebri in idiopathic aplastic anemia. J AAPOS. 2003;7:71–74. | ||
Gupta SK, Brar VS, Keshavamurthy R, Chalam KV. Aplastic anemia induced disc edema and visual loss in pregnancy: a case report. Cases J. 2008;1:322. | ||
Biousse V, Rucker JC, Vigna IC, et al. Anemia and papilledema. Am J Ophthalmol. 2003;135:437–446. | ||
Mohla A, Oworu O, Hutchinson C. Aplastic anaemia presenting with features of raised intracranial pressure. J R Soc Med. 2006;99:315–316. | ||
Quiquandon I, Morel P, Lai JL, et al. Primary Sjögren’s syndrome and aplastic anaemia. Ann Rheum Dis. 1997;56:438. | ||
Matsumoto N, Kagawa H, Ichiyoshi H, et al. Aplastic anemia complicating Sjögren’s syndrome. Intern Med. 1997;36:371–374. | ||
Satoh M, Yamagata H, Watanabe F, et al. A case of Sjögren’s syndrome complicating immune-mediated aplastic anaemia. Clin Rheumatol. 1993;12:257–260. | ||
Zagorski Z, Biziorek B, Rakowska E, Jedrzejewski D. [Zinsser-Engman-Cole syndrome (dyskeratosis congenita) with severe sicca syndrome, panuveitis and corneal perforation – a case report]. Klin Monatsbl Augenheilkd. 2001;218:455–458. German. | ||
Seki T, Kiyosawa K, Monno S, et al. Cell-mediated immune-pancytopenia complicating primary Sjögren’s syndrome. Am J Hematol. 1993;43:221–225. | ||
Koramaz I, Sonmez M, Pulathan Z, et al. Successful coronary artery bypass grafting in a patient with aplastic anemia and Sjögren’s syndrome. Saudi Med J. 2006;27:1251–1252. | ||
Akiyama M, Yanagisawa T, Yuza Y, et al. Severe aplastic anaemia complicating Sjögren syndrome in a 2-year-old girl. Eur J Pediatr. 2005;164:700–702. | ||
Coppo P, Sibilia J, Maloisel F, et al. Primary Sjögren’s syndrome associated agranulocytosis: a benign disorder? Ann Rheum Dis. 2003;62:476–478. | ||
Jones NP. Disseminated nocardia infection with subretinal abscess. Eye. 2010;24:187–188. | ||
Sasaki T, Okayama A, Eizuru Y, et al. Progressive retinitis-encephalitis due to ganciclovir-resistant cytomegalovirus associated with aplastic anemia. Intern Med. 1997;36:375–379. | ||
Robinson MR, Fine HF, Ross ML, et al. Sino-orbital-cerebral aspergillosis inimmunocompromised pediatric patients. Pediatr Infect Dis J. 2000;19:1197–1203. | ||
Saah D, Drakos PE, Elidan J, et al. Rhinocerebral aspergillosis in patients undergoing bone marrow transplantation. Ann Otol Rhinol Laryngol. 1994;103:306–310. | ||
McGill TJ, Simpson G, Healy GB. Fulminant aspergillosis of the nose and paranasal sinuses: a new clinical entity. Laryngoscope. 1980;90:748–754. | ||
Antoine GA, Gates RH, Park AO. Invasive aspergillosis in a patient with aplastic anemia receiving amphotericin B. Head Neck Surg. 1988;10:199–203. | ||
Carraro MC, Rossetti L, Gerli GC. Prevalence of retinopathy in patients with anemia and thrombocytopenia. Eur J Haematol. 2001;67:238–244. | ||
Merin S, Freund M. Retinopathy in severe anemia. Am J Ophthalmol. 1968;66:1102–1106. | ||
Aroch I, Kass PH, Bruchim Y, Ofri R. A prospective study of the association of anemia and thrombocytopenia with ocular lesions in dogs. Vet J. 2009;182:187–192. | ||
Dunst J, Becker A, Lautenschläger C, et al. Anemia and elevated systemic levels of vascular endothelial growth factor (VEGF). Strahlenther Onkol. 2002;178:436–441. | ||
Mangione KK, McKee E, Hickey M, Hofmann M. Aerobic training in a patient with nonsevere aplastic anemia: a case report. Arch Phys Med Rehabil. 2000;81:226–229. | ||
Kawagushi T, Nakakuma H. New insights into molecular pathogenesis of bone marrow failure in paroxysmal nocturnal hemoglobinuria. Int J Hematol. 2007;86:27–32. | ||
Morishita E, Nakao S, Asakura H, et al. Hypercoagulability and high lipoprotein(a) levels in patients with aplastic anemia receiving cyclosporine. Blood Coagul Fibrinolysis. 1996;7:609–614. | ||
Shishina RN, Pchelinseva TA, Sevenko TA, et al. [The state of homeostasis system in patients with aplastic anemia in the period of full-fledged clinical manifestation of disease]. Klin Lab Diagn. 2012;5:34–37. Russian. | ||
Bessmel’tsev SS, Khorshev SK, Abdulkadyrov KM, Shilova ER. [Hemorrhagic and neurological complications in aplastic anemia patients]. Ter Arkh. 1996;68:56–59. Russian. | ||
Michiels JJ. Acquired von Willebrand disease due to increasing platelet count can readily explain the paradox of thrombosis and bleeding in thrombocythemia. Clin Appl Thromb Hemost. 1999;5:147–151. | ||
Ataga KI. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica. 2009;94:1481–1484. | ||
Kodama Y, Okamoto Y, Hashiguchi T, et al. Vascular endothelial growth factor corrected for platelet count and hematocrit is associated with the clinical course of aplastic anemia in children. Int J Hematol. 2012;95:494–499. | ||
Weber M, Kröger N, Langer F, et al. Non-overt disseminated intravascular coagulation in patients during treatment with antithymocyte globulin for unrelated allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2003;31:817–822. | ||
Raich PC, Rector SC. Disseminated intravascular coagulation in paroxysmal nocturnal hemoglobinuria. Southern Med J. 1981;74:887–888. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.