Back to Journals » Clinical Ophthalmology » Volume 9
Ocular manifestations and management recommendations of lysosomal storage disorders I: mucopolysaccharidoses
Authors Fenzl C, Teramoto K, Moshirfar M ![]()
Received 29 November 2014
Accepted for publication 13 January 2015
Published 7 September 2015 Volume 2015:9 Pages 1633—1644
DOI https://doi.org/10.2147/OPTH.S78368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Carlton R Fenzl,1 Kyla Teramoto,2 Majid Moshirfar3
1John A Moran Eye Center, University of Utah, Salt Lake City, UT, USA; 2John A Burns School of Medicine, University of Hawai’i, Honolulu, HI, USA; 3Cornea and Refractive Surgery Division, Department of Ophthalmology, Francis I. Proctor Foundation, University of California, San Francisco, CA, USA
Abstract: The mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders caused by inborn errors of glycosaminoglycan (GAG) metabolism. These diseases are classified by enzyme deficiency into seven groups: type I, II, III, IV, VI, VII, and IX. GAG accumulation leads to characteristic clinical features. Some ophthalmic findings that are characteristic of MPS diseases include corneal clouding, retinal degeneration, decreased electroretinogram wave amplitude, optic atrophy, papilledema, and glaucoma. Current treatments such as hematopoietic stem cell transplantation and enzyme replacement therapy have increased the life span of many MPS patients and created the need to improve management of ocular symptoms. This article aims to provide a comprehensive review of ocular manifestations and treatment options for the various types of MPS.
Keywords: MPS, glycosaminoglycan, corneal clouding
Introduction
The mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders caused by inborn errors of glycosaminoglycan (GAG) metabolism. Lysosomes are membrane-bound organelles found in animal cells and are responsible for the degradation of proteins, nucleic acids, carbohydrates, lipids, and cellular debris. Lysosomal enzymes are synthesized in the endoplasmic reticulum, tagged with mannose-6-phosphate in the Golgi apparatus and delivered to the lysosomes. Proteins are needed to protect the enzymes from being degraded by an acidic environment maintained by an energy-dependent pump. Defects in lysosomal enzymes, cofactors, or transport proteins can lead to MPS.1
In MPS, the enzymes required to degrade GAGs, or mucopolysaccharides, are deficient. GAGs are found in various tissues and organs throughout the body. The clinical manifestations of MPS are due to accumulation of GAG in these tissues. The most typical and prominent clinical features of patients with MPS are dysmorphic facial features, such as flattened face, depressed nasal bridge, thick lips, and an enlarged mouth. Patients may also have hearing impairment, skeletal disease, cardiorespiratory disease, and significant neurological and intellectual problems.1 Previously, the diseases were identified by their clinical manifestations; however, due to biochemical advances, they are now classified by the specific enzyme deficiencies.
There are seven groups of MPS: type I, II, III, IV, VI, VII, and IX (Table 1). All are inherited in an autosomal recessive pattern, with the exception of Hunter syndrome, which follows an X-linked recessive inheritance pattern. There is a wide degree of variation in phenotype, as some are fatal within the first few months of life and others have no significant impact on life span. Some ophthalmic findings that are characteristic of MPS diseases include corneal clouding (Figure 1), retinal degeneration, decreased electroretinogram (ERG) wave amplitude, optic atrophy, papilledema, and glaucoma.1 In the past, ocular management was limited due to the shortened life span of these patients. Current treatments such as hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT) have increased the life span of many MPS patients and created the need to improve management of ocular symptoms.1 This article aims to provide a comprehensive review of ocular manifestations and treatment options for the various types of MPS. In the following sections, we will be discussing MPS according to subtype classification.
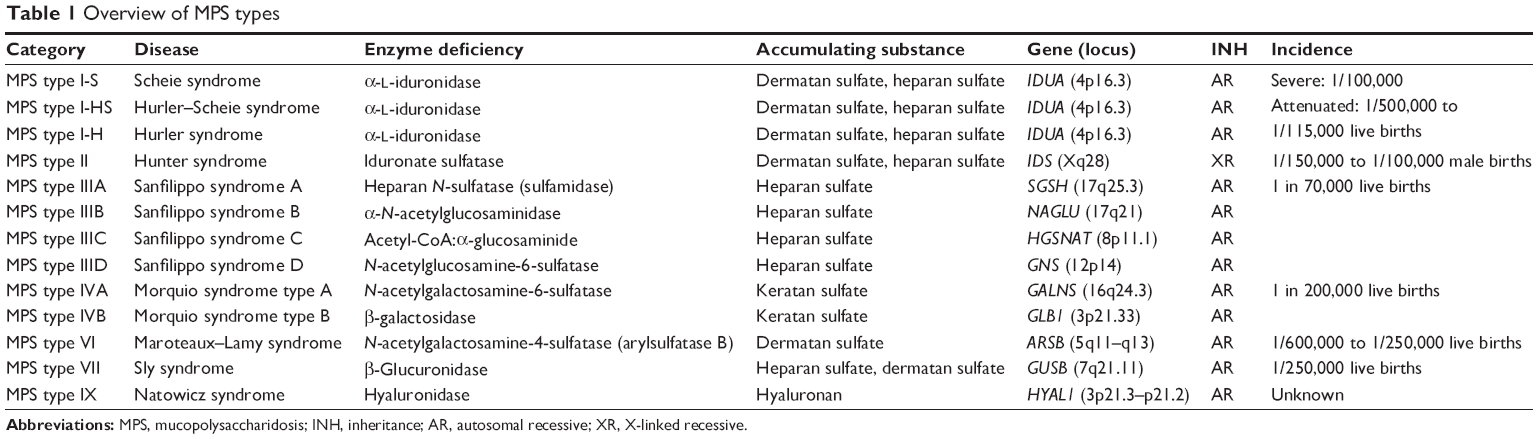 | Table 1 Overview of MPS types |
 | Figure 1 Corneal clouding. |
Pathophysiology
Systemic manifestations
Although the enzyme deficiencies causing MPS are understood, the pathophysiologic mechanisms leading to clinical manifestations need to be studied further. Clarke notes that GAGs are biologically active as free molecules and as proteoglycan complexes.2 They are necessary for many pathways, including signal transduction, sequestration of extracellular factors, and communication between cells.2 Disruption of any of these actions may contribute to clinical phenotype (Table 2).
