Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Nucleoskeleton dynamics and functions in health and disease
Authors Meinke P, Makarov A ![]() , Lê Thành P, Sadurska D, Schirmer E
, Lê Thành P, Sadurska D, Schirmer E
Received 13 October 2014
Accepted for publication 12 November 2014
Published 27 January 2015 Volume 2015:7 Pages 55—69
DOI https://doi.org/10.2147/CHC.S53821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Denis Wirtz
Peter Meinke, Alexandr A Makarov, Phú Lê Thành, Daina Sadurska, Eric C Schirmer
Wellcome Trust Centre for Cell Biology and Institute of Cell Biology, University of Edinburgh, Edinburgh, UK
Abstract: It is a common misconception to view the "cyto"-skeleton as just the filament systems in the "cyto"-plasm. In fact, the cytoskeleton extends into the nucleus where the complex network connects to chromatin, and it also connects through the plasma membrane to the cytoskeleton of adjacent cells and to the "exo"-skeleton of the extracellular matrix. This review will focus principally on the subcomplex of the cytoskeleton associated with the nucleus, often referred to as the nucleoskeleton, but in the context of its extensive interconnectivity with the rest of the nucleus and with cytoplasmic filament systems all the way to the exoskeleton. The nucleoskeleton, made principally of type-V intermediate filament lamins, connects across the double membrane system of the nuclear envelope to likely all three primary cytoplasmic filament systems. It provides structural stability to the nucleus, and also incredible flexibility. In both its core structural aspect and through specificity gained by tissue-specific partner proteins, it contributes to genome organization and regulation as well as to signal transduction, both through chemical signaling cascades and likely through mechanotransduction. Defects in the nucleoskeleton have far-ranging effects due to its interactions with cytoplasmic filament systems, from mispositioning of nuclei to disruption of cell polarity and both decreased and increased cell migration depending on the defect. Accordingly, it is not surprising that many nucleoskeletal components are linked to a wide range of human diseases from specific types of cancer to muscular dystrophies, neuropathies, dermopathies, and premature aging syndromes.
Keywords: nuclear envelope, lamin, LINC complex, cytoskeleton, nuclear envelopathies
Introduction
Cytoskeletal proteins are among the most abundant in any cell. Three principal filament systems defined by their diameter work together to largely define cell shape and stability: microfilaments (7 nm), intermediate filaments (IFs; 10 nm), and microtubules (25 nm). Additional meshworks formed by spectrin and nesprin (syne) proteins also contribute to these functions. These are supported by roughly 5% of all genes encoded in the human genome that contribute to cytoskeletal assembly, regulation, and function. They also direct a variety of more specific functions for skeletal elements such as involvement in cytokinesis, pinocytosis, and phagocytosis; intracellular transport; signaling pathways; and cell migration.1 Some functions and structures are highly tissue-specific such as the Z-bands of muscle, immune synapses, actin in the acrosomes of spermatozoa (in lower organisms), cilia, and flagella, and many others.1,2
In the context of these myriad functions, the cytoskeleton connects different cellular organelles. Organelles are delimited by membranes and often have some kind of “scaffolding” protein to stabilize their membranes. For example, the inner surface of the plasma membrane is lined by spectrin filaments, and COP proteins coat the outer surface of the endoplasmic reticulum, contributing to its structure as much as to vesicle trafficking. The nucleus stands out from other organelles because it has the only organelle membrane lined on the inside by one of the “big 3” cytoskeletal filament types – the IFs (Figure 1). The nucleus also stands out in having a uniformly spaced double membrane maintained by a specialized system that also connects the nucleoskeletal IFs across the double membrane to cytoplasmic filament systems (Figure 1).
 | Figure 1 Schematic of the nuclear envelope. |
The first nucleoskeletal components were discovered roughly 30 years ago,3–6 but our knowledge of this specialized cytoskeletal subdomain is poor compared to cytoplasmic filament systems. To begin with, the actual structure of the nucleoskeletal polymer formed by the type-V IF lamins that lines the inner nuclear membrane remains obscure. Electron microscopy of the inner nuclear envelope surface in freeze-fractured frog oocyte nuclei reveals a network of the expected ~10 nm filaments;7 however, such a structure has not been observed in any somatic cell or oocyte from a higher vertebrate. This may reflect the tendency for chromatin to be highly connected to the inner nuclear membrane (except in lower oocytes) such that it obscures the lamin filaments, but it is also possible that differences in the milieu of nuclear cytoskeletal-associated proteins and the nature of lining a membrane surface result in a different structure for the polymer.
