Back to Journals » Journal of Blood Medicine » Volume 12
Obstacles to Early Diagnosis and Treatment of Inherited von Willebrand Disease: Current Perspectives
Authors Castaman G, Linari S
Received 16 December 2020
Accepted for publication 15 January 2021
Published 22 March 2021 Volume 2021:12 Pages 165—175
DOI https://doi.org/10.2147/JBM.S232758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Giancarlo Castaman, Silvia Linari
Center for Bleeding Disorders and Coagulation, Department of Oncology, Careggi University Hospital, Florence, Italy
Correspondence: Giancarlo Castaman
Center for Bleeding Disorders and Coagulation, Department of Oncology, Careggi University Hospital, Largo G. Brambilla 3, Florence, 50134, Italy
Tel +39 055 7947587
Fax +39 055 7947794
Email [email protected]
Abstract: Von Willebrand disease (VWD), the most common inherited bleeding disorder, is highly heterogeneous, and its early diagnosis may be difficult, especially for mild cases and in qualitative von Willebrand factor (VWF) defects. Appropriate VWD diagnosis requires the combination of personal and/or family history of bleeding and abnormal VWF laboratory testing. The use of bleeding assessment tools has been helpful in standardizing bleeding history collection and quantification of bleeding symptoms to select patients who may benefit of further hemostatic testing. Type 1 and 3 VWD which represent quantitative VWD variants are relatively easy to diagnose. The diagnosis of type 2 VWD requires multiple assessments to evaluate the effects induced by the responsible abnormality on the heterogeneous functions of VWF. Sensitive and reproducible tests are needed to evaluate different VWF activities, starting from measuring VWF-platelet interaction. In the recent years, several increasingly sensitive, rapid and automated assays have been developed, but they are not widely available so far. Genetic testing for VWD diagnosis is not a common practice because VWF gene is very large and highly polymorphic and therefore it is used only in specific cases. It is evident that the early and correct VWD diagnosis allows optimal management of bleeding and situations at risk. Tranexamic acid, desmopressin, replacement therapy with plasma-derived concentrates with a variable content of VWF and FVIII, or the new recombinant VWF are the different therapeutic options available. Careful VWD classification guides treatment because desmopressin is widely used in type 1 while replacement therapy is the cornerstone of treatment for type 2 and 3 variants.
Keywords: von Willebrand disease, von Willebrand factor, bleeding history, laboratory assays, desmopressin, replacement therapy
Introduction
Von Willebrand disease (VWD) is the most common inherited bleeding disorder, characterized by an extreme variability of clinical manifestations and laboratory phenotypes. The clinical spectrum varies from subjects with dubious, mild symptoms to patients with spontaneous, severe, sometimes life-threatening bleeds. Due to the complexities of clinical and laboratory assessment, the diagnosis can be difficult, especially in mild forms, so that the disease may be under-diagnosed also among symptomatic patients.
In this paper, we analyze the actual obstacles to early diagnosis and subsequent treatment of VWD.
VWD
VWD is a bleeding disorder characterized by a deficiency and/or an abnormality of von Willebrand factor (VWF), an adhesive glycoprotein that plays an essential function in both primary and secondary hemostasis. VWF is required for platelet adhesion to the subendothelium exposed by a vascular injury and also in mediating platelet–platelet interactions together with fibrinogen, thus enhancing the hemostatic process. Furthermore, VWF is the carrier of coagulation factor VIII (FVIII), driving it to the site of vascular damage and modulating its proteolytic degradation.1,2 VWD is extremely heterogeneous from a genetic and clinical point of view. The inheritance can be autosomal dominant or recessive, although VWD usually presents with an autosomal dominant inheritance. Thus, patients often report other relatives with a history of bleeding, although severity may differ among affected members belonging to same family. In epidemiological studies, VWF deficiency is identified in about 1% of the general population, but only about 0.01% has clinically significant bleeding symptoms.3,4 The actual prevalence is probably between these two extreme values and some people could have undiagnosed VWD-related bleeding.
Biology of VWF
VWF is a large multimeric adhesive plasma glycoprotein synthesized in megakaryocytes and endothelial cells.5–7 The biosynthesis and organization of VWF involves a complex intracellular pathway and defects at any step of which may cause a decreased plasma VWF level or dysfunction. The gene coding for VWF is located on the short arm of chromosome 12 (12p13.2) and it includes 52 exons.8 VWF has a partial pseudogene located on chromosome 22 with 97% homology with the authentic VWF gene.9 The presence of this pseudogene can complicate genetic analysis and contribute to a pathophysiological mechanism of gene conversion.
The primary translation protein contains 2813 amino acids, composed by a prepeptide of 22 amino acid, a propeptide 741 amino acid long and the mature VWF subunit of 2050 amino acids. The different binding abilities of VWF are scattered over four repeated domains (D1, D2, D’, D3, A1, A2, A3, D4, C1-C6, CK).10,11 The D’-D3 domains are essential for binding to FVIII. The VWF A1 domain is important in binding VWF to platelets through the platelet receptor glycoprotein, GPIb, and contains binding sites for heparin and collagen. The A2 domain contains the cleavage site which regulates post-secretion processing of VWF size by the VWF-cleaving protease, ADAMTS13. The A3 domain contains a binding site for collagen. The C1 domain contains a unique RGDS (Arg-Gly-Asp-Ser) sequence responsible for binding to the glycoprotein GPIIb/IIIa complex on platelet surface.