 | Table 2 Overview of systemic features in MPS |
Lysosomal enzymes are responsible for degrading many substrates that are complex lipids, and therefore not soluble in water.1 It is assumed that the accumulation of these substances leads to the clinical manifestations of MPS. Mesenchymal cells, like chondrocytes, normally metabolize the extracellular matrix. Their function is affected in MPS due to deficiencies in acid hydrolases leading to disrupted cartilage formation, characteristic short stature, chest wall abnormalities, and bony malformation seen in these disorders.3 The buildup of partially degraded GAG in ligaments, tendons, joint capsules, and other soft tissue causes joint contracture and carpal tunnel syndrome in many MPS patients.4 Ziyadeh et al present a case of Hurler–Scheie syndrome (MPS I-HS) in which synostosis of the sagittal suture was present with permeability of both coronal sutures.5 Excess heparan and dermatan sulfates can affect bone maturation and lead to skull deformity.5 A study on murine MPS VII growth plates suggest that bone shortening is due to accumulation of chondroitin-4-sulfate and alteration of the Janus kinase/STAT (JAK/STAT) signaling pathway.2 The pathophysiology of the dysostosis multiplex is not fully understood, but animal models suggest that GAG accumulation promotes inflammation and chondrocyte apoptosis, which leads to the release of matrix metalloproteinases and joint degeneration.4
An in vitro study has suggested that GAG deposition alters cardiac tissue by decreasing elastin synthesis and creating a deficiency of elastin binding protein, which is necessary for elastin fiber assembly.2 Vacuoles of GAG are also present in endothelial cells, myocytes, and fibroblasts, which leads to dysfunction of the endocardium, myocardium, valves, coronary arteries, conduction system, and vessels.6 Progressive valvular thickening causes incompetence of valves.2 In addition, there appears to be increased expression of elastin-degrading proteins and activation of transcriptional regulators MMP, STAT1, and STAT3.2
Ocular manifestations
Corneal clouding is a common feature in MPS (Figure 1; Table 3). Alroy et al demonstrated that corneas affected by MPS I have an increased mean fibril diameter and irregular fibril distribution compared to normal corneas.7 In animal models of MPS disease (dog-MPS VII and cat-MPS I and VI), corneal clouding resulted from the buildup of GAGs, most likely heparan sulfate, in the stromal keratocytes. In MPS type I feline corneas, the endothelial cells were affected by secondary lysosomal inclusions causing cell vacuolization, which worsened as the animal aged. Despite these morphologic findings, the diseased corneas were shown to have normal function, and no corneal edema was seen.8 Huang et al9 presented a 14-year-old girl with Hurler syndrome (MPS I-H) with a history of bone marrow transplantation and a right penetrating keratoplasty that supported the findings in the animal study above.8 She was found to have accumulation of proteoglycans in the vacuolated stromal cells and nearby stroma.9 Her cornea also contained a large range of fibril diameters with an abnormal distribution.9
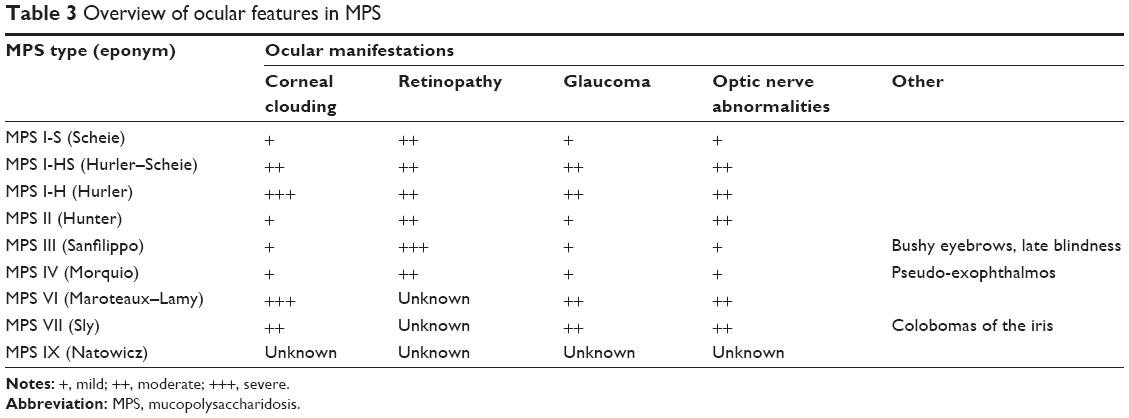 | Table 3 Overview of ocular features in MPS |
Patients may also have optic nerve swelling or atrophy. Collins et al performed an observational study, which found swelling of the optic nerve head in eight out of 14 eyes with Hurler syndrome, nine out of 21 in Hurler–Scheie, and 0 out of four in Scheie syndrome (MPS I-S). They concluded that this optic nerve swelling seen in systemic MPS likely precedes the development of optic atrophy.10 Thickening of the sclera at the lamina cribrosa can cause optic nerve head deformation and impingement of the nerve, which leads to optic nerve atrophy.11
Patients with MPS may develop glaucoma. It has been proposed that the accumulated GAGs thicken the extracellular matrix and results in constriction of the anterior chamber.12 Distortion of the meshwork in the corneoscleral junction and adjacent to the canal of Schlemm causes disruption of aqueous outflow.12 Nowaczyk et al12 suggested that although corneal clouding is an important clinical sign of glaucoma, it is also very common in MPS so glaucomatous disease may be overlooked in these patients. Also, corneal clouding makes examination of the optic nerve and gonioscopy difficult. It has been suggested that increased corneal thickness measurements should be considered when evaluating patients with corneal opacification and elevated intraocular pressure (IOP).13 Altered stromal architecture and accumulation of GAG can also falsely increase IOP.14 Fahnehjelm et al showed that corneal hysteresis and corneal resistance measurements were elevated in patients with MPS I and VI. Corrected IOPs were normal in these patients and are important to calculate to avoid unnecessary hypotensive treatment in patients with MPS.15
Patients with MPS, especially type III, may have retinopathy with pigmentary retinal degeneration and associated electroretinogram changes. Deposition of GAG within the retinal pigment epithelial cells causes photoreceptor loss as well as retinal degeneration and dysfunction.13 The electroretinogram typically shows a reduction of scotopic responses, suggesting rod degeneration. Later, B-waves will be reduced in photopic conditions, indicating cone loss.13
MPS type I
The three subtypes of MPS type I disease include Hurler, Scheie, and Hurler–Scheie syndromes. The gene affected is IDUA at locus 4p16.3.13 They are defined by a deficiency of lysosomal hydrolase α-l-iduronidase, which is required to break down heparan (Figure 2) and dermatan (Figure 3) sulfate. As a result, these metabolites build up in various tissues. The phenotypic spectrum of α-l-iduronidase deficiencies includes the mildest form, Scheie syndrome, the intermediate form, Hurler-Scheie syndrome, and Hurler syndrome, the most severe.1 The estimated incidence of severe MPS type I is one in 100,000 babies.16 Attenuated MPS type I occurs in about one in 500,000 births.16 Ocular findings are common in all forms of MPS type I.
