Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Octreotide-Resistant Acromegaly: Challenges and Solutions
Authors Corica G, Ceraudo M, Campana C, Nista F, Cocchiara F, Boschetti M, Zona G, Criminelli D, Ferone D ![]() , Gatto F
, Gatto F ![]()
Received 23 January 2020
Accepted for publication 10 March 2020
Published 5 May 2020 Volume 2020:16 Pages 379—391
DOI https://doi.org/10.2147/TCRM.S183360
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Giuliana Corica,1,2 Marco Ceraudo,3 Claudia Campana,1,2 Federica Nista,1,2 Francesco Cocchiara,2 Mara Boschetti,1,2 Gianluigi Zona,3 Diego Criminelli,3 Diego Ferone,1,2 Federico Gatto1
1Endocrinology Unit, IRCCS Ospedale Policlinico San Martino, Genoa, Italy; 2Endocrinology Unit, Department of Internal Medicine and Medical Specialties (DIMI) and Centre of Excellence for Biomedical Research (CEBR), University of Genoa, Genoa, Italy; 3Neurosurgery Unit, Department of Neurosciences (DINOGMI), IRCCS Ospedale Policlinico San Martino, University of Genoa, Genoa, Italy
Correspondence: Diego Ferone
Endocrinology Unit, IRCCS Ospedale Policlinico San Martino, Largo Rosanna Benzi, 10, Genoa 16132, Italy
Tel +39 010 3537946
Fax +39 010 3537060
Email [email protected]
Abstract: Acromegaly is a rare and severe disease caused by an increased and autonomous secretion of growth hormone (GH), thus resulting in high circulating levels of insulin-like growth factor 1 (IGF-1). Comorbidities and mortality rate are closely related to the disease duration. However, in most cases achieving biochemical control means reducing or even normalizing mortality and restoring normal life expectancy. Current treatment for acromegaly includes neurosurgery, radiotherapy and medical therapy. Transsphenoidal surgery often represents the recommended first-line treatment. First-generation somatostatin receptor ligands (SRLs) are the drug of choice in patients with persistent disease after surgery and are suggested as first-line treatment for those ineligible for surgery. However, only about half of patients treated with octreotide (or lanreotide) achieve biochemical control. Other available drugs approved for clinical use are the second-generation SRL pasireotide, the dopamine agonist cabergoline, and the GH-receptor antagonist pegvisomant. In the present paper, we revised the current literature about the management of acromegaly, aiming to highlight the most relevant and recent therapeutic strategies proposed for patients resistant to first-line medical therapy. Furthermore, we discussed the potential molecular mechanisms involved in the variable response to first-generation SRLs. Due to the availability of different medical therapies, the choice for the most appropriate drug can be currently based also on the peculiar clinical characteristics of each patient.
Keywords: acromegaly, biochemical control, medical therapy, resistance, somatostatin receptor ligands
Introduction
Acromegaly is a rare, chronic disease due to the excess of growth hormone (GH) production and secretion. This results in increased levels of insulin-like growth factor 1 (IGF-1), leading to a number of different clinical manifestations. In about 95–98% of patients, a GH-secreting pituitary adenoma causes acromegaly.1,2
The incidence is approximately 3 cases per 1 million persons per year, though more recent estimates report an increased incidence (about 11 per million person-years).3–6 As far as prevalence, some studies estimate that the disease affects 40–70 patients per million of the general population, or even more (86–240 per million).7,8
Comorbidities and mortality risk in acromegalic patients are related to time of exposure to GH and IGF-1 excess.9 On the other hand, in most cases achieving disease control means reducing/normalizing the mortality rate and restoring normal life expectancy.10 Unfortunately, the diagnosis is often delayed, preceded by a mean of 7–10 years of undiagnosed active disease.1,11 Therefore, at the time of diagnosis, many patients already show a wide range of clinical signs and symptoms.
Current treatment includes neurosurgery, radiotherapy and medical therapy, alone or combined as multimodal therapeutic strategies.12,13
In the majority of patients, transsphenoidal neurosurgery is the recommended first-line treatment, since it provides a rapid reduction of GH levels with a relatively low complication rates.14
Three different classes of drugs are currently available for medical treatment: somatostatin receptor ligands (SRLs), dopamine agonists (DAs) and the GH-receptor antagonist (GHRA) pegvisomant (PEG).15 First-generation SRLs, such as octreotide (OCT) LAR (long-acting release) and lanreotide (LAN) Autogel, are recommended in patients with persistent disease after surgery, and as first-line treatment for those ineligible for surgery, whereas their role in neo-adjuvant settings is still debated.14,16
Radiotherapy remains an option in aggressive resistant tumors.17
The aims of treatment in patients with acromegaly are GH and/or IGF-1 levels normalization, tumor mass control and preservation of the remaining pituitary function, amelioration of signs and symptoms, management of comorbidities, as well as reduction of mortality.14,18
To date, despite the use of novel surgical approaches, cure or long-term biochemical control is achieved in fewer than 65% of patients which underwent neurosurgery, and only approximately 55% of patients treated with SRLs reaches the control of hormone unbalance.14,19-21
Objectives and Methodology
We conducted a literature review in order to find the most recent and relevant reports about challenges and therapeutic strategies for the treatment of acromegaly resistant to first-line medical therapy. An additional aim of this review is to highlight the molecular mechanisms leading to resistance to SRL treatment.
Definition of Biochemical Control
Majority of clinical trials have used a GH cut-off of 2.5 μg/L (safe random GH levels <2.5 μg/L), while only the most recent studies have applied more stringent thresholds.20,22 Current recommendations indicate as treatment targets the normalization of age-adjusted serum levels for IGF-1 and random GH levels <1.0 μg/L. In case of patients treated with PEG, IGF-1 is the only useful parameter.18
However, it should be noted that GH and IGF-1 measurements could vary considerably depending on the different assays and reference intervals used in the different laboratories.23–25 Therefore, the most recent Consensus recommends the use of the same assay for a given patient over time and that the aforementioned assay needs to adhere to accepted performance standards.14,26
Furthermore, GH and IGF-1 values may show discrepancies related to gender, glucose balance, GH receptor polymorphisms and other clinical parameters or biological factors.27 As an example, young female patients may present with high GH levels and normal IGF-1, probably due to an oestrogen-related mechanism. On the other hand, the phenotype with elevated IGF-1 and normal GH values is more frequent in patients with glucose metabolism impairment.28
First-Line Medical Treatment and Biochemical Control
Data on biochemical control of acromegaly treated with first-generation SRLs are highly variable in the different clinical studies over the years. According to a meta-analysis published in 2005, 57–58% of patients treated with OCT achieved safe GH values, while IGF-1 levels were normal in about 55–67% of cases.20
Subsequent studies reported the normalization of IGF-1 in 38–85% of patients and GH levels <2.5 μg/L in 33–75% subjects treated with conventional doses of OCT LAR (20–30 mg/4 weeks).29 Similar results were obtained in patients treated with LAN Autogel at conventional doses of 60, 90 or 120 mg/4 weeks. Indeed, some authors reported safe GH levels in 48–64% of patients and normal IGF-1 levels in 47–61%, while others showed a higher percentage of biochemical control (38–80% for GH and 39–80% for IGF-1).30–33
Concerning the effect on tumor mass, a significant shrinkage (>20–25% volume reduction) was observed in about 63% of patients treated with LAN Autogel and 66% of those treated with OCT LAR.34,35
Therefore, the efficacy of OCT LAR and LAN Autogel is considered superimposable.33,36
Interestingly, more recent prospective studies evaluating the outcome of OCT and LAN treatment have reported significantly lower response rates: only 20–30% of patients treated with OCT and 30–50% of those treated with LAN seem to reach the biochemical control.37–39 In line with these findings, real-life studies conducted in referral centers show a disease control in about 40% of patients.40,41
The lower efficacy of first-generation SRLs observed over time could be explained by the preselection of patients (eg based on the responsiveness to short-term SRL-therapy), thus leading to a possible overestimation of the efficacy of long-term SRL treatment in acromegaly.42
Definition of Resistance to Somatostatin Receptor Ligands and Partial Disease Control
As Colao and co-workers suggested, resistance to SRL therapy can be defined as failure to achieve biochemical control (safe GH levels and normal age-adjusted IGF-1 levels) associated with tumor shrinkage <20% compared with baseline, or even an increased tumor volume.13 A treatment duration of at least 12 months and a correct dose titration (maximum tolerated dose) should be reached before considering a patient uncontrolled.29 Resistance is partial when SRL therapy results in a reduction of IGF-1 levels by more than 50% compared to baseline, without normalization.13,41 It should be considered that, although most studies report that biochemical control and tumor reduction are significantly associated, in some patients dissociation has been recorded.43,44
Predictors of Somatostatin Receptor Ligands Resistance
In the recent years, many in vitro and in vivo studies have focused on the factors that can influence the response to first-generation SRL treatment, identifying a number of potential clinical, histopathological and molecular markers of resistance.36,45
As for the clinical determinants, gender, age and GH and IGF-1 levels at diagnosis have been correlated to the response to SRL treatment.28,46 Particularly, younger male patients are more often resistant to SRL therapy, as well as patients with higher GH and IGF-1 levels at diagnosis.