The cleavage of the signal peptide prompts the VWF subunits to dimerize at the C-terminal region through intermolecular disulfide bridges in the endoplasmic reticulum. Then, the acidic pH and high calcium concentration in the Golgi induce the building of VWF multimers through bridging through disulfide residues between D3 domains. The propeptide remains non-covalently bound to the VWF multimer inside the cell until fully cleavage allows for the separate release into plasma. VWF is stored in α-granules of platelets or endothelial Weibel-Palade bodies11 and secreted in plasma or abluminally to subendothelium through a constitutive and a regulated pathway. The subsequent proteolysis by ADAMTS13 produces variably sized multimers,12 whose clearance occurs later by macrophages in the liver and spleen.13
VWD Clinical Manifestations and Classification
The quantitative deficiency and/or qualitative abnormality of VWF lead to a variable risk of bleeding, depending on severity of the deficiency of VWF and FVIII. VWD clinical manifestations include predominantly mucosal bleeding (epistaxis, menorrhagia, gastrointestinal bleeds), surgery or trauma-related bleeding and joint bleeding, especially for patients with more severe FVIII deficiency.
VWD is classified into three main types categorizing quantitative (type 1 and 3) or qualitative (type 2) VWF abnormalities (Table 1).14,15
 |
Table 1 Classification of Von Willebrand Disease |
Unlike type 3, type 1 VWD (60–70% of cases) presents milder VWF deficiency (10–30 U/dL), with normal or slightly reduced FVIII levels. The inheritance transmission is autosomal dominant and missense amino acid changes represent the majority of causative mutations. Bleeding manifestations are mild. VWF is virtually absent in patients with type 3 (1–2% of cases), and FVIII levels are usually very low. Most of these patients are homozygous or compound heterozygous for null alleles in the VWF gene.16 The remaining 20–25% of patients have Type 2VWD, further divided into four subtypes which take into account distinct pathophysiological mechanisms and laboratory features.14,17
Typically, type 2A is caused by mutations resulting in a decreased binding to platelets caused by the absence of significant reduction of high and intermediate-molecular-weight VWF multimers and. In type 2B, the mutant VWF shows an increased affinity for GpIb on platelet surface, which induces a rapid clearance of VWF. Type 2M is characterized by a decreased in or absent binding to GpIb, with an apparently intact VWF multimer structure. Type 2N (Normandy) VWF presents variable binding ability for FVIII with a shortened FVIII half-life. Most type 2 are usually inherited as a dominant trait with high penetrance and expressivity apart from type 2N which is transmitted in a recessive manner.
VWD Treatment
The correction of the dual hemostatic defect represents the aim of treatment in VWD. Bleeding history together with FVIII and VWF and levels in plasma allow to define the bleeding risk in each patient. When FVIII level is greater than 40–50 U/dL, the risk of serious bleeding is limited and tranexamic acid alone may allow to control most minor bleeds. In patients with a more severe defect or VWF as in qualitative variants, VWF and FVIII plasma levels must be corrected to allow for effective hemostasis. The available therapeutic strategies are based on increasing VWF and FVIII by releasing VWF from stores in endothelial cells triggered with desmopressin (DDAVP) or replacing missing VWF with plasma-derived (pd) coagulation factor concentrates containing FVIII and VWF.
DDAVP (1-deamino-8-D-arginine-vasopressin) is a synthetic analog of the antidiuretic hormone vasopressin18,19 which can be given by intravenous, subcutaneous, or intra-nasal route. In responsive patients, a peak of the released FVIII and VWF > 50 U/dL is usually obtained 60 minutes after its administration. Usually, 0.3 µg/kg is used for intravenous or subcutaneous administration, and 300 µg in adults and 150 µg in children with intranasal spray. DDAVP is cheap and carries no risk of viral transmission; however, progressive reduction in magnitude of response (tachyphylaxis) is observed after repeated closely spaced doses due to the cellular depletion of VWF/FVIII. DDAVP is mostly efficacious in patients with type 1 VWD with normal VWF content in cells and baseline VWF and FVIII levels >10 U/dL.20 In type 2B, DDAVP causes a transient occurrence or aggravation of thrombocytopenia and is considered contraindicated21 while heterogeneous patterns are observed in other type 2 varieties.14,22 Type 3 patients do not show any significant increase post-administration.
Plasma-derived VWF-FVIII (pd-VWF/FVIII) concentrates are the treatment of choice when DDAVP is not useful.14,22 Several intermediate and high-purity pd-VWF/FVIII concentrates are available (Table 2). All these concentrates proved effective and safe in VWD although they have different VWF and FVIII content and show variable lack of high molecular weight (HMW) multimers.23 There are several national or international guidelines on the dosages to be used according to different clinical situations.14,24,25 A significant accumulation of FVIII may occur after repeated infusions of pd-VWF/FVIII since FVIII endogenously synthesized is stabilized by the infused VWF thus adding to that exogenously provided.26,27 This phenomenon may lead to the risk of deep vein thrombosis or cardiovascular complications, especially if other pro-thrombotic risk factors are present.28 Thus, daily monitoring of FVIII:C to maintain plasma levels below 150 U/dL is recommended to prevent thrombotic risk especially during major surgery. Also, a pd-VWF high purity concentrate containing little FVIII (Wilfactin ®; LBF, Les Ulis, France) could be chosen in patients with mild FVIII reduction. In this case, the co-administration of a priming dose of FVIII is recommended in patients with basal FVIII:C levels <30 U/dL, when immediate hemostasis is required, since 6–8 hours are necessary for endogenous FVIII to reach levels higher than 50–60 U/dL after infusion.29 Recently, a human recombinant VWF (r-VWF) Vonicog alpha has been produced by using a genetically engineered Chinese Hamster Ovary (CHO) cell line co-expressing VWF and FVIII genes and purified by immune-affinity chromatography and approved in some countries.30 Vonicog alpha is a >99% pure r-VWF molecule with the full range of VWF multimers because ADAMTS13 is not present during the manufacturing process. Therefore, also the ultra-large multimers (ULMs) are present, which are the most active forms,31 similarly to those observed after release in plasma from Weibel-Palade bodies of endothelial cells. These ULMs are however rapidly cleaved by ADAMTS-13 when infused for treatment. Several studies in VWD patients have demonstrated the efficacy and safety for treating spontaneous bleeding and during surgery.32–34 Two Phase III studies are assessing its safety and efficacy in children and for prophylaxis.