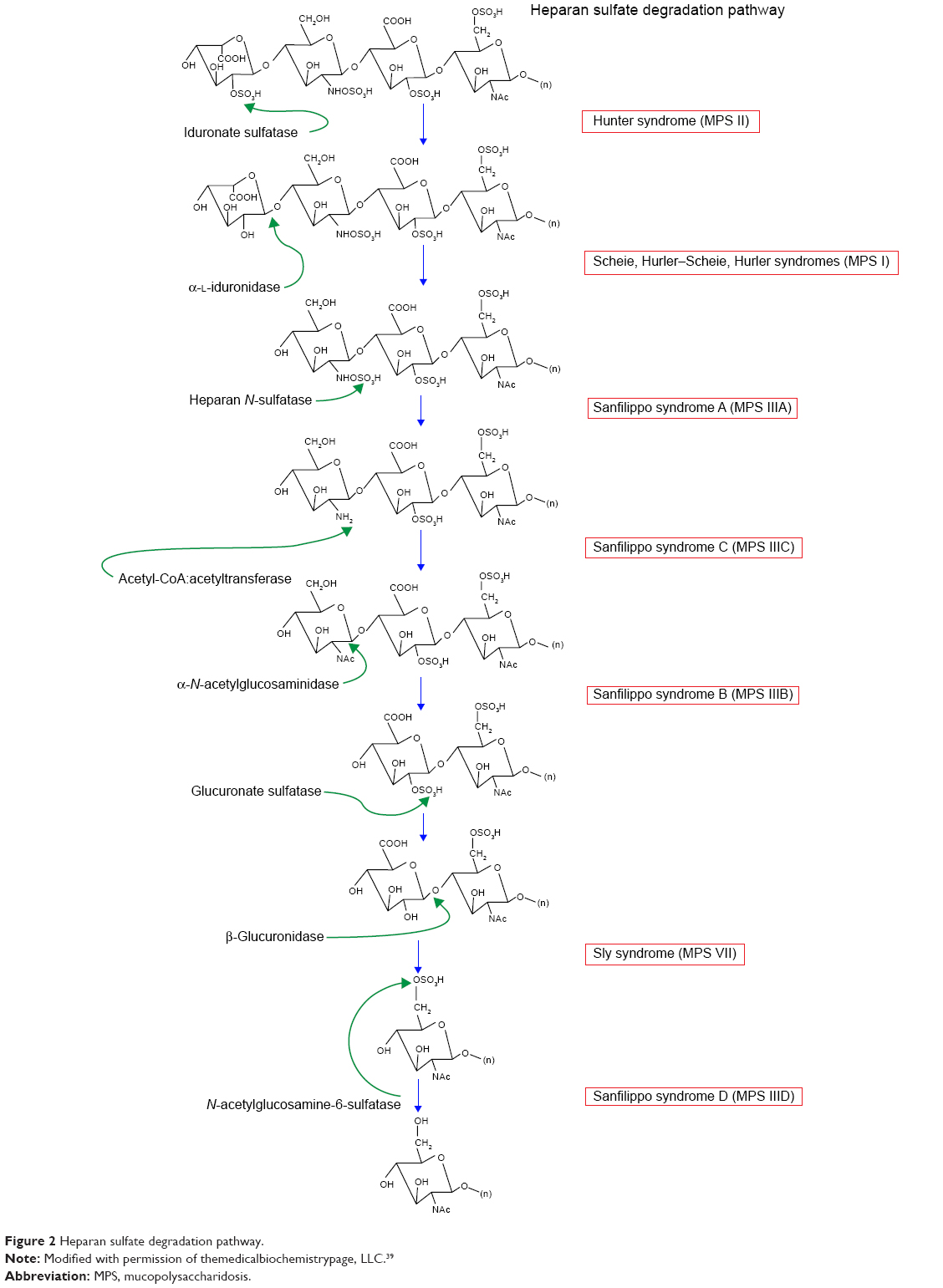 | Figure 2 Heparan sulfate degradation pathway. |
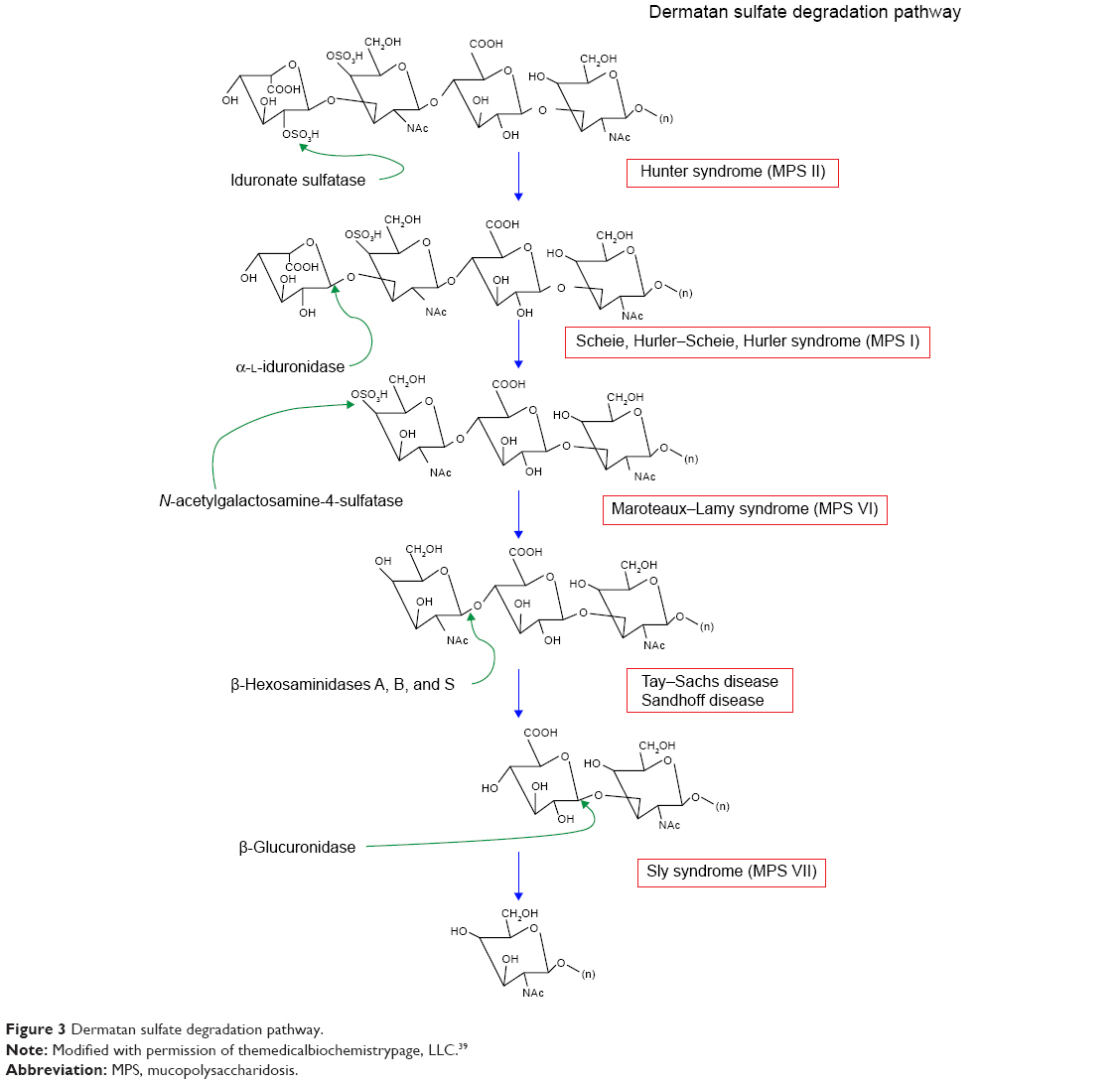 | Figure 3 Dermatan sulfate degradation pathway. |
MPS I: Scheie syndrome
Patients with Scheie syndrome can present with symptoms of shortness of breath, joint pain and stiffness, and poor vision. They often have characteristic facial changes, and mild respiratory and cardiac disease. They usually have a normal life span and intelligence. Other manifestations include hepatosplenomegaly (Figure 4), umbilical and inguinal hernia, aortic valve stenosis, cord compression, middle ear disease, deafness, and dental caries. Patients with MPS I-S may have mild corneal opacification requiring transplant in early adulthood and retinal pigment epithelium degeneration. Optic nerve atrophy is uncommon.1
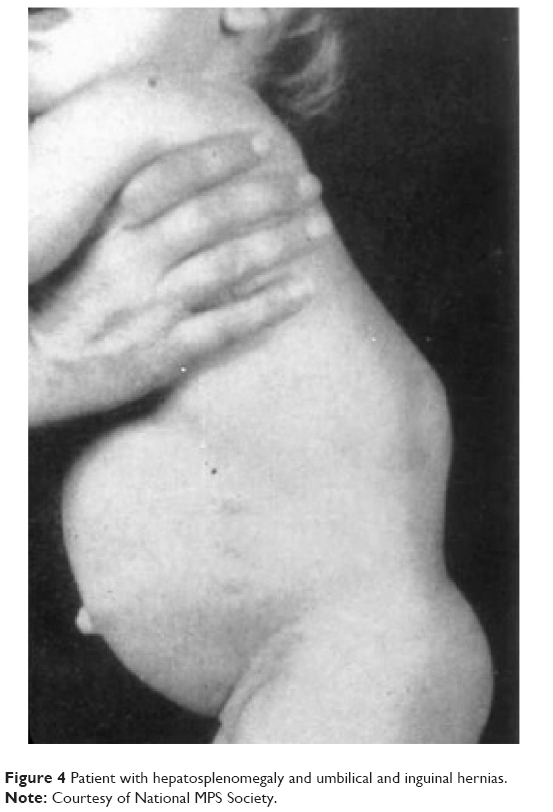 | Figure 4 Patient with hepatosplenomegaly and umbilical and inguinal hernias. |
MPS I: Hurler–Scheie syndrome
This intermediate form usually manifests with mild facial changes and premature death due to cardiorespiratory disease. As with Scheie syndrome, intelligence is often unaffected. These patients may have diffuse corneal thickening, retinopathy, and both chronic angle-closure and open-angle glaucoma. In addition, patients commonly present with optic nerve head swelling and atrophy.1
Aragona et al did a study evaluating the cornea in a patient with MPS I-HS. Confocal microscopic evaluation showed basal epithelial cells with diffuse or granular hyperreflectivity, highly reflective keratocytes, and barely visible endothelial cells indicating degeneration.17 Histopathologic changes included superficial cells with apical microfolds, small vesicles, and intercellular junctions.17 The wing cells and basal cells also showed small vesicles. The stroma had irregularly oriented lamellae, vesicle-filled keratocytes, and intercellular granular material.17 These changes are indicative of the accumulation of GAGs.
MPS I: Hurler syndrome
Clinical manifestations usually develop over the first year of life. Some of the characteristic systemic manifestations include coarse facies, multiple hernias, dysostosis multiplex, hepatosplenomegaly, and intellectual impairment. Ophthalmic findings include retinal pigmentary degeneration, optic nerve head swelling, glaucoma, and corneal clouding. Corneal clouding is the most common. Huang et al presented a 14-year-old girl with a history of bone marrow transplantation and a right penetrating keratoplasty.9 Her corneal abnormalities were not corrected after her bone marrow transplantation at the age of 2 years.9 Bone marrow transplant has improved prognoses for patients with Hurler syndrome; however, its effect on corneal opacity remains unclear.