Moreover, considering their biological functions, the rationale for SRL treatment in acromegaly is the relatively high expression of somatostatin receptors (SSTs), particularly the subtype 2 (SST2), on adenoma cell membrane.45
In this light, best responses to in vitro and in vivo SRL treatment have been observed in tumors with high expression of SST2, evaluated at both mRNA and protein levels.47–49
Furthermore, GH-secreting adenomas display two distinct morphological patterns of cytoplasmic secretory granules, namely the densely and sparsely granulated adenoma subtypes.50,51 The densely granulated tumors show a higher expression of SST2 than the sparsely granulated ones, and a better response to SRLs.52,53 On the other hand, sparsely granulated pituitary adenomas show a lower SST2 expression and a worse response to therapy in terms of GH and/or IGF-1 reduction.54 Furthermore, sparsely granulated adenomas are more frequently represented by macroadenomas, and can display a more aggressive behaviour.46 The two entities above described are also correlated with signal on T2-weighted magnetic resonance images (MRI).55 Indeed, the sparsely granulated phenotype usually appears as a hyperintense lesion in T2-weighted MRI, while the densely granulated phenotype is more commonly detected in hypointense tumors. Accordingly, T2-hyperintense adenomas show a lower response rate to SRL treatment, in either GH and IGF-1 decrease or tumor shrinkage, compared to T2-hypointense tumors.56
Besides clinical, radiological parameters and SST2 expression, the expression of other SSTs can be involved in the resistance to therapy. In this light, the expression of SST5 seems to improve the SST2-activated pathways after selective activation of the ligand, as well as playing an autonomous role in reducing GH secretion in somatotropinoma cells.41,57 However, tumors with a lower SST2/SST5 seem to be associated with poor response to first-generation SRL therapy.58 Furthermore, studies from Duran-Prado and colleagues reported the potential existence of two SST5-truncated variants (termed SST5TMD4 and SST5TMD5) in pituitary adenomas. Interestingly, the SST5TMD4 variant was found particularly abundant in OCT-resistant somatotropinomas, thus suggesting its possible role in the attenuated response observed in some cases.59,60
Looking beyond the mere membrane receptor expression, other molecules can affect the responsiveness to SST2-targeting drugs. Indeed, similarly to all G-protein coupled receptors, ligand-bound SST2 undergoes a complex intracellular trafficking, involving receptor desensitization, internalization as well as recycling/degradation.41,61-63 In this context, the expression of molecules such as filamin A (FLNA) and β-arrestins have been demonstrated to affect the receptor’s function.61,64,65 In more detail, the cytoskeletal protein FLNA has emerged as a key modulator of SST2 signaling and expression, while lower levels of β-arrestin 1 and a high SST2/β-arrestin 1 ratio seem to correlate with a better biochemical response to SRL.45 However, recent data from Coelho and colleagues did not confirm previous findings about both FLNA and β-arrestin 1 in somatotroph adenomas, thus suggesting the need for additional studies in order to better elucidate the role of these molecules as molecular markers of the in vivo responsiveness to SRL treatment.66,67
Furthermore, the expression of e-cadherin, a molecule involved in the epithelial-to-mesenchymal transition pathway, has been directly correlated with the responsiveness to SRL treatment, while e-cadherin loss has been associated with a more aggressive clinical behaviour of the adenoma.68,69
Finally, a number of gene mutations have been linked with the variable response to SRL therapy in acromegaly. Indeed, about 30–40% of GH secreting adenomas present mutations of the GNAS1, encoding the alpha subunit of the Gs protein coupled to the GHRH receptor (GSP mutations). GSP mutations result in a constitutive activation of adenylyl cyclase with consequent production of cyclic AMP, thus providing greater sensitivity to first-generation SRLs compared to wild-type tumors.13,70,71
Another mutation involved in the response to SRL treatment targets the gene encoding for the aryl hydrocarbon receptor-interacting protein (AIP), a molecule that interacts with the aryl hydrocarbon receptors and that can stimulate the expression of zinc finger protein-1 ZAC1.72 Mutations in the AIP gene are described in the context of FIPA (familial isolated pituitary adenomas), and in some sporadic cases related to germline mutations.73,74 Affected patients are generally young males, often harbouring tumors with a more aggressive behaviour and sparsely granulated features.46 Mutations of the AIP gene or low expression of wild-type AIP are related to a poor response of somatotropinomas to first-generation SRLs, both in terms of biochemical control and tumor volume reduction.75
Current Strategies of Treatment
As previously described, the efficacy of the two first-generation SRLs, OCT and LAN, is comparable. Therefore, the choice between one of these two drugs depends on other factors, such as a preferred administration route (eg intramuscular vs deep subcutaneous injection), formulation (powder for suspension vs prefilled syringe), or associated costs.14 Currently, different therapeutic options are available when biochemical control is not achieved with these two drugs (see Table 1). Particularly, different approaches are suggested for patients partially responders to first-generation SRLs compared to those completely resistant.
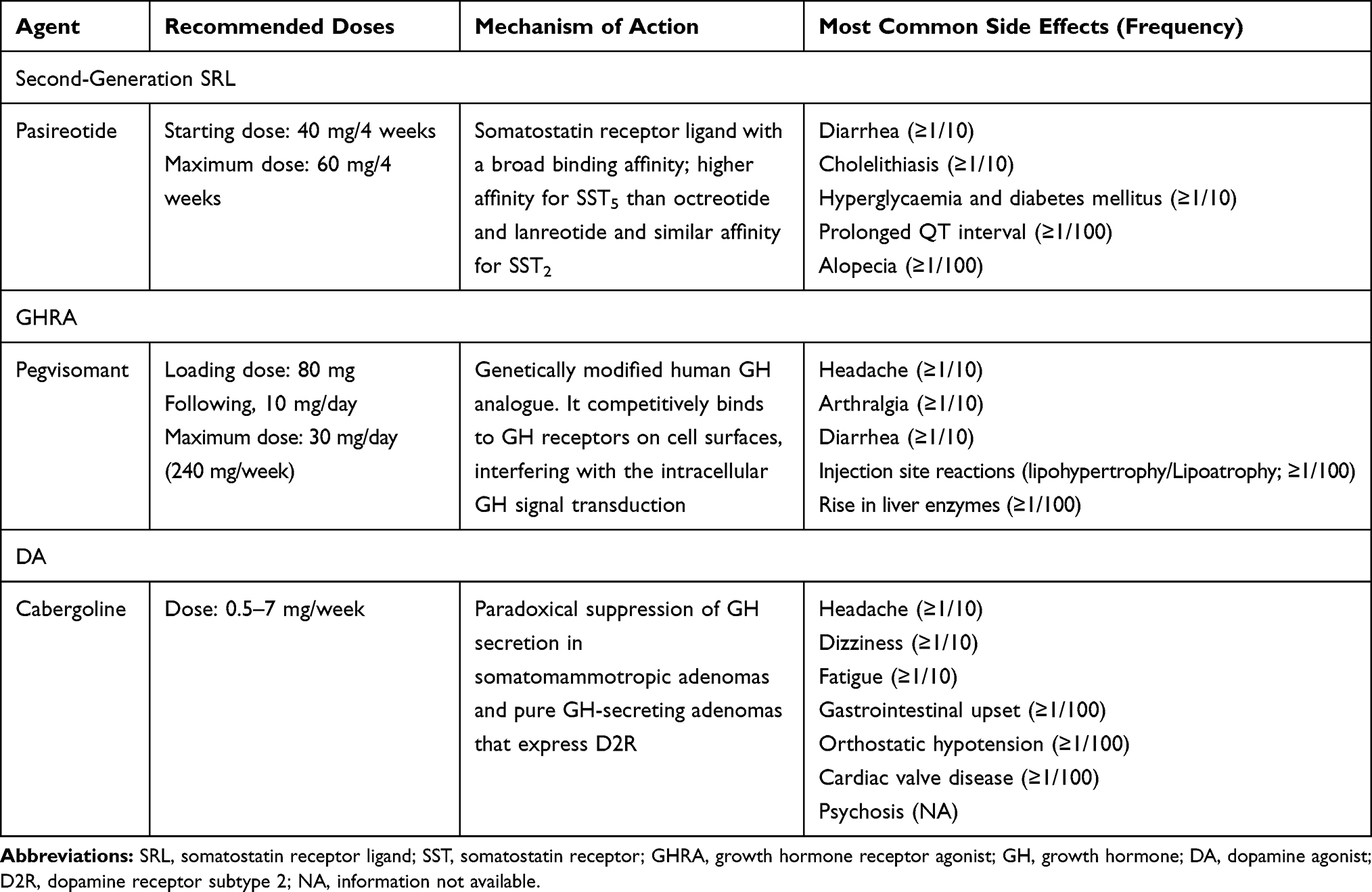 |
Table 1 Available Drugs in Second-Line Medical Treatment of Acromegaly |
Partial Response: Increase Dose and/or Frequency of First-Generation Somatostatin Receptor Ligands
Adequate titration of first-generation SRLs can improve biochemical control of acromegaly.