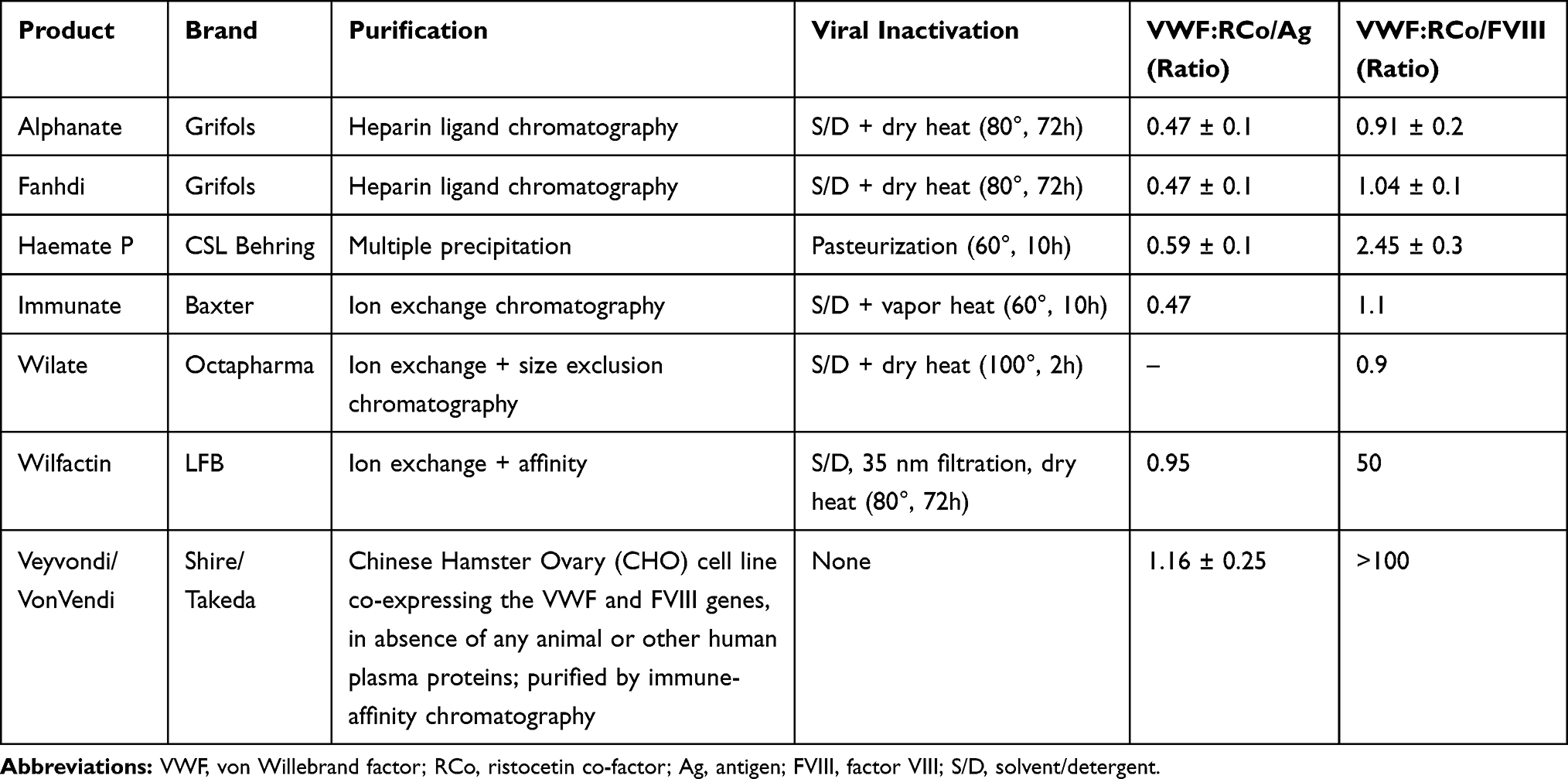 |
Table 2 Pd-VWF/FVIII Concentrates Licensed in Europe |
VWD Diagnosis
The diagnosis of VWD may be complex, especially in mild form, due to the extreme clinical variability, the difficult standardization of diagnostic tests, the presence of physiological and pathological variables influencing the plasma level of VWF. VWF plasma levels vary with age, increase during pregnancy, physical activity, inflammation, infections and cancer. Furthermore, plasma VWF levels are influenced by blood group with VWF levels 25–30% lower in type-0 individuals than in those with other blood groups.35 Subjects with blood group 0 show a VWF accelerated clearance36 and an enhanced ADAMTS13-mediated proteolysis.37 Thus, low VWF level alone does not suggest the presence of VWD and the cut-off for the diagnosis especially in type 1 is still discussed.38 However, it has been proposed as a diagnostic threshold the presence of VWF level <30 U/dL14,24 because an increased risk of bleeding is evident39 and the chance of detecting a causative mutation is very high.40 Subjects with levels between 30 and 50 U/dL may represent a difficult challenge.41,42 These subjects, and especially the females, may have increased bleeding scores, but inconsistent linkage to VWF locus and mutations identified roughly in 50% only and they have mostly blood group 0.41,42 The category “Low VWF” has been designed for these very frequent individuals.41 From the therapeutic point of view, these patients can be safely and effectively managed by using antifibrinolytics and/or DDAVP.43
Therefore, the definite diagnosis of VWD requires the combination of personal history of bleeding, family history of bleeding or diagnosed VWD and abnormal laboratory testing for VWD.
