Back to Journals » Clinical Ophthalmology » Volume 8
Ocular findings in two siblings with Joubert syndrome
Received 3 December 2013
Accepted for publication 24 December 2013
Published 15 January 2014 Volume 2014:8 Pages 229—233
DOI https://doi.org/10.2147/OPTH.S58672
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Shinji Makino, Hironobu Tampo
Department of Ophthalmology, Jichi Medical University, Shimotsuke, Tochigi, Japan
Abstract: We describe two brothers with Joubert syndrome (JS). JS diagnosis was made on the basis of neurological findings and the presence of the characteristic "molar tooth sign", which was subsequently confirmed by magnetic resonance imaging. Both brothers demonstrated ptosis, hypotropia, exotropia, and horizontal pendular nystagmus. The younger brother had mild chorioretinal discoloration at the peripapillary region in both eyes, and a small coloboma at the inferior region of his right optic disc. The elder brother had coloboma in his right eye and a colobomatous optic disc in his left eye. Optical coherence tomography showed that the foveal architecture was preserved in both patients. We discuss the ocular findings, including those from optical coherence tomography, in JS, which has recently been recognized as ciliopathy.
Keywords: strabismus, coloboma, optical coherence tomography, ciliopathy
Introduction
Joubert syndrome (JS) is a rare genetic disorder that was first described in 1969.1 It is characterized by episodic hyperpnea, abnormal eye movements, ataxia, and developmental delay with the pathognomonic finding of a “molar tooth sign” (MTS) on magnetic resonance imaging (MRI) of the brain.1–3 MTS refers to the abnormal structural features that include cerebellar vermis hypoplasia, deepened interpeduncular fossa, and elongated, horizontally oriented, thickened superior cerebellar peduncles. Since MTS is not specific for JS, other clinical features are also used to define the subtypes of JS and JS-related disorders. Although the term “JS-related disorders” describes conditions that apply to MTS and the clinical features of JS, there are other clinical manifestations involving the brain (occipital encephalocele, corpus callosum agenesis), eyes (coloboma, retinal dystrophy, nystagmus, oculomotor apraxia), kidneys (nephronophthisis, cystic dysplasia), liver (hepatic fibrosis), and limbs (polydactyly).4,5 In particular, these include other diseases, such as isolated nephronophthisis, Senior–Løken syndrome, and Bardet–Biedl syndrome, as well as cerebellar and brainstem congenital malformations and cerebro–oculo–renal syndromes.
Recently, dysfunction of the subcellular organelle, the primary cilium, was shown to be the pathogenic basis of this clinically and genetically heterogeneous disorder. Therefore, this means JS can be classified as part of an expanding group of disorders collectively called ciliopathies.6–8
Ocular and oculomotor abnormalities are common in JS and are helpful in making a diagnosis.3 Between 70% and 100% of JS patients have been found to have such abnormalities. In these patients, the abnormal eye movements vary and include nystagmus, strabismus, oculomotor apraxia, and vertical gaze palsy. Retinal problems include congenital retinal dystrophy, pigmentary retinopathy, fundus flavimaculatus, chorioretinal coloboma, and perimacular and retinal blindness. Several detailed reports have been published on JS patients.9–18 To the best of our knowledge, however, there have been few reports in the literature describing fundus photographs in these patients. Furthermore, there have been no cases examined by optical coherence tomography (OCT). Herein, we describe ocular findings in two siblings with JS.
Case report
Case 1
A 6-year-old boy presented with ptosis and strabismus of his left eye along with an abnormal head posture. He had delayed motor and language development with episodic hyperpnea–apnea spells and hypotonia in infancy. JS diagnosis was made on the basis of the characteristic MTS on MRI (Figure 1) and neurological findings observed in the pediatric clinic. He showed mild facial dysmorphism in the form of forehead prominence and bilateral epicanthic folds. He had ptosis, hypotropia, and exotropia in his left eye (Figure 2). His abnormal head posture included a head tilt to the left and turning his face to the left. On ophthalmic examination, the best-corrected visual acuity in his right eye was 0.8 with +0.25 D –1.00 D ×90°, and 0.5 with +0.50 D –2.00 D ×60° in his left eye. The prism cover test demonstrated that there was 20 prism diopter hypotropia and 35 prism diopter exotropia of his left eye. The patient was unable to maintain his eyes in a central position by convergence, and he had horizontal pendular nystagmus. Slit-lamp examination showed no other abnormalities in either of his eyes. Funduscopy showed mild chorioretinal discoloration at the peripapillary region in both eyes (Figure 3, arrows). Additionally, a small coloboma was detected at the inferior region of his right optic disc (Figure 3, arrowhead). OCT (RS-3000; NIDEK, Gamagori, Japan) showed that the foveal architecture was preserved in both eyes, although there was poor recording due to nystagmus (Figure 4).