The two SRLs have different pharmacokinetic profiles, and this aspect may influence the response rate in case of dose increase or higher frequency of administration.76 OCT LAR shows a peculiar biphasic release: after an initial peak within 1 hr, plasma concentration decreases in the following 12 hrs after administration, remains at sub-therapeutic levels for about a week and then increases with a subsequent second-release phase, showing a sustained drug release reaching a plateau between days 14–42.77,78 Plasma concentrations reach a steady state after three injections. Currently, OCT LAR is commercially available at three different dosages (10, 20, and 30 mg), with a maximum allowed dose of 40 mg (two 20 mg injections) every four weeks.79
LAN Autogel half-life is approximately 23–29 days and steady state is reached after four injections. The approved initial dose is 90 mg/4 weeks, with dose titration allowed down to 60 or up to 120 mg.1 Of note, administration frequency can be modulated according to the patient’s response (eg every three to five weeks).79
High-dose OCT LAR (40 mg/4 weeks) may be effective in patients with inadequately controlled acromegaly at standard dose regimens.80 These patients are generally younger, with higher GH levels and larger adenomas at diagnosis compared to those showing a better response to SRL therapy. Colao and colleagues showed that a dose increase up to 40 mg/4 weeks induces a further reduction of GH and IGF-1 levels in patients partially responders to 30 mg at the same interval between injections.80 In the aforementioned study, dose increase led to an additional 35% of disease control compared to standard dosages, with a safety profile similar to the standard regimen.80 Of note, a formulation of OCT LAR 40 mg is not currently available, and this aspect could have an impact on patients’ compliance. Furthermore, Giustina and colleagues showed that increasing the dose of OCT LAR up to 60 mg/4 weeks may be beneficial for patients partially resistant to standard dosages, while shortening the interval between drug administration (30 mg/21 days) seems to be less effective compared to the high-dose schedule.81
As for LAN Autogel, a recent study has shown that increasing dose (LAN 180 mg/4 weeks) or frequency (LAN 120 mg/3 weeks) of administration normalizes IGF-1 levels in about one-third of patients with acromegaly not adequately controlled with conventional drug regimens.82
Partial Response: Add Cabergoline to First-Generation Somatostatin Receptor Ligands
The expression of dopamine receptor subtype 2 (D2R) by GH-secreting adenomas represents the rationale for the use of dopamine agonists in the treatment of acromegaly, irrespective of an active prolactin secretion by the adenoma. The sensitivity to cabergoline (CAB) treatment is variable and substantially unrelated to drug dosages, with no treatment escape or tachyphylaxis reported. The adverse events directly related to cabergoline therapy are usually mild and generally improve after early administrations. A foresight is to take the drug at bedtime, with food, and to perform a slow dose escalation.83
A meta-analysis of 2011 showed that, in uncontrolled patients, IGF-1 normalization is achieved in about half of patients when CAB is added to first-generation SRLs. Similarly to monotherapy, the addition of CAB to SRL treatment seems particularly beneficial in patients with moderately elevated IGF-I levels.84,85 In more detail, the most recent Consensus Statement on medical therapy of acromegaly suggests to consider CAB as first-line medical therapy, or in addition to first-generation SRLs, in patients with IGF-1 levels <2.5 times the upper limit of normality (ULN), with the greatest benefit seen in those with IGF-1 levels ≤1.5 x ULN.14
Patient Not Controlled, Tumor Concern: Switch to Pasireotide
Pasireotide (PAS) LAR is a second-generation SRL that shows a 39-fold higher binding affinity for SST5 and a slightly lower affinity to SST2 compared with OCT.86 Besides the different receptor binding affinity, PAS exhibits different functional properties compared to OCT when binding SSTs, and particularly SST2. These differences include SST pathway activation and modulation of receptors’ phosphorylation, internalization and trafficking, involving a number of molecules that regulate membrane receptor functions, such as β-arrestins.87
In a prospective, randomized, double blind, Phase III study carried out in patients with medically naïve acromegaly, PAS LAR demonstrated a significantly superior efficacy over OCT in reaching biochemical control (GH <2.5 μg/L and normal age-adjusted IGF-1), although the two compounds showed a similar efficacy in reaching safe GH levels.88 Interestingly, this latter finding is in line with in vitro studies, showing that the direct effect of PAS and OCT in reducing GH secretion from somatotroph cells is superimposable.89–91
Furthermore, the PAOLA study, carried out in patients inadequately controlled after treatment with first-generation SRLs, proved PAS superiority versus continued treatment with OCT LAR or LAN Autogel in achieving biochemical control and tumor volume reduction. During the above mentioned clinical trials, PAS was substantially well tolerated, but nearly 70% of patients exhibited hyperglycemia-related adverse events.92
In a real-life multicenter retrospective study, about 50% of patients partially controlled with first-generation SRLs achieved normalization of IGF-1 levels after switching to PAS treatment, even at low doses.93 PAS improved significantly also clinical symptoms such as headache, however confirmed an impairment of glucose metabolism in more than 60% of patients.93
A retrospective analysis of current data suggests that pre-existing hyperglycemia may be predictive of the development of PAS-associated hyperglycemia.94 Current recommendations suggest to carefully screen and monitor for hyperglycemia-related adverse events those patients considered for treatment with PAS. In this light, patients with a normal glucose profile but uncontrolled disease under first-line medical therapy are more suitable for PAS.14
In conclusion, switching to PAS may improve biochemical control in inadequately controlled patients and lead to a greater tumor volume reduction.
In a recent review article, Coopmans and colleagues suggest use of PAS in young patients with tumor growth during first-generation SRLs or PEG therapy, in patients with headache not responsive to first-generation SRL therapy and in those with PEG-related side effects.95
Patient Not Controlled, Impaired Glucose Metabolism: Switch to Pegvisomant
When SRLs fail to achieve biochemical control of acromegaly, PEG represents a valid treatment alternative, showing a good efficacy and a satisfactory safety profile.96 PEG is a pegylated form of a modified human GH analog which competitively blocks GH receptor, thus preventing the action of native GH and inducing a dose-dependent reduction of IGF-1 levels. Due to the peculiar mechanism of action of this compound, biochemical control is evaluated using IGF-1 levels alone, since PEG does not directly target GH secretion at pituitary level. In this light, the reduction of tumor mass is not a goal of PEG treatment as well.