Bleeding Assessment Tools
Mild bleeding events may frequently occur also in subjects without a specific bleeding disorder. Furthermore, reporting and interpreting bleeding episodes may be subjective. The development of bleeding assessment tools (BATs) has allowed to standardize bleeding history collection and to assign a quantitative score to bleeding symptoms.44 These questionnaires generate a cumulative score of different bleeding symptoms according to their severity and frequency. The ISTH-BAT is designed for both pediatric and adult subjects and it evaluates 12 different bleeding symptoms, scoring from −1 (eg, no bleeding after invasive procedures) to +4 (eg, blood transfusion required after invasive procedures) according to the presence and severity of a specific symptom. An ISTH-BAT score ≥3 in children, ≥4 in males, ≥6 in females is considered positive for the presence of a significant bleeding history.45 BAT is an important mean to gather a detailed and standardized bleeding history thus selecting patients requiring additional hemostatic evaluation.46
Laboratory Testing for VWD
Among the hemostatic screening tests, the aPTT (activated partial thromboplastin time) and PFA-100 (Platelet Function Analyzer) may be used as an initial screen for possible VWD patients. However, the aPTT may be prolonged only in those patients in whom FVIII plasma level is reduced (Type 3 or Type 2N VWD). The PFA-100 mimicks in vivo primary hemostasis after small vessel wall injury and has a high sensitivity (90%) when VWF is significantly reduced (<20–25 U/dL) and thus most patients with type 2 are also identified.47 However, PFA-100 is normal in type 2N and in many patients with VWF levels only slightly reduced. Furthermore, a complete blood count is also required to assess the presence of thrombocytopenia which may suggest type 2B VWD.
The appropriate VWD diagnosis is based on several laboratory assays, which explore the different functions of VWF14 (Table 3). Laboratory testing for VWD initially includes VWF antigen determination (VWF:Ag), a VWF-platelet binding test, usually VWF-ristocetin cofactor activity assay (VWF:RCo) or less widely used newer tests (VWF:GPIbM, VWF:GPIbR), and the measurement of coagulant activity of FVIII (FVIII:C).
 |
Table 3 Laboratory Pattern in Von Willebrand Disease |
Type 1 and 3 VWD are characterized by equally low or absent VWF:Ag and VWF-platelet binding activity. The qualitative type 2 variants require additional tests to make an appropriate diagnosis, like VWF:collagen binding activity (VWF:CB), platelet-rich plasma agglutination at different ristocetin concentrations (RIPA), VWF-FVIII binding (VWF:FVIIIB) and VWF propeptide (VWFpp). The assessment of VWF multimer pattern may be also needed.48 VWD laboratory diagnosis can therefore be complex not only for the number of tests that may be necessary, but also for the poor reproducibility for some of them. Furthermore, some tests are still limited to a few selected laboratories.
VWF:Ag measurement is usually carried out with automated ELISA or latex immunoassays (LIA).49 Although these tests have a high reproducibility, they are of limited value for type 2 VWD in which sometimes they could be normal and must be used with other functional assays.
VWF:RCo activity is the time-honored assay for assessing the interaction of VWF with its platelet GpIb receptor. Ristocetin induces a conformational change in VWF, thus exposing the A1 domain which binds to GpIb and produces platelet agglutination proportional to the function of VWF. A VWF:RCo/VWF:Ag ratio <0.6 suggests the presence of a qualitatively abnormal VWF (type 2 VWD).24 However, traditional VWF:RCo assays using normal fresh or formalin-fixed platelets have low inter-laboratories reproducibility,50 high coefficient of variation and poor sensitivity for very low VWF values. The optimal ristocetin concentration may be another limitation of the test,51 as well as also the erroneous low levels in presence with p.P1467S and p.D1472H VWF polymorphisms.52
The search for assays with less variability and increased sensitivity than VWF:RCo has prompted the development of new tests. VWF:GPIbR uses recombinant GPIbα fragments (rGPIb) adhered to microparticles, eliminating the use of whole platelets.53 Instead, VWF:GPIbM uses recombinant GPIbα fragments containing two gain-of-function variants to induce spontaneous binding of VWF without ristocetin,54 but introducing a non-physiologic mutant GPIb receptor. Both assays have an excellent coefficient of variation, a more sensitive lower limit of detection, and excellent correlation with VWF:RCo,55 besides not being affected by some common VWF variants.56
FVIII:C assay must also be performed because VWF is the carrier for FVIII and usually a reduction of VWF causes also a variable reduction of FVIII. FVIII:C/VWF:Ag ratio is around 1 in normal subjects while in type 2N it is decreased (<0.5). In type 3 patients, FVIII:C is typically <10 U/dL.