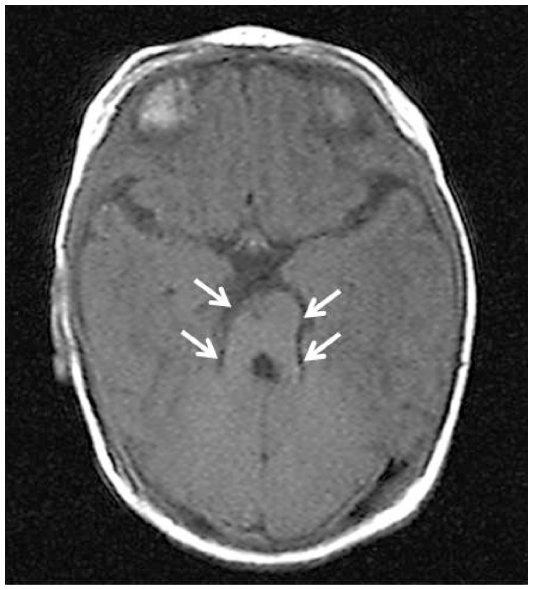 | Figure 1 Case 1: cranial MRI showing “molar tooth sign” (arrows), which is a classic finding in Joubert syndrome. |
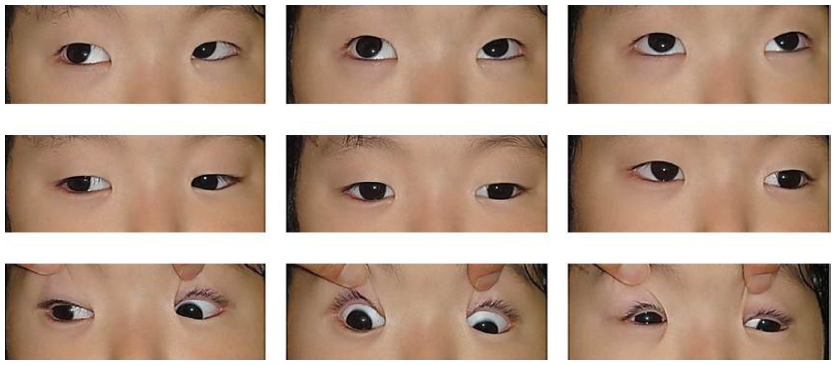 | Figure 2 Case 1: ptosis, hypotropia, and exotropia in the patient’s left eye. |
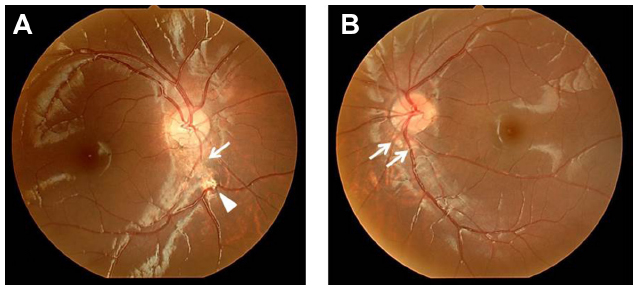 | Figure 3 Case 1: funduscopy findings. |
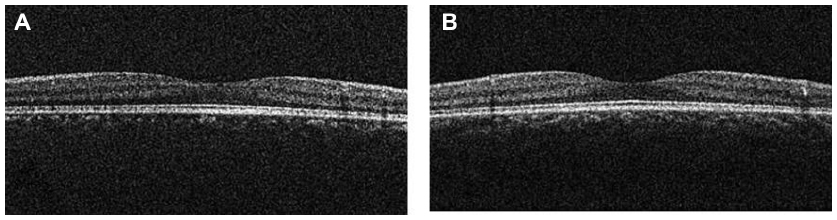 | Figure 4 Case 1: optical coherence tomography showed that the foveal architecture was preserved in both eyes. |
Case 2 (elder brother of Case 1)
An 11-year-old boy presented with ptosis and strabismus of his left eye along with an abnormal head posture. He had delayed motor and language development with episodic hyperpnea–apnea spells and hypotonia in infancy. JS diagnosis was made on the basis of the characteristic MTS on MRI and neurological findings observed in the pediatric clinic. He showed mild facial dysmorphism in the form of forehead prominence and bilateral epicanthic folds. He had ptosis, hypotropia, and exotropia in his left eye, and he was unable to maintain his eyes in a central position by convergence. He also had horizontal pendular nystagmus. On ophthalmic examination, best-corrected visual acuity in his right eye was 1.2 with −1.00 D, and was 1.2 with −1.25 D −1.50 D ×50° in his left eye. Slit-lamp examination showed no abnormalities in either of his eyes. Funduscopy showed coloboma in his right eye and a