The ACROSTUDY, a world-wide non-interventional post-marketing surveillance study, shows the normalization of IGF-1 levels in about 65–70% of patients treated with PEG.22,97 Moreover, this outcome is associated with a low rate of pituitary tumor growth and a relatively low percentage of drug-related adverse events, such as significant impairment of liver function or lipodystrophy due to injection site reactions.98
In early clinical studies, the reported efficacy of PEG was equal to or greater than 90%. This difference observed between clinical studies and real-life data can be explained by multiple factors like suboptimal dose titration during daily clinical practice, poor compliance of patients dealing with daily injections as well as assays variability in the measurement of IGF-1.99
However, a number of clinical studies have shown that younger, female patients and subjects with higher BMI and IGF-1 values at diagnosis need more PEG to reach biochemical control, and seem to be more resistant to treatment.100–102
The recent Consensus on acromegaly treatment recommends the use of PEG at a dosage of 10–30 mg per day, titrated up to the highest tolerated dose (max 240 mg/week).
Of note, PEG use is indicated in patients resistant to therapy with first-generation SRLs, particularly in diabetic patients because of its positive impact on glucose metabolism.14,103
Combination Therapy
The combination of medical therapies aims to achieve a greater efficacy compared to monotherapy, to reduce the dosage of each individual drug (and therefore the associated side effects), to increase patient compliance as well as to reduce overall medical costs.12
As above discussed in this review, the combination between first-generation SRLs and CAB can represent a suitable option in SRL-partial responders showing slightly elevated IGF-1.
A valid therapeutic strategy in patients with a partial response to SRL presenting a clinically relevant residual tumor and impaired glucose metabolism is to add PEG to SRLs, thus taking advantage of the different biological mechanisms of these drugs.14 This combination has been shown to have high efficacy and a good safety profile.104 Furthermore, PEG seems to have the potential to counterbalance the slight detrimental effects of SRLs on glucose metabolism.
A study from Neggers and colleagues of 2014 showed that the addition of PEG led to the normalization of IGF-1 values in 97% of patients with uncontrolled acromegaly during treatment with the maximum allowed dose of SRLs.104 Noteworthy, other studies demonstrated that, although taking into account a significant inter-individual variability, SRL and PEG combination might allow a dose reduction (about 50%) of one of the two compounds compared to the monotherapy, without affecting patients’ biochemical control.105,106
Interestingly, although few multicenter studies have reported lower efficacy rates, Van de Lely et al demonstrated that the combination between LAN Autogel and PEG led to the normalization of IGF-1 values in 58% of patients after 28 weeks of treatment and in 79% of subjects when considering the lowest IGF-1 value observed during the study period.107 Furthermore, Trainer et al reported similar results investigating the combination OCT LAR plus PEG.108
As for the safety profile, some reports describe a higher percentage of transient elevation in liver enzymes (>2–3 fold) in patients receiving SRL plus PEG combination (11–15%) compared to PEG monotherapy (1.5–5.2%).97,104,109
Few data are currently available focusing on the effect of SRL plus PEG combination on tumor volume. However, the majority of patients seem to have stable tumor volume, while 15–20% show a significant volume reduction (>20% vs baseline).104,109,110
Nowadays few reports have deeply investigated the potential role of the combination therapy with CAB and PEG. In a prospective study by Higham and colleagues, the combination of low-dose PEG (10 mg/day) and CAB (0.5 mg/day) led to the normalization of IGF-1 levels in 68% of patients. Interestingly, after CAB withdrawal only 26% of patients had normal IGF-1 levels, thus suggesting that the combination therapy was more effective than monotherapy with either drug.111
Furthermore, in a small group of patients partially resistant to SRLs and with persistent mild IGF-1 elevation on PEG monotherapy, addition of CAB normalized IGF-1 in 28% of patients and decreased IGF-1 in 64%. The association of PEG and CAB proved to be more effective in female patients with lower baseline IGF-1 levels, lower body weight and higher baseline PRL concentrations.112
Finally, the combination of PAS LAR and PEG has been recently explored in a prospective open-label trial investigating the efficacy and safety of this combination in patients previously controlled after combination therapy with first-generation SRLs and PEG. In order to carefully evaluate the impact of this novel combination therapy, at baseline the dose of PEG was reduced by 50% up to 12 weeks. Then, based on IGF-1 levels after 12 weeks, patients were switched to PAS LAR 60 mg monotherapy or PAS LAR 60 mg plus the 50% reduced PEG dose. At the end of the core study (24 weeks), IGF-1 levels were reduced into the reference range in 73.8% of patients and PEG dose was reduced by 66.1%.113
These results were confirmed by an extension phase, evaluating biochemical control and safety up to 48 weeks.114 Frequency of diabetes mellitus increased from 68% at 24 weeks to 77% at 48 weeks, suggesting that PEG treatment does not have a positive impact on PAS-induced hyperglycemia. Based on these data, Coopmans et al suggest switching from first-generation SRLs plus PEG therapy to the combination of PAS LAR plus PEG in non-diabetic patients, which show symptoms of active acromegaly or pituitary tumor size increase, aiming to reduce PEG doses.95
Non-Medical Secondary Treatment
Second Surgery for Uncontrolled Acromegaly Patients
The role of a second surgery in the management of acromegaly is still not well established. As for primary surgery, the overall remission rate after a re-operation is highly variable between series (ranging from 8% to 59%).115,116 This is probably due to the lack of standardized recommendations for a second intervention and no clear stratification between surgically resectable and unresectable tumors. As an example, Abe and colleagues, based on Lüdecke’s classification system and intraoperative GH measurement, reported a biochemical remission rate after a second surgery of 88.9% in resectable adenomas and 57.1% including not resectable tumors.117,118 On the other hand, looking to a big series of 140 patients from Nomikos et al, the overall remission rate was 27%, going up to 39% if only resectable tumor were considered.119 Interestingly, in a small series of Kurosaki and colleagues, using intraoperative GH measurement as a tool to confirm radical tumor removal, the overall remission rate was 56.3%, and 81.3% for the subgroup of resectable adenomas.120 Yamada et al have identified cavernous sinus invasion, tumor segmentation, age (>40 years-old), GH and IGF-1 levels as predictive factors for the surgical outcome, while pretreatment with adjuvant therapy, sex, tumor size and tumor fibrosis were not.115 In their series, the overall remission rate was 59%. Mathioudakis and Salvatori, combining data from previous series, estimated an overall remission rate of 34% and a remission rate for resectable adenomas of 52%.116 These data show that in acromegaly success rate of a second surgery is similar to first surgery, particularly in selected cases with favourable predictive factors.121,122 A number of surgeons feel that re-operation may be difficult for the presence of scarred tissue or the absence of the normal anatomical planes and landmarks, leading to a complication rate of secondary transsphenoidal surgery greater than that of primary surgery.115,117,123 Indeed, the incidence of major complication seems to be slightly higher in re-operated cases: meningitis 1.8–6%, cerebrospinal fluid leak/fistula 2–9%, vascular injury 0.1–6%, ophthalmopathy 6% and hormone deficiencies 1.9%.116 However, with no increase in mortality rate and minimal increase in morbidity, secondary surgery appears as a reasonable option in case of recurrence or persistence of acromegaly.121 Indeed, the management of tumor persistence or recurrence after first surgery is still controversial. Current practice in most reference centers for pituitary diseases is to propose medical management and/or radiotherapy to patients failing to achieve biochemical remission after initial surgical resection.121 Indications for a second surgery are not well established yet, but re-operation is usually reserved for debulking purposes to increase the likelihood of remission with adjuvant medical therapies or when relief of mass effect on the optic chiasm is needed.116 Second surgery can be also performed in presence of recurrent or regrowing tumors at magnetic resonance imaging, in patients with an unsatisfactory response to any adjuvant medical therapy tested, in subjects which develop severe adverse events during pharmacological treatment or in case of cost-related issues.115,124 Therefore, comparing the cost-effectiveness of medical therapy, radiation and surgery, a second surgery should be considered as a possible option in all eligible cases.121 Further re-operations could be proposed in the presence of the same conditions of secondary surgery, always after multidisciplinary discussion.