The VWF multimer pattern analysis is useful to identify type 2 cases, although the traditional procedure with agarose gel electrophoresis is technically difficult and time consuming.57 Recently a semiautomated rapid, and sensitive method58 has been made available. All size multimers are present in Type 1 VWD, although reduced in concentration, while they are absent in type 3. The absence of HMW multimers is observed in type 2A and 2B VWD, while a normal pattern is present in type 2M and 2N.59
VWF:CB measures the VWF ability to bind to exposed collagen at the site of vascular injury. This ability correlates with the presence of HMW multimers, and patients with type 2A or 2B VWD have reduced VWF:CB.60 In Type 1, VWF:CB/VWF:Ag ratio is approximately 1, while a VWF:CB/VWF:Ag ratio <0.6 suggests the deficiency of HMW multimers as in type 2A or more rarely a specific collagen binding defect without loss of HMW multimers. The ratio is normal in type 2 M which has a normal VWF multimer pattern. VWF:CB is usually tested by an ELISA but recently an automated and rapid chemiluminescence method has become available.61 Typically, a mixture of type I or type III collagen is used because it increases specificity.62 The A3 VWF domain binds type I and type III collagen, while the A1 domain binds type IV and type VI collagen, but assays testing this latter interaction are not widely available.63
The VWF-platelet binding assessed in platelet-rich-plasma at different ristocetin concentrations (RIPA) may help in distinguishing type 2B from type 2A because in type 2 B (and platelet-type VWD) platelet aggregation occurs even at low-dose ristocetin concentration (≤0.6 mg/m).14
The VWF-FVIII binding (VWF:FVIIIB) test is required to diagnose type 2N, characterized by an abnormal VWF-FVIII binding.64 The ELISA microplate assay explores the capacity of patient’s plasma VWF to bind to recombinant FVIII.
The VWF propeptide (VWFpp) is in a 1:1 ratio with VWF in plasma.65 Its measurement is used as a surrogate marker of VWF synthesis and secretion. An increased VWFpp/VWF:Ag ratio identifies those patients with a shortened VWF half-life from plasma, commonly referred as Type 1 C (clearance), like the Vicenza variant. VWFpp/VWF:Ag ratio is usually slightly elevated increased in type 2A and 2B. The assay is still not widely performed.
Finally, genetic analysis for VWD diagnosis is not common practice because of the large size of the VWF gene and the time required. Furthermore, VWF is highly polymorphic.14,66 Therefore, it is used in specific cases, where it may have implications for diagnosis, patient’s management or counseling.14 Genetic testing in type 1 VWD is poorly informative, because an average of 62% of subjects shows a sequence variant in VWF gene. In particular, when VWF is <30 U/dL, the detection rate of identifiable sequence variants is around 80%67 and in patients with VWF:Ag >30 U/dL, mutations in VWF gene are not consistently identified.40,68 Mutations are more detectable in type 2 and type 3 VWD. Mutations causing type 2A usually are located in A2 domain, near the site of ADAMTS13 cleavage, or in the N or C terminal multimerization domains.69 In type 2B and 2M, mutations are mainly identified in the A1 domain and in type 2N mutations are located in the VWF D’ and D3 domains. Identifying mutations responsible for type 2 B rule out the possible diagnosis of platelet-type VWD. Identifying mutations responsible for type 2 N is helpful to exclude the possible diagnosis of mild/moderate hemophilia A. Mutations responsible for type 1 and 3 are scattered over the entire VWF gene. In type 3 VWD, genetic analysis may help to determine the risk of alloantibodies development with possible anaphylactic reactions upon treatment, a complication more common in presence of homozygous large gene deletions.70 The online ISTH VWF database includes most of the mutations and polymorphisms so far identified (http://www.vwf.group.shef.ac.uk).16
Diagnostic Flow-Chart and Therapeutic Management
VWD is a frequent and very heterogeneous bleeding disorder. Bleeding severity increases from type 1 to 3 and treatment differs. Several laboratory assays are available to properly diagnose VWD. In clinical practice, a flow-chart can be very useful to support a laboratory diagnosis.71 This algorithm starts with the evaluation of bleeding history in the patient and take advantage of the different laboratory assays used in different steps for a possible diagnosis of VWD (Figure 1).
 |
Figure 1 Algorithm for laboratory diagnosis of von Willebrand disease, modified with permission of Nancy International Ltd Subsidiary AME Publishing Company, from Von Willebrand disease in the United States: perspective from the Zimmerman program, Flood VH, Abshire TC, Christopherson PA, et al, volume 3, 2018]; permission conveyed through Copyright Clearance Center, Inc.54 |
When not all laboratory assays are available for a definitive VWD diagnosis, minimal diagnostic criteria may be considered an increased BS and VWF:RCo <40 U/dL. Based on the significance of these two criteria, an adequate VWD management may be provided (Figure 2).72
 |
Figure 2 Clinical spectrum of VWD: implications for management.Notes: Copyright ©2019. Taylor & Francis Online. Adapted from Castaman G, Linari S. Advances in diagnosis of VWD. Expert Opinion on Orphan Drugs. 2019;7(4):147-155.59 |
Conclusion
VWD is a common inherited bleeding disorder; however, its early diagnosis still may be complex. Several reasons are responsible for these diagnostic and consequently therapeutic difficulties. The first difficulty lies in understanding the significance of the reported bleeding. Mild bleeding symptoms may occur also in normal subjects and interpretation of hemorrhagic events erratic. For this reason, the use of BATs helps to standardize bleeding history collection and to quantify bleeding frequency and severity. ISTH-BAT represents a valid tool to identify patients for further laboratory evaluation. An important issue in early VWD diagnosis is represented by those individuals with slightly reduced VWF levels, which may vary over time and be significantly influenced by several physiological variables. The definition “low VWF” should be restricted to subjects with consistent VWF levels ranging from 30 to 50 U/dL and personal and/or family evidence of bleeding symptoms. Therefore, again a careful evaluation of bleeding history is of utmost significance in the diagnostic and prognostic work-up of a patient with suspected VWD or low VWF.