colobomatous optic disc in his left eye (Figure 5, arrows). OCT showed that the foveal architecture was preserved in both eyes (Figure 6). In addition, on the basis of the Early Treatment Diabetic Retinopathy Study sectors, the thickness of center (central fovea), inner ring (1–3 mm from the central fovea), and outer ring (3–6 mm from the central fovea) in his right eye were 283 μm, 327.5 μm, and 272.0 μm, respectively. In his left eye, these measurements were 284 μm, 315.0 μm, and 270.0 μm, respectively.
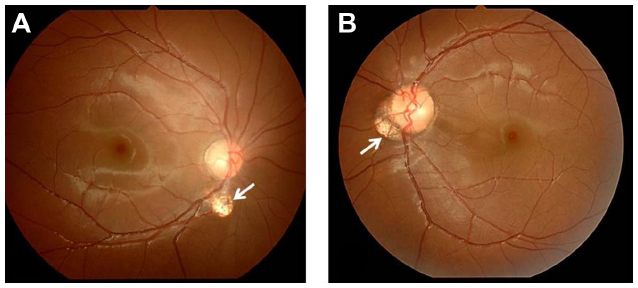 | Figure 5 Case 2: funduscopy findings. |
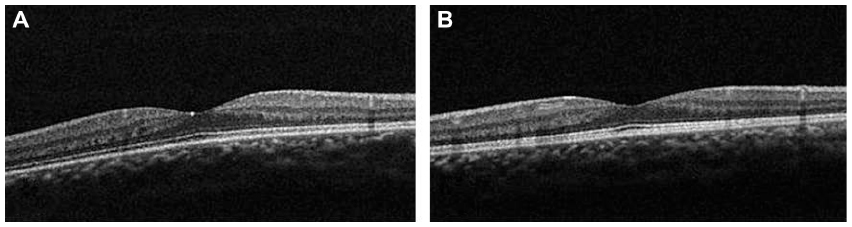 | Figure 6 Case 2: optical coherence tomography showed that the foveal architecture was preserved in both eyes. |
Discussion
The incidence of JS has been estimated to be between 1/80,000 and 1/100,000 live births.8
The cardinal neurological features of JS are hypotonia that evolves into ataxia and developmental delay. The syndrome is often associated with intellectual disability, altered respiratory pattern in the neonatal period, and abnormal ocular movements.3–5,8 Primary position nystagmus is also commonly observed. In addition, strabismus is not uncommon and presents as either horizontal or vertical misalignment. Horizontal strabismus can be an exodeviation or esodeviation, fixed or alternating in nature.11,12,14 In the current cases, the type of strabismus proved to be similar in both brothers.