Beyond corneal clouding, there have been other ocular manifestations of Hurler syndrome noted in the literature. Collins et al observed optic nerve head swelling in eight out of 14 eyes with Hurler syndrome, nine out of 21 in Hurler–Scheie syndrome, and 0 out of four in Scheie syndrome.10 Glaucoma is another known manifestation of MPS type I disease. Of the three children in a case series by Nowaczyk et al with Hurler syndrome and glaucoma, one was managed with medical therapy, and the other two required goniotomies to achieve normal IOP.12
MPS type II: Hunter syndrome
The incidence of Hunter syndrome is thought to be one in every 100,000 to 150,000 male births.16 It is unique among the MPS disorders in that it is inherited in an X-linked recessive pattern. The enzymatic deficiency is of iduronate-2-sulfatase, which results in accumulation of dermatan and heparan sulfate.1 It is on gene IDS at locus Xq28.13 Systemic manifestations consist of abdominal hernias, viral upper respiratory infections, and recurrent otitis media. These findings are fairly common in the 1-year-old age group and may not alarm clinicians until later sequelae arise, such as learning disabilities, abnormal facial features, white skin lesions, declining cardiac function, obstructive airway disease, or multiple joint stiffness.1,16 Seizures and craniosynostosis are also common in patients with MPS II.1,5 Eighty-four percent of affected males have some neurologic involvement.18
Ocular manifestations of Hunter syndrome include exophthalmos, hypertelorism, disk swelling and optic atrophy, and retinopathy. Optic nerve swelling is present in about 20% of affected patients and optic atrophy in 11%.18 Corneal changes are usually mild and do not significantly impair vision.1 Discrete corneal lesions may be found on slit-lamp examination.18 Other ocular findings that have been reported in patients with MPS II include bilateral uveal effusions, peripheral pigment epithelial changes, and radial parafoveal folds.1
MPS type III: Sanfilippo syndromes A, B, C, and D
The prevalence of Sanfilippo syndrome is suspected to be higher than that of any other MPS. Incidence for all subtypes of MPS type III is estimated at one in 70,000.16 Sanfilippo type A is the most common and severe form of the disease. The individual subtypes of MPS III are characterized by enzyme deficiency. Types A, B, C, and D are caused by deficiency in heparin sulfamidase, α-N-acetylglucosaminidase, acetyl-CoA:α-glucosaminide, and N-acetylgalactosamine-6-sulfatase, respectively. The proposed genes affected are SGSH (17q25.3), NAGLU (17q21), HGSNAT (8p11.1), and GNS (12p14), respectively.13 All subtypes result in an accumulation of heparan sulfate.