Severe quantitative VWF deficiencies are easy to be identified diagnose, while type 2 qualitative abnormalities are more challenging from the diagnostic point of view. Therefore, several assays s are required to evaluate the whole spectrum of VWF activities and interactions with specialized cellular receptors. These assays are becoming more and more specific, accurate and available also for non-specialized laboratories. The first step is to assess the ability of VWF to bind platelets. New tests are now increasingly available, which improve sensitivity and avoid laboratory artifacts. However, all the necessary assays are not widely available and VWF:RCo still represents the standard for measuring VWF activity, despite it does not reflect the range of physiologic VWF activities.
The correct typing of VWD allows the identification of the most appropriate treatment for the patient. For this purpose, DDAVP trial is also useful to identify the most suitable patient candidates. Type 1 patients are the ideal candidates for its use which could be considered also in some type 2 M and 2 N cases on the basis of the results of the infusion test. Type 2 B cases should not be treated with the compound because of the risk of thrombocytopenia. Replacement therapy is the cornerstone of treatment for type 2 A and B and 3 patients. Genetic testing is particularly indicated when the identification of a specific mutation guides clinical and laboratory monitoring after treatment (eg, assessing risk for alloantibody with use of factor concentrates), or for genetic counseling (eg, discriminating between mild/moderate hemophilia A versus type 2N VWD). Laboratory methods are being developed to permit rapid testing and more accurate measurements improving sensitivity, but some of them are still not widely used. Therefore, bleeding history still represents the crucial key in order to induce an early diagnosis and consequent optimal treatment of affected patients.
Disclosure
Dr Giancarlo Castaman reports personal fees from CSL Behring, personal fees from Kedrion, personal fees from Grifols, personal fees from Takeda, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. De Meyer SF, Deckmyn H, Vanhoorelbeke K. von Willebrand factor to the rescue. Blood. 2009;113:5049–5057. doi:10.1182/blood-2008-10-165621
2. Yee A, Kretz CA. Von Willebrand factor: form for function. Semin Thromb Hemost. 2014;40:17–27. doi:10.1055/s-0033-1363155
3. De Jong A, Eikenboom J. Developments in the diagnostic procedures for von Willebrand disease. J Thromb Haemost. 2016;14:449–460. doi:10.1111/jth.13243
4. Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69:454–459. doi:10.1182/blood.V69.2.454.454
5. Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi:10.1146/annurev.biochem.67.1.395
6. Wagner DD, Marder VJ. Biosynthesis of von Willebrand protein by human endothelial cells. Identification of a large precursor polypeptide chain. J Biol Chem. 1983;258:2065–2067. doi:10.1016/S0021-9258(18)32879-5
7. Sporn LA, Chavin SI, Marder VJ, Wagner DD. Biosynthesis of von Willebrand protein by human megakaryocytes. J Clin Invest. 1985;76:1102–1106. doi:10.1172/JCI112064
8. Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem. 1989;264:19514–19527. doi:10.1016/S0021-9258(19)47144-5
9. Mancuso DJ, Tuley EA, Westfield LA, et al. Human von Willebrand factor gene and pseudogene: structural analysis and differentiation by polymerase chain reaction. Biochemistry. 1991;30:253–269. doi:10.1021/bi00215a036
10. Zhou YF, Eng ET, Zhu J, et al. Sequence and structure relationships within von Willebrand factor. Blood. 2012;120:449–458. doi:10.1182/blood-2012-01-405134
11. Haberichter SL. Biosynthesis and organization of von Willebrand factor. In: Federici AB, Lee CA, Berntorp EE, editors. Von Willebrand Disease: Basic and Clinical Aspects. Oxford: Wiley-Blackwell; 2011:7–29.
12. Dong JF. Cleavage of ultra-large von Willebrand factor by ADAMTS-13 under flow conditions. J Thromb Haemost. 2005;3:1710–1716. doi:10.1111/j.1538-7836.2005.01360.x
13. Sadler JE. Low von Willebrand factor: sometimes a risk factor and sometimes a disease. Hematology Am Soc Hematol Educ Program. 2009;2009:106–112. doi:10.1182/asheducation-2009.1.106
14. Castaman G, Goodeve A, Eikenboom JC; on behalf of the European Group on von Willebrand disease (EUVWD). Principles of care for diagnosis and treatment of von Willebrand disease. Haematologica. 2013;98:667–674. doi:10.3324/haematol.2012.077263
15. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4:2103–2114. doi:10.1111/j.1538-7836.2006.02146.x
16. Hampshire DJ, Goodeve AC. The International Society on Thrombosis and Haemostasis von Willebrand disease database: an update. Semin Thromb Hemost. 2011;37:470–479. doi:10.1055/s-0031-1281031
17. Favaloro EJ. Von Willebrand disease: local diagnosis and management of a globally distributed bleeding disorder. Semin Thromb Hemost. 2011;37:425–426. doi:10.1055/s-0031-1280567
18. Rodeghiero F, Castaman G, Tosetto A. How I treat von Willebrand disease. Blood. 2009;114(6):1158–1165. doi:10.1182/blood-2009-01-153296
19. Leissinger C, Carcao M, Gill JC, Journeycake J, Singleton T, Valentino L. Desmopressin (DDAVP) in the management of patients with congenital bleeding disorders. Haemophilia. 2014;20(2):158–167. doi:10.1111/hae.12254
20. Castaman G, Lethagen S, Federici AB, et al. Response to desmopressin is influenced by the genotype and the phenotype in type 1 von Willebrand disease (VWD): results from the European study MCMDM-1 VWD. Blood. 2008;111:3531–3539. doi:10.1182/blood-2007-08-109231
21. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood. 2009;113(3):526–534. doi:10.1182/blood-2008-04-152280
22. Mannucci PM. New therapies for von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2019;2019:590–595. doi:10.1182/hematology.2019000368
23. Batlle J, Lopez-Fernandez MF, Fraga EL, et al. Von Willebrand factor/factor VIII concentrates in the treatment of von Willebrand disease. Blood Coagul Fibrinolysis. 2009;20:89–100. doi:10.1097/MBC.0b013e3283254570
24. Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14(2):171–232. doi:10.1111/j.1365-2516.2007.01643.x
25. Laffan MA, Lester W, O’Donnell JS, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol. 2014;167(4):453–465. doi:10.1111/bjh.13064
26. Franchini M. Surgical prophylaxis in von Willebrand’s disease: a difficult balance to manage. Blood Transfus. 2008;6(Suppl 2):33–38.