The retina is one of the organs that is most frequently involved in JS, mostly in the form of retinal dystrophy that occurs due to progressive degeneration of the photoreceptor cells.3–5,10 Colobomas can be unilateral or bilateral, and mostly affect the posterior segment of the eye. Recently, JS has been classified into six phenotypic subgroups: pure JS; JS with ocular defect; JS with renal defect; JS with oculo-renal defects; JS with hepatic defect; and JS with orofaciodigital defects.5 Although colobomas represent a rare cause of visual impairment in all JS clinical subgroups, in the JS-with-hepatic defect subgroup, the frequency of colobomas can be over 30%. Our cases were classified as JS with ocular defect.
JS is further classified into two groups on the basis of the presence or absence of retinal dystrophy.10 Patients with retinal dystrophy have a higher prevalence of multicystic renal disease, with these patients appearing to have decreased survival rates compared with those of patients without retinal dystrophy. Although electroretinograms were not available in the current study, our cases did not exhibit any retinal dystrophy or other organ abnormalities.
Despite the tremendous acceleration in gene recovery enabled by next-generation sequencing techniques, the extremely complex JS genetic bases are still only partly understood. Recently, 21 causative genes have been identified, with autosomal or X-linked recessive inheritance.8 Since causative genes have been shown to encode proteins that localize to the primary cilium, JS has been classified as one of a number of the so-called ciliopathies. The primary cilium is a nonmotile type of cilium that is found on nearly every cell of the body. Photoreceptors contain a primary cilium known as the connecting cilium, which is a slender structure that connects the outer and inner segment. In addition to its structural role, the photoreceptor cilium provides a critical transport function.6 Several genes (AHI1, INPP5E, ARL13B, and CC2D2A) have been shown to be nearly exclusively mutated in patients presenting with either pure JS or JS with retinal involvement.8,19 Westfall et al20 reported that AHI1 is required for the maintenance of photoreceptor outer segments, as it ensures that there is proper preservation of polarized vesicular trafficking to the cilia. When AHI1 is absent in the mouse model, retinal degeneration occurs.
We initially speculated that some microstructural abnormalities due to ciliopathy existed in the present cases. OCT revealed that, with the exception of coloboma, there were no retinal microstructural changes or thinning of the retina observed in our patients; however, the resolution of OCT cannot rule out histological alterations of ciliopathy at early age. Electroretinograms were not available in the current study. Further studies with additional cases will need to be evaluated in order to prove our speculation. Moreover, definitive characterization of JS will require that additional genetic screening be undertaken.
Disclosure
The authors report no conflicts of interest in this work.
References
Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. | |
Boltshauser E, Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977;8:57–66. | |
Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14:583–590; discussion 590–591. | |
Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Human Genet. 2007;15:511–521. | |
Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. | |
Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007;28:113–125. | |
Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. | |
Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12:894–905. | |
Lambert SR, Kriss A, Gresty M, Benton S, Taylor D. Joubert syndrome. Arch Ophthalmol. 1989;107:709–713. | |
Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43:726–731. | |
Maria BL, Hoang KB, Tusa RJ, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–430. | |
Maria BL, Quisling RG, Rosainz LC, et al. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14:368–376. | |
Hodgkins PR, Harris CM, Shawkat FS, et al. Joubert syndrome: long-term follow-up. Dev Med Child Neurol. 2004;46:694–699. | |
Khan AO, Oystreck DT, Seidahmed MZ, et al. Ophthalmic features of Joubert syndrome. Ophthalmology. 2008;115:2286–2289. | |
Sturm V, Leiba H, Menke MN, et al. Ophthalmological findings in Joubert syndrome. Eye (Lond). 2010;24:222–225. | |
Keskinbora KH. Ocular and oculomotor findings of Joubert syndrome. J Pediatr Ophthalmol Strabismus. 2008;45:5–6. | |
Lau FH, Yu CB, Yip WW, Fan DS. Serous retinal detachment in Joubert syndrome. J Pediatr Ophthalmol Strabismus. 2009;46:362–364. | |
Gill H, Muthusamy B, Atan D, Williams C, Ellis M. Joubert syndrome presenting with motor delay and oculomotor apraxia. Case Rep Pediatr. 2011;2011:262641. | |
Parisi MA, Doherty D, Eckert ML, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43:334–339. | |
Westfall JE, Hoyt C, Liu Q, et al. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1 J Neurosci. 2010;30:8759–8768. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.