Radiation Therapy
The main indication for radiation therapy in the current treatment algorithm of acromegaly is the presence of a residual or recurrent adenoma after transsphenoidal surgery, growing irrespective of appropriate surgical management and medical therapy. Different types of radiation therapy can be performed. Fractionated radiotherapy refers to radiation therapy delivered over multiple small doses in multiple sittings and has been used as adjuvant treatment in acromegaly for many years.124,125 Stereotactic radiosurgery (SRS) is defined as the highly precise delivery of radiation in a single session targeted at the tumor, minimizing the dose received by surrounding critical neural structures, such as the optic nerve.126 The overall remission rate of fractionated radiotherapy and SRS is variable, with a biochemical remission rate ranging from 5% up to 79% for fractionated radiotherapy, and about 60% after 10 years for SRS, as reported in a recent series by Ding et al.124,127 While the control of tumor growth is similar between the two techniques, with 95% of stable tumors observed at 5 years, stereotactic radiosurgery seems to result in a faster biochemical control compared to fractionated radiation.125 Since SRS presents lower risk of complications, such as hypopituitarism, currently this approach is preferred to fractionated radiotherapy.127 Radiation therapy should be performed in the following conditions: tumor growth despite surgery and medical therapy; disease activity (eg high IGF-1 values) with residual tumor after maximal surgical removal, no possibility for re-operation and lack of response to adjuvant medical therapy; patients not eligible for surgical intervention and resistant to medical treatment; patients refusing surgical and medical treatments. In case the residual tumor is at least 3–5 mm far from the optic chiasm, SRS is usually the preferred option, while if the tumor is closer than 3–5 mm to the optic chiasm or other critical anatomical structures, fractionated radiation is burdened by lower treatment-related detrimental effects compared to radiosurgery.125
Conclusions
The management of patients with acromegaly not controlled with first-line medical therapy is challenging. New therapeutic strategies are now possible due to the availability of different drugs and the approval of their combination for clinical purposes (see Figure 1). Other compounds are currently under investigation and could represent additional therapeutic tools in the next future. Furthermore, in the last decade, considerable scientific progresses have been achieved to identify a number of different biomarkers correlated to the efficacy of the different drugs. These findings could help clinicians to perform a real patients’ tailored therapy, based on the whole clinical characteristics of each subject. Identifying the best therapy for each patient means improving the outcome and reducing overall health care costs.
 |
Figure 1 Proposed algorithm for second-line medical treatment in acromegaly.Notes: *As adjuvant therapy and/or neoadjuvanttreatment; aIf significant tumor shrinkage after neoadjuvant SRL treatment, consider surgery; bNon-diabetic patients; cPAS + PEG if risk related to tumor concern and uncontrolled disease is greater than the worsening of glucose unbalance.Abbreviations: SRL, somatostatin receptor ligand; MRI, magnetic resonance imaging; CAB, cabergoline; PEG, pegvisomant; PAS, pasireotide. |
Finally, it is strongly recommended that the management of patients with acromegaly is performed in the setting of a multidisciplinary team (pituitary unit) including a number of different dedicated specialists, besides skilled endocrinologists.
Disclosure
DF has been a speaker for, participated on advisory boards and received research grants from Novartis, Pfizer and Ipsen. FG has been a speaker for Novartis, participated on advisory boards and/or received personal fees from Novartis, AMCo Ltd and IONIS Pharmaceuticals. The other authors report no conflicts of interest in this work.
References
1. Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119(11):3189–3202. doi:10.1172/JCI39375
2. Pita-Gutierrez F, Pertega-Diaz S, Pita-Fernandez S, et al. Place of preoperative treatment of acromegaly with somatostatin analog on surgical outcome: a systematic review and meta-analysis. PLoS One. 2013;8(4):e61523. doi:10.1371/journal.pone.0061523
3. Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary. 1999;2(1):29–41. doi:10.1023/A:1009965803750
4. Melmed S. Medical progress: acromegaly. N Engl J Med. 2006;355(24):2558–2573. doi:10.1056/NEJMra062453
5. Christofides EA. Clinical importance of achieving biochemical control with medical therapy in adult patients with acromegaly. Patient Prefer Adherence. 2016;10:1217–1225. doi:10.2147/PPA.S102302
6. Gatto F, Trifiro G, Lapi F, et al. Epidemiology of acromegaly in Italy: analysis from a large longitudinal primary care database. Endocrine. 2018;61(3):533–541. doi:10.1007/s12020-018-1630-4
7. Ben-Shlomo A, Sheppard MC, Stephens JM, Pulgar S, Melmed S. Clinical, quality of life, and economic value of acromegaly disease control. Pituitary. 2011;14(3):284–294. doi:10.1007/s11102-011-0310-7
8. Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72(3):377–382.
9. Abreu A, Tovar AP, Castellanos R, et al. Challenges in the diagnosis and management of acromegaly: a focus on comorbidities. Pituitary. 2016;19(4):448–457.
10. Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008;159(2):89–95. doi:10.1530/EJE-08-0267
11. Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (Oxf). 2010;72(2):203–208. doi:10.1111/cen.2010.72.issue-2
12. Katznelson L, Laws ER
13. Colao A, Auriemma RS, Lombardi G, Pivonello R. Resistance to somatostatin analogs in acromegaly. Endocr Rev. 2011;32(2):247–271. doi:10.1210/er.2010-0002
14. Melmed S, Bronstein MD, Chanson P, et al. A consensus statement on acromegaly therapeutic outcomes. Nat Rev Endocrinol. 2018;14(9):552–561. doi:10.1038/s41574-018-0058-5
15. McKeage K. Pasireotide in acromegaly: a review. Drugs. 2015;75(9):1039–1048. doi:10.1007/s40265-015-0413-y
16. Bacigaluppi S, Gatto F, Anania P, et al. Impact of pre-treatment with somatostatin analogs on surgical management of acromegalic patients referred to a single center. Endocrine. 2016;51(3):524–533. doi:10.1007/s12020-015-0619-5
17. Abu Dabrh AM, Asi N, Farah WH, et al. Radiotherapy versus radiosurgery in treating patients with acromegaly: a systematic review and meta-analysis. Endocr Pract. 2015;21(8):943–956. doi:10.4158/EP14574.OR
18. Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: a consensus on the medical treatment of acromegaly. Nat Rev Endocrinol. 2014;10(4):243–248. doi:10.1038/nrendo.2014.21
19. Murray RD, Melmed S. A critical analysis of clinically available somatostatin analog formulations for therapy of acromegaly. J Clin Endocrinol Metab. 2008;93(8):2957–2968. doi:10.1210/jc.2008-0027
20. Carmichael JD, Bonert VS, Nuno M, Ly D, Melmed S. Acromegaly clinical trial methodology impact on reported biochemical efficacy rates of somatostatin receptor ligand treatments: a meta-analysis. J Clin Endocrinol Metab. 2014;99(5):1825–1833. doi:10.1210/jc.2013-3757
21. Babu H, Ortega A, Nuno M, et al. Long-term endocrine outcomes following endoscopic endonasal transsphenoidal surgery for acromegaly and associated prognostic factors. Neurosurgery. 2017;81(2):357–366. doi:10.1093/neuros/nyx020
22. Giustina A, Arnaldi G, Bogazzi F, et al. Pegvisomant in acromegaly: an update. J Endocrinol Invest. 2017;40(6):577–589. doi:10.1007/s40618-017-0614-1
23. Pokrajac A, Wark G, Ellis AR, Wear J, Wieringa GE, Trainer PJ. Variation in GH and IGF-I assays limits the applicability of international consensus criteria to local practice. Clin Endocrinol (Oxf). 2007;67(1):65–70. doi:10.1111/cen.2007.67.issue-1
24. Arafat AM, Mohlig M, Weickert MO, et al. Growth hormone response during oral glucose tolerance test: the impact of assay method on the estimation of reference values in patients with acromegaly and in healthy controls, and the role of gender, age, and body mass index. J Clin Endocrinol Metab. 2008;93(4):1254–1262. doi:10.1210/jc.2007-2084
25. Clemmons DR. IGF-I assays: current assay methodologies and their limitations. Pituitary. 2007;10(2):121–128. doi:10.1007/s11102-007-0032-z
26. Clemmons DR. Consensus statement on the standardization and evaluation of growth hormone and insulin-like growth factor assays. Clin Chem. 2011;57(4):555–559. doi:10.1373/clinchem.2010.150631
27. Schilbach K, Strasburger CJ, Bidlingmaier M. Biochemical investigations in diagnosis and follow up of acromegaly. Pituitary. 2017;20(1):33–45. doi:10.1007/s11102-017-0792-z
28. Paragliola RM, Corsello SM, Salvatori R. Somatostatin receptor ligands in acromegaly: clinical response and factors predicting resistance. Pituitary. 2017;20(1):109–115. doi:10.1007/s11102-016-0768-4
29. Fleseriu M. Clinical efficacy and safety results for dose escalation of somatostatin receptor ligands in patients with acromegaly: a literature review. Pituitary. 2011;14(2):184–193. doi:10.1007/s11102-010-0282-z
30. Freda PU, Katznelson L, van der Lely AJ, Reyes CM, Zhao S, Rabinowitz D. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005;90(8):4465–4473. doi:10.1210/jc.2005-0260
31. Roelfsema F, Biermasz NR, Pereira AM, Romijn JA. Therapeutic options in the management of acromegaly: focus on lanreotide Autogel. Biol Targets Ther. 2008;2(3):463–479.