27. O’Donnell JS, Lavin M. Perioperative management of patients with von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2019;2019:604–609. doi:10.1182/hematology.2019000065
28. Coppola A, Franchini M, Makris M, Santagostino E, Di Minno G, Pannucci PM. Thrombotic adverse events to coagulation factor concentrates for treatment of patients with haemophilia and von Willebrand disease: a systematic review of prospective studies. Haemophilia. 2012;18(3):e173–e187. doi:10.1111/j.1365-2516.2012.02758.x
29. Borel-Derlon A, Federici AB, Roussel-Robert V, et al. Pharmacokinetic studies on Wilfactin, a von Willebrand factor concentrate with a low FVIII content treated with three virus-inactivation/removal methods. J Thromb Haemost. 2005;3(10):2219–2227. doi:10.1111/j.1538-7836.2005.01435.x
30. Turecek PL, Mitterer A, Matthiessen HP, et al. Development of a plasma- and albumin-free recombinant von Willebrand factor. Hamostaseologie. 2009;29(Suppl 1):S32–S38. doi:10.1055/s-0037-1617202
31. Stockschlaeder M, Schneppenheim R, Budde U. Update on von Willebrand factor multimers: focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul Fibrinolysis. 2014;25:206–216. doi:10.1097/MBC.0000000000000065
32. Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective trial. Blood. 2013;122(5):648–657. doi:10.1182/blood-2013-01-479527
33. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015;126(17):2038–2046. doi:10.1182/blood-2015-02-629873
34. Peyvandi F, Mamaev A, Wang JD, et al. Phase 3 study of recombinant von Willebrand factor in patients with severe von Willebrand disease who are undergoing elective surgery. J Thromb Haemost. 2019;17(1):52–62. doi:10.1111/jth.14313
35. Albanez S, Ogiwara K, Michels A, et al. Aging and ABO blood type influence von Willebrand factor and factor VIII levels through interrelated mechanisms. J Thromb Haemost. 2016;14:953–963. doi:10.1111/jth.13294
36. Gallinaro L, Cattini MG, Sztukowska M, et al. A shorter von Willebrand factor survival in 0 blood group subjects explains how AB0 determinants influence plasma von Willebrand factor. Blood. 2008;111:3540–3545. doi:10.1182/blood-2007-11-122945
37. Bowen DJ. An influence of AB0 group on the rate of proteolysis of von Willebrand factor by ADAMTS13. J Thromb Haemost. 2003;1:33–40. doi:10.1046/j.1538-7836.2003.00007.x
38. Sadler JE. Slippery criteria for von Willebrand disease type 1. J Thromb Haemost. 2004;2:1720–1723. doi:10.1111/j.1538-7836.2004.00933.x
39. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost. 2006;4:766–773. doi:10.1111/j.1538-7836.2006.01847.x
40. Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease(MCMDM-1VWD). Blood. 2007;109:112–121. doi:10.1182/blood-2006-05-020784
41. Flood VH, Christopherson PA, Gill JC, et al. Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood. 2016;127:2481–2488. doi:10.1182/blood-2015-10-673681
42. Lavin M, Aguila S, Schneppenheim S, et al. Novel insights into the clinical phenotype and pathophysiology underlying low VWF levels. Blood. 2017;130:2344–2353. doi:10.1182/blood-2017-05-786699
43. Lavin M, O’Donnell JS. How I treat low von Willebrand factor levels. Blood. 2019;133:795–804. doi:10.1182/blood-2018-10-844936
44. Rydz N, James PD. The evolution and value of bleeding assessment tools: the evolution and value of bleeding assessment tools. J Thromb Haemost. 2012;10:2223–2229. doi:10.1111/j.1538-7836.2012.04923.x
45. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders: ISTH/SSC bleeding assessment tool. J Thromb Haemost. 2010;8:2063–2065. doi:10.1111/j.1538-7836.2010.03975.x
46. Spradbrow J, Letourneau S, Grabell J, et al. Bleeding assessment tools to predict von Willebrand disease: utility of individual bleeding symptoms. Res Pract Thromb Haemost. 2020;4:92–99. doi:10.1002/rth2.12256
47. Favaloro EJ. The Platelet Function Analyser (PFA)-100 and von Willebrand disease: a story well over 16 years in the making. Haemophilia. 2015;21:642–645. doi:10.1111/hae.12710
48. Sharma R, Haberichter SL. New advances in the diagnosis of von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2019;2019:596–600. doi:10.1182/hematology.2019000064
49. Castaman G, Tosetto A, Cappelletti A, et al. Validation of a rapid test (VWF-LIA) for the quantitative determination of the von Willebrand factor antigen in type 1 von Willebrand disease diagnosis within the European multicenter study MCMDM-1VWD. Thromb Res. 2010;126:227–231. doi:10.1016/j.thromres.2010.06.013
50. Meijer P, Haverkate F. An external quality assessment program for von Willebrand factor laboratory analysis: an overview from the European concerted action on thrombosis and disabilities foundation. Semin Thromb Haemost. 2006;32:485–491. doi:10.1055/s-2006-947862