Clinically, the subtypes of MPS III are indistinguishable. These patients present with severe learning and behavioral disturbances along with mild systemic features. Accumulation of heparan sulfate causes mild facial malformation, hirsutism, arthritis, nerve damage, and eventual progression to dementia. Patients may also have behavioral disturbance, sleep problems, middle ear disease, dental caries, hernia, and joint contractures.1 Most patients do not live past their teenage years, though some patients have survived into their 30s.
One prominent ocular finding in MPS III patients is retinopathy with pigmentary retinal degeneration and associated electroretinogram changes.1 Patients with Sanfilippo syndrome usually do not develop significant corneal opacification, though rare cases have been documented. Optic atrophy and disk swelling and bushy eyebrows may be present in some patients.1
MPS type IV: Morquio syndromes A and B
The incidence of Morquio syndrome has been measured as one in 200,000 live births.16 Originally, MPS IV was divided into MPS IVA and MPS IVB. MPS IVB is now thought to be a variant of GM1-gangliosidosis due to mutated GLB1 at locus 3p21.33.13,16 MPS IVA is caused by mutations in GALNS (16q24.3) and deficient N-acetylgalactosamine-6-sulfatase leading to a buildup of keratan sulfate (Figure 5). Accumulation of this compound does not reach high enough levels to cause problems until 1 to 3 years of age.16 A characteristic feature of Morquio syndrome is skeletal deformities like joint laxity, and kyphoscoliosis resulting in a shortened torso.19 These patients may develop deafness and middle ear disease, restrictive respiratory disease, hernia, and cardiac valve lesions.1 As a result, patients may experience shortness of breath and trouble hearing, as well as poor vision. Notably, patients with this type of MPS have normal mental function.
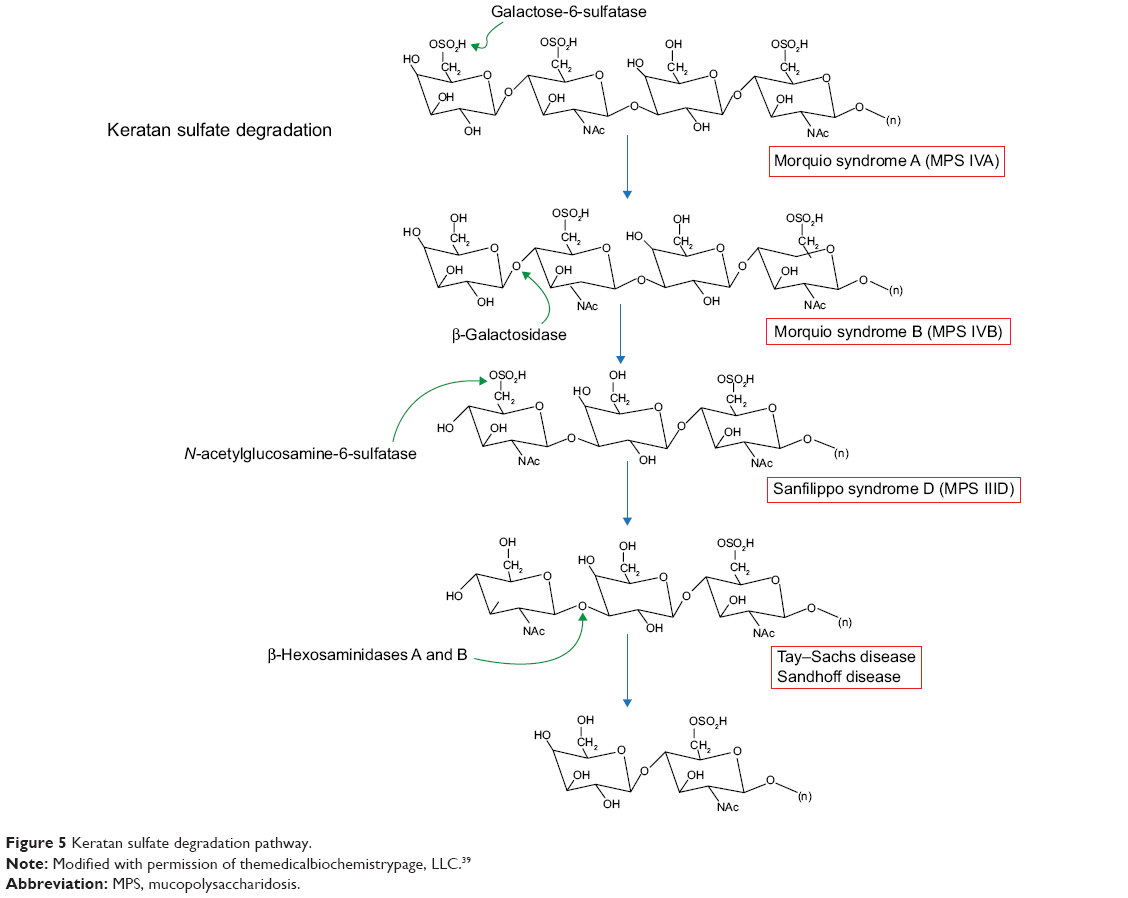 | Figure 5 Keratan sulfate degradation pathway. |
Patients with Morquio syndrome display mild corneal opacification, though severe clouding can be present,20 and pseudo-exophthalmos from shallow orbits.20 Transplantation as therapy for opacification results in reaccumulation and reinfiltration in the graft, but can improve quality of life.20 Corneal exposure keratopathy can result from pseudo-exophthalmos and is implicated in causing peripheral corneal vascularization in some patients. Retinopathy also occurs.1,20
MPS type VI: Maroteaux–Lamy syndrome
Another very rare form of MPS is called Maroteaux–Lamy syndrome. With an incidence somewhere between one case per 250,000 to 600,000 live births, few patients have been diagnosed with this condition worldwide. Like the other lysosomal storage diseases, it is inherited in an autosomal recessive pattern. MPS type VI is caused by deficiencies of N-acetylgalactosamine-4-sulfatase leading to an excess of dermatan sulfate.1 The affected gene is ARSB at locus 5q11.13 The signs of MPS VI include coarse facies, umbilical hernia, restriction of joint movement, cardiac disease, hepatosplenomegaly, and macroglossia. However, patients with this subtype usually lack delayed mental development or retardation. Patients may also present with hydrocephaly and spinal stenosis.1
Progressive corneal opacification is one of the most important ocular manifestations of MPS VI and causes the majority of visual morbidity in this population. It can be effectively treated with corneal transplant.1 Glaucoma and elevated IOP have been reported in MPS I and MPS VI. As addressed previously, it can be difficult to evaluate these patients for glaucoma since corneal opacification interferes with examination of the disk as well as visualization of anterior segment structures with gonioscopy. Retinopathy is not associated with MPS VI. Patients with Maroteaux–Lamy syndrome may have optic atrophy and swelling or papilledema associated with hydrocephalus.1
MPS type VII: Sly syndrome
Sly Syndrome is a rare disorder that is estimated to occur in one in 250,000 live births.16 It is caused by deficiencies of β-glucuronidase leading to inability to break down heparan and dermatan sulfate. The GUSB gene at locus 7q11 is likely mutated.13 In its rare form, MPS VII causes hydrops fetalis and survival is less than a few months. More commonly, patients with Sly syndrome survive into their teenage or young adult years.16 Major systemic signs and symptoms include coarse facies, macrocephaly, frontal prominence, depressed nasal bridge, spinal kyphosis, pectus excavatum or pectus carinatum, upper airway obstruction, cardiomyopathy, hepatosplenomegaly, hernia, hydrocephalus, and learning difficulties.1
The major causes of ocular morbidity are corneal opacification, which is generally milder than in other forms of MPS, and optic nerve malformation. Penetrating keratoplasty may be necessary to treat this opacification.1
MPS type IX: Natowicz syndrome