32. Chanson P, Borson-Chazot F, Kuhn JM, et al. Control of IGF-I levels with titrated dosing of lanreotide Autogel over 48 weeks in patients with acromegaly. Clin Endocrinol (Oxf). 2008;69(2):299–305. doi:10.1111/j.1365-2265.2008.03208.x
33. Tutuncu Y, Berker D, Isik S, et al. Comparison of octreotide LAR and lanreotide autogel as post-operative medical treatment in acromegaly. Pituitary. 2012;15(3):398–404. doi:10.1007/s11102-011-0335-y
34. Mazziotti G, Giustina A. Effects of lanreotide SR and Autogel on tumor mass in patients with acromegaly: a systematic review. Pituitary. 2010;13(1):60–67. doi:10.1007/s11102-009-0169-z
35. Giustina A, Mazziotti G, Torri V, Spinello M, Floriani I, Melmed S. Meta-analysis on the effects of octreotide on tumor mass in acromegaly. PLoS One. 2012;7(5):e36411. doi:10.1371/journal.pone.0036411
36. Cuevas-Ramos D, Fleseriu M. Somatostatin receptor ligands and resistance to treatment in pituitary adenomas. J Mol Endocrinol. 2014;52(3):R223–R240. doi:10.1530/JME-14-0011
37. Mercado M, Borges F, Bouterfa H, et al. A prospective, multicentre study to investigate the efficacy, safety and tolerability of octreotide LAR (long-acting repeatable octreotide) in the primary therapy of patients with acromegaly. Clin Endocrinol (Oxf). 2007;66(6):859–868. doi:10.1111/cen.2007.66.issue-6
38. Melmed S, Cook D, Schopohl J, Goth MI, Lam KS, Marek J. Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension. Pituitary. 2010;13(1):18–28. doi:10.1007/s11102-009-0191-1
39. Shimatsu A, Teramoto A, Hizuka N, Kitai K, Ramis J, Chihara K. Efficacy, safety, and pharmacokinetics of sustained-release lanreotide (lanreotide Autogel) in Japanese patients with acromegaly or pituitary gigantism. Endocr J. 2013;60(5):651–663. doi:10.1507/endocrj.EJ12-0417
40. Colao A, Auriemma RS, Pivonello R, Kasuki L, Gadelha MR. Interpreting biochemical control response rates with first-generation somatostatin analogues in acromegaly. Pituitary. 2016;19(3):235–247. doi:10.1007/s11102-015-0684-z
41. Gadelha MR, Wildemberg LE, Bronstein MD, Gatto F, Ferone D. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary. 2017;20(1):100–108. doi:10.1007/s11102-017-0791-0
42. Gatto F, Campana C, Cocchiara F, et al. Current perspectives on the impact of clinical disease and biochemical control on comorbidities and quality of life in acromegaly. Rev Endocr Metab Disord. 2019;20:365–381. doi:10.1007/s11154-019-09506-y
43. Besser GM, Burman P, Daly AF. Predictors and rates of treatment-resistant tumor growth in acromegaly. Eur J Endocrinol. 2005;153(2):187–193. doi:10.1530/eje.1.01968
44. Casarini AP, Pinto EM, Jallad RS, Giorgi RR, Giannella-Neto D, Bronstein MD. Dissociation between tumor shrinkage and hormonal response during somatostatin analog treatment in an acromegalic patient: preferential expression of somatostatin receptor subtype 3. J Endocrinol Invest. 2006;29(9):826–830. doi:10.1007/BF03347378
45. Gatto F, Biermasz NR, Feelders RA, et al. Low beta-arrestin expression correlates with the responsiveness to long-term somatostatin analog treatment in acromegaly. Eur J Endocrinol. 2016;174(5):651–662. doi:10.1530/EJE-15-0391
46. Gadelha MR, Kasuki L, Korbonits M. Novel pathway for somatostatin analogs in patients with acromegaly. Trends Endocrinol Metab. 2013;24(5):238–246. doi:10.1016/j.tem.2012.11.007
47. Gatto F, Feelders RA, van der Pas R, et al. Immunoreactivity score using an anti-sst2A receptor monoclonal antibody strongly predicts the biochemical response to adjuvant treatment with somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2013;98(1):E66–E71. doi:10.1210/jc.2012-2609
48. Wildemberg LE, Neto LV, Costa DF, et al. Low somatostatin receptor subtype 2, but not dopamine receptor subtype 2 expression predicts the lack of biochemical response of somatotropinomas to treatment with somatostatin analogs. J Endocrinol Invest. 2013;36(1):38–43. doi:10.3275/8305
49. Ferone D, de Herder WW, Pivonello R, et al. Correlation of in vitro and in vivo somatotropic adenoma responsiveness to somatostatin analogs and dopamine agonists with immunohistochemical evaluation of somatostatin and dopamine receptors and electron microscopy. J Clin Endocrinol Metab. 2008;93(4):1412–1417. doi:10.1210/jc.2007-1358
50. Obari A, Sano T, Ohyama K, et al. Clinicopathological features of growth hormone-producing pituitary adenomas: difference among various types defined by cytokeratin distribution pattern including a transitional form. Endocr Pathol. 2008;19(2):82–91. doi:10.1007/s12022-008-9029-z
51. Mayr B, Buslei R, Theodoropoulou M, Stalla GK, Buchfelder M, Schofl C. Molecular and functional properties of densely and sparsely granulated GH-producing pituitary adenomas. Eur J Endocrinol. 2013;169(4):391–400. doi:10.1530/EJE-13-0134
52. Kato M, Inoshita N, Sugiyama T, et al. Differential expression of genes related to drug responsiveness between sparsely and densely granulated somatotroph adenomas. Endocr J. 2012;59(3):221–228. doi:10.1507/endocrj.EJ11-0177
53. Brzana J, Yedinak CG, Gultekin SH, Delashaw JB, Fleseriu M. Growth hormone granulation pattern and somatostatin receptor subtype 2A correlate with postoperative somatostatin receptor ligand response in acromegaly: a large single center experience. Pituitary. 2013;16(4):490–498. doi:10.1007/s11102-012-0445-1
54. Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol (Oxf). 2012;76(1):96–102. doi:10.1111/cen.2011.76.issue-1
55. Heck A, Ringstad G, Fougner SL, et al. Intensity of pituitary adenoma on T2-weighted magnetic resonance imaging predicts the response to octreotide treatment in newly diagnosed acromegaly. Clin Endocrinol (Oxf). 2012;77(1):72–78. doi:10.1111/j.1365-2265.2011.04286.x
56. Potorac I, Petrossians P, Daly AF, et al. T2-weighted MRI signal predicts hormone and tumor responses to somatostatin analogs in acromegaly. Endocr Relat Cancer. 2016;23(11):871–881. doi:10.1530/ERC-16-0356
57. Grant M, Alturaihi H, Jaquet P, Collier B, Kumar U. Cell growth inhibition and functioning of human somatostatin receptor type 2 are modulated by receptor heterodimerization. Mol Endocrinol. 2008;22(10):2278–2292. doi:10.1210/me.2007-0334
58. Taboada GF, Luque RM, Neto LV, et al. Quantitative analysis of somatostatin receptor subtypes (1-5) gene expression levels in somatotropinomas and correlation to in vivo hormonal and tumor volume responses to treatment with octreotide LAR. Eur J Endocrinol. 2008;158(3):295–303. doi:10.1530/EJE-07-0562
59. Duran-Prado M, Gahete MD, Martinez-Fuentes AJ, et al. Identification and characterization of two novel truncated but functional isoforms of the somatostatin receptor subtype 5 differentially present in pituitary tumors. J Clin Endocrinol Metab. 2009;94(7):2634–2643. doi:10.1210/jc.2008-2564
60. Duran-Prado M, Saveanu A, Luque RM, et al. A potential inhibitory role for the new truncated variant of somatostatin receptor 5, sst5TMD4, in pituitary adenomas poorly responsive to somatostatin analogs. J Clin Endocrinol Metab. 2010;95(5):2497–2502. doi:10.1210/jc.2009-2247