51. Vanhoorelke K, Cauwenberghs N, Vauterin S, et al. A reliable and reproducible ELISA method to measure ristocetin cofactor activity of von Willebrand factor. Thromb Haemost. 2000;83:107–113. doi:10.1055/s-0037-1613765
52. Flood VH, Gill JC, Morateck PA, et al. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood. 2010;116:280–286. doi:10.1182/blood-2009-10-249102
53. Verfaillie CJ, De Witte E, Devreese KM. Validation of a new panel of automated chemiluminescence assays for von Willebrand factor antigen and activity in the screening for von Willebrand disease. Int J Lab Hematol. 2013;35:555–565. doi:10.1111/ijlh.12087
54. Flood VH, Abshire TC, Christopherson PA, et al. Von Willebrand disease in the United States: perspective from the Zimmerman program. Ann Blood. 2018;3:7. doi:10.21037/aob.2017.12.05
55. Bodò I, Eikenboom J, Montgomery R, Patzke J, Schneppenheim R, Di Paola J. von Willebrand factor Subcommittee of the Standardization and Scientific Committee of the International Society for Thrombosis and Haemostasis. Platelet-dependent von Willebrand factor activity. Nomenclature and methodology: communication from the SSC of the ISTH. J Thromb Haemost. 2015;13(7):1345–1350. doi:10.1111/jth.12964
56. Boender J, Eikenboom J, van der Bom JC, et al. Clinically relevant differences between assays for von Willebrand factor activity. J Thromb Haemost. 2018;16(12):2413–2424.
57. Ruggeri ZM, Zimmerman TS. Variant von Willebrand’s disease: characterization of two subtypes by analysis of multimeric composition of FVIII/von Willebrand factor in plasma and platelets. J Clin Invest. 1980;65:1318–1325. doi:10.1172/JCI109795
58. Bowyer AE, Goodfellow KJ, Seidel H, et al. Evaluation of a semiautomated von Willebrand factor multimer assay, the hydragel von Willebrand multimer, by two European Centers. Res Pract Thromb Haemost. 2018;2:790–799. doi:10.1002/rth2.12141
59. Castaman G, Linari S. Advances in diagnosis of von Willebrand disease. Exp Opin Orphan Drugs. 2019;7:147–155. doi:10.1080/21678707.2019.1609352
60. Favaloro EJ, Grispo L, Exner T, et al. Development of a simple collagen based ELISA assay aids in the diagnosis of, and permits sensitive discrimination between type I and II, von Willebrand’s disease. Blood Coagul Fibrinolysis. 1991;2:285–291. doi:10.1097/00001721-199104000-00011
61. Jousselme E, Jourdy Y, Rugeri L, et al. Comparison of an automated chemiluminescent assay to a manual ELISA assay for determination of von Willebrand Factor collagen binding activity on VWD plasma patients previously diagnosed through molecular analysis of VWF. Int J Lab Hematol. 2018;40:77–83. doi:10.1111/ijlh.12743
62. Keesler DA, Flood VH. Current issues in diagnosis and treatment of von Willebrand disease. Res Pract Thromb Haemost. 2018;2:34–41. doi:10.1002/rth2.12064
63. Flood VH, Gill JC, Christopherson PA, et al. Critical von Willebrand factor A1 domain residues influence type VI collagen binding. J Thromb Haemost. 2012;10:1417–1424. doi:10.1111/j.1538-7836.2012.04746.x
64. Nishino M, Girma JP, Rothschild C, et al. New variant of von Willebrand disease with defective binding to factor VIII. Blood. 1989;74:1591–1599. doi:10.1182/blood.V74.5.1591.1591
65. Haberichter SL, Balistreri M, Christopherson R, et al. Assay of the von Willlebrand factor (VWF) propeptide to identify type 1 von Willebrand disease patients with decreased VWF survival. Blood. 2006;108:3344–3351. doi:10.1182/blood-2006-04-015065
66. Bellissimo DB, Christopherson PA, Flood VH, et al. VWF mutations and new sequence variations identified in healthy controls are more frequent in the African-American population. Blood. 2012;119:2135–2140. doi:10.1182/blood-2011-10-384610
67. Sharma R, Flood VH. Advances in the diagnosis and treatment of von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2017;2017(1):379–384. doi:10.1182/asheducation-2017.1.379
68. James PD, Notley C, Hegardon C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109:145–154. doi:10.1182/blood-2006-05-021105
69. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev. 2010;24:123–134. doi:10.1016/j.blre.2010.03.003
70. Christopherson PA, Perry CL, Bellissimo DB, et al. Genotype-phenotype relationship and the role of alloantibodies in type 3 VWD in the Zimmerman Program. Blood. 2017;130(suppl 1):19.
71. Boender J, Eikenboom J, van der Bom JG, et al. Clinically relevant differences between assays for von Willebrand factor activity. J Thromb Haemost. 2018;16:2413–2424.
72. Castaman G. How I treat von Willebrand disease. Thromb Res. 2020;196:618–625. doi:10.1016/j.thromres.2020.07.051
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.