Epidemiologic data for Natowicz syndrome are not available because it is extremely uncommon. It is thought to be due to deficiency of hyaluronidase and accumulation of hyaluronan. The HYAL1 gene at locus 3p21 is affected.13 The first description comes from a 1996 case report by Natowicz et al. A 14-year-old girl presented with frequent episodes of otitis media, soft tissue masses, short stature, and mildly dysmorphic craniofacial features. She had full range of motion in all joints, no organomegaly, and normal ophthalmologic and neurologic examinations. She was found to have pathologic storage of mucopolysaccharide in lysosomes of histiocytes and fibroblasts, markedly elevated plasma hyaluronan concentration, and complete deficiency of hyaluronidase activity.21 Imundo et al described three affected children from a single consanguineous family that presented with knee or hip pain and swelling.22 Enzymatic analysis showed hyaluronoglucosaminidase 1 (HYAL1) deficiency, which degrades hyaluronan, and confirmed the diagnosis of MPS IX.22 These three patients had isolated joint disease and lacked the findings of a bifid uvula, cleft palate, and recurrent otitis media that Natowicz reported.21,22 Patients with MPS IX appear to have normal intelligence and mental development.22 Ocular manifestations of this disease have not been studied due to its extremely low prevalence, though no cases to date report ocular abnormalities with this condition.
Treatment options and management recommendations
Treatment of the MPS disorders depends on the severity of clinical manifestations and usually requires a multidisciplinary approach. Pediatricians, orthopedic surgeons, cardiologists, radiologists, neurologists, anesthesiologists, and ophthalmologists must work together to achieve the best outcome for the patient.13
HSCT was initially proposed as a way to target and prevent or slow down pathologic responses in all organ systems. It was first performed for MPS in a 9-month-old patient with Hurler syndrome in 1980.13 Since then, it has been used in patients with severe neuronopathic MPS (MPS type I) to increase life expectancy and provide some improvement in symptoms. It is considered optional or investigational in other types.13 It is most effective when initiated early, before 2 years of age. This process works by introducing enzymatically normal bone marrow cells from a matched donor. The cells then travel through the body and secrete the deficient enzyme. The clinical response depends upon the concentration of donor cells and the penetration level of the enzyme into recipient tissues.23 Recently, patients have undergone HSCT with unrelated donor umbilical cord blood with promising outcomes.13 HSCT appears to stabilize or improve visual acuity, corneal opacification, and optic nerve swelling in MPS patients.23 It appears to have no effect on retinal degeneration.14 Risks of HSCT include the development of cataract, epithelial punctate keratopathy, and dry eye syndrome, as well as graft versus host disease causing conjunctivitis, keratoconjunctivitis sicca, corneal epithelial defects, and pseudomembrane formation.14
ERT uses recombinant DNA in mammalian cell lines to replace the enzyme deficiency through intravenous infusion. ERT is available for MPS I-H, MPS II, and MPS VI with laronidase (Aldurazyme; Genzyme, Cambridge, MA, USA), idursulfase (Elaprase; Shire Human Genetic Therapies, Lexington, MA, USA), and galsulfase (Naglazyme; BioMarin Pharmaceutical Inc, Novato, CA, USA), respectively.13 Membrane receptors are necessary for intracellular transport of the enzyme. It has been shown that receptor expression levels vary among tissue, with large concentrations in the heart, lung, and kidney and lower levels in muscle and brain tissue.13 These penetration problems at privileged sites and the fact that the enzyme does not cross the blood–brain barrier may explain the lack of effect of ERT on neurologic symptoms. ERT is recommended for use in patients with mild manifestations of MPS, as patients with more severe phenotypes are given the option of HSCT.13 ERT may improve symptoms such as hepatosplenomegaly, urinary GAG excretion, respiratory function, and joint and cardiovascular involvement if initiated early in the disease process.13 In general, ERT is associated with stabilization of corneal clouding. Visual acuity is more difficult to assess because of multiple comorbid ocular conditions that may be present including glaucoma, optic nerve edema, optic atrophy, and retinal degeneration. It is worth noting that patients who receive ERT are often older in age than those receiving HSCT. Beginning ERT at an earlier age may have a greater effect on ocular findings.23,24 The effects of ERT on optic nerve edema and atrophy is inconclusive, with both stabilization and worsening of disease observed.25 Of note, some patients may develop antibodies against the protein that could affect therapy.13 Further study is needed to determine the effects of ERT on ocular pathology related to MPS, especially in those patients in whom ERT is started early in life.