61. Gatto F, Hofland LJ. The role of somatostatin and dopamine D2 receptors in endocrine tumors. Endocr Relat Cancer. 2011;18(6):R233–R251. doi:10.1530/ERC-10-0334
62. Treppiedi D, Mangili F, Giardino E, et al. Cytoskeleton protein Filamin A is required for efficient somatostatin receptor type 2 internalization and recycling through Rab5 and Rab4 sorting endosomes in tumor somatotroph cells. Neuroendocrinology. 2019. doi:10.1159/000503791
63. Treppiedi D, Jobin ML, Peverelli E, et al. Single-molecule microscopy reveals dynamic FLNA interactions governing SSTR2 clustering and internalization. Endocrinology. 2018;159(8):2953–2965. doi:10.1210/en.2018-00368
64. Gatto F, Feelders R, van der Pas R, et al. beta-Arrestin 1 and 2 and G protein-coupled receptor kinase 2 expression in pituitary adenomas: role in the regulation of response to somatostatin analogue treatment in patients with acromegaly. Endocrinology. 2013;154(12):4715–4725. doi:10.1210/en.2013-1672
65. Peverelli E, Giardino E, Treppiedi D, et al. Filamin A (FLNA) plays an essential role in somatostatin receptor 2 (SST2) signaling and stabilization after agonist stimulation in human and rat somatotroph tumor cells. Endocrinology. 2014;155(8):2932–2941. doi:10.1210/en.2014-1063
66. Coelho MCA, Vasquez ML, Wildemberg LE, et al. Clinical significance of filamin A in patients with acromegaly and its association with somatostatin and dopamine receptor profiles. Sci Rep. 2019;9(1):1122. doi:10.1038/s41598-018-37692-3
67. Coelho MCA, Vasquez ML, Wildemberg LE, et al. Molecular evidence and clinical importance of beta-arrestins expression in patients with acromegaly. J Cell Mol Med. 2018;22(4):2110–2116. doi:10.1111/jcmm.13427
68. Venegas-Moreno E, Flores-Martinez A, Dios E, et al. E-cadherin expression is associated with somatostatin analogue response in acromegaly. J Cell Mol Med. 2019;23(5):3088–3096. doi:10.1111/jcmm.13851
69. Fougner SL, Lekva T, Borota OC, Hald JK, Bollerslev J, Berg JP. The expression of E-cadherin in somatotroph pituitary adenomas is related to tumor size, invasiveness, and somatostatin analog response. J Clin Endocrinol Metab. 2010;95(5):2334–2342. doi:10.1210/jc.2009-2197
70. Peverelli E, Mantovani G, Lania AG, Spada A. cAMP in the pituitary: an old messenger for multiple signals. J Mol Endocrinol. 2014;52(1):R67–R77. doi:10.1530/JME-13-0172
71. Barlier A, Gunz G, Zamora AJ, et al. Pronostic and therapeutic consequences of Gs alpha mutations in somatotroph adenomas. J Clin Endocrinol Metab. 1998;83(5):1604–1610. doi:10.1210/jcem.83.5.4797
72. Chahal HS, Trivellin G, Leontiou CA, et al. Somatostatin analogs modulate AIP in somatotroph adenomas: the role of the ZAC1 pathway. J Clin Endocrinol Metab. 2012;97(8):E1411–E1420. doi:10.1210/jc.2012-1111
73. Chahal HS, Chapple JP, Frohman LA, Grossman AB, Korbonits M. Clinical, genetic and molecular characterization of patients with familial isolated pituitary adenomas (FIPA). Trends Endocrinol Metab. 2010;21(7):419–427. doi:10.1016/j.tem.2010.02.007
74. Cazabat L, Bouligand J, Salenave S, et al. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab. 2012;97(4):E663–E670. doi:10.1210/jc.2011-2291
75. Kasuki L, Vieira Neto L, Wildemberg LE, et al. AIP expression in sporadic somatotropinomas is a predictor of the response to octreotide LAR therapy independent of SSTR2 expression. Endocr Relat Cancer. 2012;19(3):L25–L29.
76. Astruc B, Marbach P, Bouterfa H, et al. Long-acting octreotide and prolonged-release lanreotide formulations have different pharmacokinetic profiles. J Clin Pharmacol. 2005;45(7):836–844.
77. McKeage K, Cheer S, Wagstaff AJ. Octreotide long-acting release (LAR): a review of its use in the management of acromegaly. Drugs. 2003;63(22):2473–2499.
78. Lancranjan I, Bruns C, Grass P, et al. Sandostatin LAR: a promising therapeutic tool in the management of acromegalic patients. Metabolism. 1996;45(8 Suppl 1):67–71.
79. Gatto F, Barbieri F, Arvigo M, et al. Biological and biochemical basis of the differential efficacy of first and second generation somatostatin receptor ligands in neuroendocrine neoplasms. Int J Mol Sci. 2019;20(16):3940.
80. Colao A, Pivonello R, Auriemma RS, Galdiero M, Savastano S, Lombardi G. Beneficial effect of dose escalation of octreotide-LAR as first-line therapy in patients with acromegaly. Eur J Endocrinol. 2007;157(5):579–587.
81. Giustina A, Bonadonna S, Bugari G, et al. High-dose intramuscular octreotide in patients with acromegaly inadequately controlled on conventional somatostatin analogue therapy: a randomised controlled trial. Eur J Endocrinol. 2009;161(2):331–338.
82. Giustina A, Mazziotti G, Cannavo S, et al. High-dose and high-frequency lanreotide autogel in acromegaly: a randomized, multicenter study. J Clin Endocrinol Metab. 2017;102(7):2454–2464.
83. Auriemma RS, Pivonello R, Ferreri L, Priscitelli P, Colao A. Cabergoline use for pituitary tumors and valvular disorders. Endocrinol Metab Clin North Am. 2015;44(1):89–97.
84. Sandret L, Maison P, Chanson P. Place of cabergoline in acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2011;96(5):1327–1335.
85. Kuhn E, Chanson P. Cabergoline in acromegaly. Pituitary. 2017;20(1):121–128.
86. Schmid HA. Pasireotide (SOM230): development, mechanism of action and potential applications. Mol Cell Endocrinol. 2008;286(1–2):69–74.
87. Poll F, Lehmann D, Illing S, et al. Pasireotide and octreotide stimulate distinct patterns of sst2A somatostatin receptor phosphorylation. Mol Endocrinol. 2010;24(2):436–446.
88. Colao A, Bronstein MD, Freda P, et al. Pasireotide versus octreotide in acromegaly: a head-to-head superiority study. J Clin Endocrinol Metab. 2014;99(3):791–799.
89. Gatto F, Feelders RA, Franck SE, et al. In vitro head-to-head comparison between octreotide and pasireotide in GH-secreting pituitary adenomas. J Clin Endocrinol Metab. 2017;102(6):2009–2018.
90. Hofland LJ, van der Hoek J, van Koetsveld PM, et al. The novel somatostatin analog SOM230 is a potent inhibitor of hormone release by growth hormone- and prolactin-secreting pituitary adenomas in vitro. J Clin Endocrinol Metab. 2004;89(4):1577–1585.
91. Ibanez-Costa A, Rivero-Cortes E, Vazquez-Borrego MC, et al. Octreotide and pasireotide (dis)similarly inhibit pituitary tumor cells in vitro. J Endocrinol. 2016;231(2):135–145.
92. Gadelha MR, Bronstein MD, Brue T, et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, Phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(11):875–884.
93. Shimon I, Adnan Z, Gorshtein A, et al. Efficacy and safety of long-acting pasireotide in patients with somatostatin-resistant acromegaly: a multicenter study. Endocrine. 2018;62(2):448–455.
94. Schmid HA, Brue T, Colao A, et al. Effect of pasireotide on glucose- and growth hormone-related biomarkers in patients with inadequately controlled acromegaly. Endocrine. 2016;53(1):210–219.