Reports have varied regarding the evolution of corneal opacification after HSCT or ERT.13,23 This determination was often based on the subjective judgment of the clinician. There have recently been studies that demonstrate that Pentacam (Pentacam Rotating Scheimpflug Camera; Oculus, Wetzlar, Germany) densitometry or iris recognition cameras can provide a reliable and objective determination of opacification.26,27 This will be useful both in research settings and clinical practice to determine the effectiveness of treatments in the future.
Management of corneal opacification in MPS depends on the severity of symptoms. Corneal transplantation may be helpful, but, without systemic treatment, the graft may opacify due to GAG deposition in donor keratocytes.13 Effectiveness is dependent on the type of GAG deposited and the severity of the disease. Fifty percent of the mucopolysaccharides in a normal cornea are keratan sulfate, 25% are chondroitin sulfate, and 25% are chondroitin-4-sulfate.28 Dermatan sulfate is present in healed corneal wounds, rejected grafts, and post-viral opacification.28 Käsmann-Kellner et al suggest that perforating keratoplasty may have better long-term results in diseases with dermatan sulfate accumulation than in diseases with keratan sulfate accumulation.20 Re-opacification may develop as early as 1 year after surgery.25 Deep anterior lamellar keratoplasty has been recommended over penetrating keratoplasty due to decreased risk of rejection. Endothelial involvement occurs only in late stages of the disease, and an intact Descemet’s membrane may act as a barrier and prevent stromal recurrence of disease in the graft.13
Limbal stem cell transplant in conjunction with keratoplasty may be effective in MPS patients. This would ensure a source of epithelial cells and delay recurrence of corneal opacification.13 However, visual outcome may be limited despite successful keratoplasty due to coexisting retinopathy, optic nerve dysfunction, and cortical impairment.13
Management of glaucoma is also difficult in the MPS patient due to the challenge of following the progression of the disease and because data regarding the effectiveness of antiglaucoma therapy are limited.25 As mentioned previously, IOP may be falsely elevated due to increased corneal thickness or altered corneal resistance. Interestingly, a study by Kottler et al found no significant difference in central corneal thickness in patients with MPS II and VI when compared with healthy subjects.29 In general, pachymetry measurements should be included in the evaluation for glaucoma in MPS patients. Many patients are young children with developmental delay, making visual field testing challenging and further adding to the difficulty in monitoring progression. Trabeculectomies have been performed with mixed results.30 There has been one published case report of an MPS I-H patient whose IOP returned to normal after HSCT.31 Risks and benefits must be reviewed on an individual basis when considering treatment for glaucoma in an MPS patient.
Management of optic nerve disease is dependent on the cause of the optic nerve pathology. HSCT has shown positive results in improving optic disk pathology. In one study, 30% of patients demonstrated optic nerve edema associated with increased intracranial pressure, all of which resolved after bone marrow transplantation and with no sign of optic disk atrophy afterward.32 Another study showed that 70% of MPS I patients had normal optic nerves after HSCT.33 The results for ERT seem to be mixed, with one report of reversed papilledema and improvement of visual acuity.34 Another report showed no improvement in papilledema after ERT,35 and an ultrasonographic study demonstrated no change in the morphological findings of the optic nerve.11
Data regarding the effects of therapy on retinal pathology are sparse and not very promising. One study showed little or no improvement with bone marrow transplantation treatment.36 Another study showed initial improvement in ERG in 81% of MPS patients after bone marrow transplantation, with a progressive worsening after 1 year.32 However, novel approaches to treatment are being developed using gene therapy to manage retinal pathology in MPS with positive results in animal models. Trials in canines showed a sustained increase in the action of β-glucuronidase in retinal pigment epithelium cells after transduction with viruses containing β-glucuronidase cDNA.37
Many MPS patients are hyperopic. There are multiple possible mechanisms to explain this phenomenon. One is that thickening of the sclera leads to reduced axial length and hypermetropia.38 Another is that the increased rigidity of the cornea and sclera from GAG accumulation induce a flattening effect upon the cornea, reducing its refractive power.38 Patients become hypermetropic even after receiving systemic treatments. Since these changes occur early in life, it is important to evaluate and correct the refractive error in patients with MPS early and regularly to avoid amblyopia and strabismus.
Ocular evaluation and management of patients with MPS should be comprehensive. Fahnehjelm et al recommend seeing the patient every 6 to 12 months during preschool age and then annually until they reach age 18 years.14 Patients and caretakers should be educated to seek medical attention if there is any significant change in vision or symptoms. While progress has been made in terms of systemic treatments for MPS patients, more data are needed to determine their effect on ocular manifestations, particularly in retinal and posterior segment pathology. Further studies on ERT are needed to confirm its effects on the eye and improve methods of delivery. Additionally, the relationship of corneal clouding, central corneal thickness, and IOP measurements needs to be more precisely delineated.
Conclusion
The MPS are a group of disorders that require special attention from ophthalmologists. The ocular manifestations of these diseases can affect all parts of the eye and can be challenging to evaluate and manage. As systemic treatments increase life expectancy for MPS patients, ophthalmologists must be prepared to appropriately manage ocular features of MPS and ensure that these patients have the best possible quality of life.
Disclosure
The authors report no conflicts of interest in this work.
References
Ashworth JL, Biswas S, Wraith E, Lloyd IC. Mucopolysaccharidoses and the eye. Surv Ophthalmol. 2006;51(1):1–17. | ||
Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50 Suppl 5:v13–v18. | ||
Kumar V, Abbas AK, Fausto N, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 8th ed. Philadelphia: Saunders; 2010. | ||
Cimaz R, La Torre F. Mucopolysaccharidoses. Curr Rheumatol Rep. 2014;16:389. | ||
Ziyadeh J, Le Merrer M, Robert M, Arnaud E, Valayannapoulos V, Di Rocco F. Mucopolysaccharidosis type I and craniosynostosis. Acta Neurochir (Wien). 2013;155:1973–1976. | ||
Golda A, Jurecka A, Tylki-Szymanska A. Cardiovascular manifestations of mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome). Int J Cardiol. 2012;158:6–11. | ||
Alroy J, Haskins M, Birk DE. Altered corneal stromal matrix organization is associated with mucopolysaccharidosis I, III and VI. Exp Eye Res. 1999;68:523–530. | ||
Mollard RJ, Telegan P, Haskins M, Aguirre G. Corneal endothelium in mucopolysaccharide storage disorders. Morphologic studies in animal models. Cornea. 1996;15(1):25–34. | ||
Huang Y, Bron AJ, Meek KM, Vellodi A, McDonald B. Ultrastructural study of the cornea in a bone marrow-transplanted Hurler syndrome patient. Exp Eye Res. 1996;62:377–387. | ||
Collins ML, Traboulsi El, Maumenee IH. Optic nerve head swelling and optic atrophy in the systemic mucopolysaccharidoses. Ophthalmology. 1990;97(11):1445–1449. | ||
Schumacher RG, Brzezinska R, Schulze-Frenking G, Pitz S. Sonographic ocular findings in patients with mucopolysaccharidoses I, II and VI. Pediatr Radiol. 2008;38:543–550. | ||
Nowaczyk MJ, Clarke JT, Morin JD. Glaucoma as an early complication of Hurler’s disease. Arch Dis Child. 1988;63:1091–1093. | ||
Ganesh A, Bruwer Z, Al-Thihli K. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol. 2013;24(5):379–388. | ||
Fahnehjelm KT, Ashworth JL, Pitz S, et al. Clinical guidelines for diagnosing and managing ocular manifestations in children with mucopolysaccharidosis. Acta Ophthalmol. 2012;90:595–602. | ||
Fahnehjelm KT, Chen E, Winiarski J. Corneal hysteresis in mucopolysaccharidosis I and VI. Acta Ophthalmol. 2012;5:445–448. | ||
“Mucopolysaccharidoses Fact Sheet”. National Institute of Neurological Disorders and Stroke (NINDS) [webpage on the Internet]. Available from www.ninds.nih.gov/disorders/mucopolysaccharidoses/detail_mucopolysaccharidoses.htm. Accessed September 1, 2014. | ||
Aragona P, Wylegala E, Wroblewska-Czajka E, et al. Clinical, confocal, and morphological investigations on the cornea in human mucopolysaccharidosis IH-S. Cornea. 2014;33(1):35–42. | ||
Scarpa M. Mucopolysaccharidosis type II. In: Pagon RA, Adam MP, Ardinger HH, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington; 1993–2015. Available from: www.ncbi.nlm.nih.gov/books/NBK1116/. Accessed November 16, 2014. | ||
Wraith JE. Mucopolysaccharidoses and oligosaccharidoses. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn Metabolic Diseases: Diagnosis and Treatment. 5th ed. New York, NY: Springer; 2012;39:497–507. | ||
Käsmann-Kellner B, Weindler J, Pfau B, Ruprecht KW. Ocular changes in mucopolysaccharidosis IV A (Morquio A syndrome) and long-term results of perforating keratoplasty. Ophthalmologica. 1999;213(3):200–205. | ||
Natowicz MR, Short MP, Wang Y, et al. Clinical and biochemical manifestations of hyaluronidase deficiency. N Engl J Med. 1996;335:1029–1033. | ||
Imundo L, Leduc CA, Guha S, et al. A complete deficiency of Hyaluronoglucosaminidase 1 (HYAL1) presenting as familial juvenile idiopathic arthritis. J Inherit Metab Dis. 2011;34:1013–1022. | ||
Summers CG, Fahnehjelm KT, Pitz S, et al. Systemic therapies for mucopolysaccharidosis: ocular changes following haematopoietic stem cell transplantation or enzyme replacement therapy – a review. Clin Experiment Ophthalmol. 2010;38:34–42. | ||
Pitz S, Ogun O, Bajbouj M, Arash L, Schulze-Frenking G, Beck M. Ocular changes in patients with mucopolysaccharidosis I receiving enzyme replacement therapy: a 4-year experience. Arch Ophthalmol. 2007;125(10):1353–1356. | ||
Ferrari S, Ponzin D, Ashworth JL, et al. Diagnosis and management of ophthalmological features in patients with mucopolysaccharidosis. Br J Ophthalmol. 2011;95:613–619. | ||
Elflein HM, Hofherr T, Berisha-Ramadani F, et al. Measuring corneal clouding in patients suffering from mucopolysaccharidosis with the Pentacam densitometry programme. Br J Ophthalmol. 2013;97(7): 829–833. | ||
Aslam TM, Shakir S, Wong J, Au L, Ashworth J. Use of iris recognition camera technology for the quantification of corneal opacification in mucopolysaccharidoses. Br J Ophthalmol. 2012;96(12):1466–1468. | ||
Bothun ED, Decanini A, Summers CG, Orchard PJ, Tolar J. Outcome of penetrating keratoplasty for mucopolysaccharidoses. Arch Ophthalmol. 2011;129(2):138–144. | ||
Kottler U, Demir D, Schmidtmann I, Beck M, Pitz S. Central corneal thickness in mucopolysaccharidosis II and VI. Cornea. 2010;29(3):260–262. | ||
Mullaney P, Awad AH, Millar L. Glaucoma in mucopolysaccharidosis 1–H/S. J Pediatr Ophthalmol Strabismus. 1996;33(2):127–131. | ||
Christiansen SP, Smith TJ, Henslee-Downey PJ. Normal intraocular pressure after a bone marrow transplant in glaucoma associated with mucopolysaccharidosis type I-H. Am J Ophthalmol. 1990;109(2):230–231. | ||
Gullingsrud EO, Krivit W, Summers CG. Ocular abnormalities in the mucopolysaccharidoses after bone marrow transplantation. Longer follow-up. Ophthalmology. 1998;105(6):1099–1105. | ||
Connell P, McCreery K, Doyle A, Darcy F, O’Meara A, Brosnahan D. Central corneal thickness and its relationship to intraocular pressure in mucopolysaccararidoses-1 following bone marrow transplantation. J AAPOS. 2008;12(1):7–10. | ||
Koseoglu ST, Harmatz P, Turbeville S, Nicely H. Reversed papilledema in an MPS VI patient with galsulfase (Naglazyme) therapy. Int Ophthalmol. 2009;29(4):267–269. | ||
Pitz S, Ogun O, Arash L, Miebach E, Beck M. Does enzyme replacement therapy influence the ocular changes in type VI mucopolysaccharidosis? Graefes Arch Clin Exp Ophthalmol. 2009;247(7):975–980. | ||
Tzetzi D, Hamilton R, Robinson PH, Dutton GN. Negative ERGs in mucopolysaccharidoses (MPS) Hurler-Scheie (I-H/S) and Hurler (I-H)-syndromes. Doc Ophthalmol. 2007;114:153–158. | ||
Verdugo ME, Scarpino V, Moullier P, Haskins ME, Aguirre GD, Ray J. Adenoviral vector-mediated beta-glucuronidase cDNA transfer to treat MPS VII RPE in vitro. Curr Eye Res. 2001;23(5):357–367. | ||
Fahnehjelm KT, Törnquist AL, Winiarski J. Ocular axial length and corneal refraction in children with mucopolysaccharidosis (MPS I-Hurler). Acta Ophthalmol. 2012;90:287–290. | ||
The medical biochemistry page [website on the Internet]. [updated May 14, 2015]. Available from: http://themedicalbiochemistrypage.org/. Accessed May 9, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.