95. Coopmans EC, Muhammad A, van der Lely AJ, Janssen J, Neggers S. How to position pasireotide LAR treatment in acromegaly. J Clin Endocrinol Metab. 2019;104(6):1978–1988.
96. Grottoli S, Maffei P, Bogazzi F, et al. ACROSTUDY: the Italian experience. Endocrine. 2015;48(1):334–341.
97. Freda PU, Gordon MB, Kelepouris N, Jonsson P, Koltowska-Haggstrom M, van der Lely AJ. Long-term treatment with pegvisomant as monotherapy in patients with acromegaly: experience from ACROSTUDY. Endocr Pract. 2015;21(3):264–274.
98. van der Lely AJ, Biller BM, Brue T, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97(5):1589–1597.
99. Buchfelder M, van der Lely AJ, Biller BMK, et al. Long-term treatment with pegvisomant: observations from 2090 acromegaly patients in ACROSTUDY. Eur J Endocrinol. 2018;179(6):419–427.
100. Ragonese M, Grottoli S, Maffei P, et al. How to improve effectiveness of pegvisomant treatment in acromegalic patients. J Endocrinol Invest. 2018;41(5):575–581. doi:10.1007/s40618-017-0773-0
101. Sievers C, Baur DM, Schwanke A, et al. Prediction of therapy response in acromegalic patients under pegvisomant therapy within the German ACROSTUDY cohort. Pituitary. 2015;18(6):916–923. doi:10.1007/s11102-015-0673-2
102. Franck SE, Korevaar TIM, Petrossians P, et al. A multivariable prediction model for pegvisomant dosing: monotherapy and in combination with long-acting somatostatin analogues. Eur J Endocrinol. 2017;176(4):421–431. doi:10.1530/EJE-16-0956
103. Feola T, Cozzolino A, Simonelli I, et al. Pegvisomant improves glucose metabolism in acromegaly: a meta-analysis of prospective interventional studies. J Clin Endocrinol Metab. 2019;104(7):2892–2902. doi:10.1210/jc.2018-02281
104. Neggers SJ, Franck SE, de Rooij FW, et al. Long-term efficacy and safety of pegvisomant in combination with long-acting somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2014;99(10):3644–3652. doi:10.1210/jc.2014-2032
105. Neggers SJ, de Herder WW, Feelders RA, van der Lely AJ. Conversion of daily pegvisomant to weekly pegvisomant combined with long-acting somatostatin analogs, in controlled acromegaly patients. Pituitary. 2011;14(3):253–258.
106. Madsen M, Poulsen PL, Orskov H, Moller N, Jorgensen JO. Cotreatment with pegvisomant and a somatostatin analog (SA) in SA-responsive acromegalic patients. J Clin Endocrinol Metab. 2011;96(8):2405–2413. doi:10.1210/jc.2011-0654
107. van der Lely AJ, Bernabeu I, Cap J, et al. Coadministration of lanreotide Autogel and pegvisomant normalizes IGF1 levels and is well tolerated in patients with acromegaly partially controlled by somatostatin analogs alone. Eur J Endocrinol. 2011;164(3):325–333. doi:10.1530/EJE-10-0867
108. Trainer PJ, Ezzat S, D’Souza GA, Layton G, Strasburger CJ. A randomized, controlled, multicentre trial comparing pegvisomant alone with combination therapy of pegvisomant and long-acting octreotide in patients with acromegaly. Clin Endocrinol (Oxf). 2009;71(4):549–557. doi:10.1111/cen.2009.71.issue-4
109. Neggers SJ, van Aken MO, Janssen JA, Feelders RA, de Herder WW, van der Lely AJ. Long-term efficacy and safety of combined treatment of somatostatin analogs and pegvisomant in acromegaly. J Clin Endocrinol Metab. 2007;92(12):4598–4601. doi:10.1210/jc.2007-1234
110. Bianchi A, Valentini F, Iuorio R, et al. Long-term treatment of somatostatin analog-refractory growth hormone-secreting pituitary tumors with pegvisomant alone or combined with long-acting somatostatin analogs: a retrospective analysis of clinical practice and outcomes. J Exp Clin Cancer Res. 2013;32:40. doi:10.1186/1756-9966-32-40
111. Higham CE, Atkinson AB, Aylwin S, et al. Effective combination treatment with cabergoline and low-dose pegvisomant in active acromegaly: a prospective clinical trial. J Clin Endocrinol Metab. 2012;97(4):1187–1193. doi:10.1210/jc.2011-2603
112. Bernabeu I, Alvarez-Escola C, Paniagua AE, et al. Pegvisomant and cabergoline combination therapy in acromegaly. Pituitary. 2013;16(1):101–108. doi:10.1007/s11102-012-0382-z
113. Muhammad A, van der Lely AJ, Delhanty PJD, et al. Efficacy and safety of switching to pasireotide in patients with acromegaly controlled with pegvisomant and first-generation somatostatin analogues (PAPE Study). J Clin Endocrinol Metab. 2018;103(2):586–595. doi:10.1210/jc.2017-02017
114. Muhammad A, Coopmans EC, Delhanty PJD, et al. Efficacy and safety of switching to pasireotide in acromegaly patients controlled with pegvisomant and somatostatin analogues: PAPE extension study. Eur J Endocrinol. 2018;179(5):269–277. doi:10.1530/EJE-18-0353
115. Yamada S, Fukuhara N, Oyama K, Takeshita A, Takeuchi Y. Repeat transsphenoidal surgery for the treatment of remaining or recurring pituitary tumors in acromegaly. Neurosurgery. 2010;67(4):949–956. doi:10.1227/NEU.0b013e3181ec4379
116. Mathioudakis N, Salvatori R. Management options for persistent postoperative acromegaly. Neurosurg Clin N Am. 2012;23(4):621–638. doi:10.1016/j.nec.2012.06.005
117. Abe T, Ludecke DK. Recent results of secondary transnasal surgery for residual or recurring acromegaly. Neurosurgery. 1998;42(5):
118. Ludecke DK. Value of transcavernous surgery in the treatment of pituitary adenomas. Eur J Endocrinol. 1995;133(2):147–148. doi:10.1530/eje.0.1330147
119. Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur J Endocrinol. 2005;152(3):379–387. doi:10.1530/eje.1.01863
120. Kurosaki M, Luedecke DK, Abe T. Effectiveness of secondary transnasal surgery in GH-secreting pituitary macroadenomas. Endocr J. 2003;50(5):635–642. doi:10.1507/endocrj.50.635
121. Wilson TJ, McKean EL, Barkan AL, Chandler WF, Sullivan SE. Repeat endoscopic transsphenoidal surgery for acromegaly: remission and complications. Pituitary. 2013;16(4):459–464. doi:10.1007/s11102-012-0457-x
122. Almeida JP, Ruiz-Trevino AS, Liang B, et al. Reoperation for growth hormone-secreting pituitary adenomas: report on an endonasal endoscopic series with a systematic review and meta-analysis of the literature. J Neurosurg. 2018;129(2):404–416. doi:10.3171/2017.2.JNS162673
123. Long H, Beauregard H, Somma M, Comtois R, Serri O, Hardy J. Surgical outcome after repeated transsphenoidal surgery in acromegaly. J Neurosurg. 1996;85(2):239–247. doi:10.3171/jns.1996.85.2.0239
124. Del Porto LA, Liubinas SV, Kaye AH. Treatment of persistent and recurrent acromegaly. J Clin Neurosci. 2011;18(2):181–190. doi:10.1016/j.jocn.2010.10.003
125. Shih HA, Loeffler JS. Radiation therapy in acromegaly. Rev Endocr Metab Disord. 2008;9(1):59–65. doi:10.1007/s11154-007-9065-x
126. Kim EH, Oh MC, Chang JH, et al. Postoperative gamma knife radiosurgery for cavernous sinus-invading growth hormone-secreting pituitary adenomas. World Neurosurg. 2018;110:e534–e545. doi:10.1016/j.wneu.2017.11.043
127. Ding D, GU M, MR P, et al. Stereotactic radiosurgery for acromegaly: an international multicenter retrospective cohort study. Neurosurgery. 2019;84(3):717–725. doi:10.1093/neuros/nyy178
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.