Although many functions have been ascertained for cytoplasmic filament-associated proteins, there are few clearly indicated thus far for the nuclear IF lamins. The best characterized are the SUN and nesprin families, forming a complex termed the LInker of Nucleoskeleton and Cytoskeleton (LINC) complex,8 which spans the double membrane and connects the nucleoskeleton to the cytoplasmic cytoskeletal filament systems (Figure 1). Both SUN and nesprin proteins are nuclear envelope transmembrane (NET) proteins. The recent identification of a large number of NETs by proteomics9–12 paves the way to determine the likely many nuclear-cytoskeletal-associated protein functions. Many NETs are highly tissue-specific,13 thus potentially enabling tissue-specific nucleoskeletal functions. Indeed, the use of multiple tissue libraries in 2-hybrid studies has identified over 600 partners for lamins including many of these NETs,14,15 thus increasing the likelihood that they could mediate such tissue-specific functions. Although only a small subset of identified NETs has been directly tested, nearly all those bind both lamins and chromatin,16 thus also connecting the genome to the cytoskeleton (Figure 1).
The importance of these many nucleoskeletal functions is underscored by the association of many diseases with mutations in nucleoskeletal proteins. More mutations causing distinct diseases have now been linked to LMNA than any other gene in the human genome.17 Moreover, many NETs have also been linked to disease, particularly those involved in the LINC complex.18 Interestingly, these diseases are extremely wide ranging, including muscular dystrophies, neuropathies, lipodystrophies, dermopathy, osteopoikilosis, and multisystemic disorders such as the premature aging progeroid syndromes.17,19 This makes for a conundrum, because, often, mutations in the same widely expressed protein can cause multiple diseases with distinct tissue pathologies. Thus, it is likely that additionally more tissue-specific nuclear envelope proteins are also involved.20
Nuclear versus cytoplasmic filament systems
Similar to cytoplasmic filaments, the nucleoskeleton plays a central role in nuclear structure and stability. Thus, one might assume that the two related and connected systems would be similar in organization. Yet, the nucleoskeleton comprises just IFs. To understand the logic of this evolutionary choice, it is necessary to consider the different properties of the three central filament systems (Figure 2A).
 | Figure 2 Cytoskeletal systems. |
Cytoplasmic filament systems
The building blocks for microfilaments (actin) and microtubules (tubulin) are soluble globular proteins that assemble into linear arrays (Figure 2B). In contrast, the building blocks for IF assembly are poorly soluble because roughly half of the protein mass is a series of heptad repeats formed into a linear coiled-coil dimer of 48–52 nm length. This increases the strength and stability but also renders them less dynamic than actin and tubulin that are polarized and dynamically assemble and disassemble driven by nucleotide binding and hydrolysis. The coiled-coil domain is broken into four parts by the linker regions, which may explain in part why IFs are also flexible. IF dimers next assemble into multiple head-to-tail linear arrays, which then layer over one another in a staggered fashion using interactions from both the coiled coils and the globular head and tail domains within and between arrays.21,22 The arrays layer like the individual fibers of a rope until there are 32 linear arrays/fibers in cross section with intermolecular interactions in all directions throughout to form a 10-nm filament. This complex structure renders IFs less dynamic23,24 and apolar.
These characteristics all contribute to significant differences in the biophysical behavior of the various filament systems under stress. Microfilaments are stabilized by tension and tend to resist stretch/strain, but break easily under compression forces. Microtubules are more resistant to a compression force, but break easily under stretch/strain. In contrast, IFs resist both compression and stretch/strain forces that tear microfilaments and microtubules apart.25 Thus, IFs are, at the same time, the most stable, strongest, and most elastic of the filament systems.
The filament systems also differ from the standpoint of tissue specificity. The three tubulin isoforms are universal, while actin has one universal isoform and four with specialized/tissue-specific functions such as forming Z-bands in the muscle. In contrast, IFs have over 70 distinct genes, many encoding multiple splice forms, and all except lamins are highly tissue-specific.1 For example, myogenic and neuronal precursors both express vimentin, but this is replaced by desmin in mature muscle and neurofilaments in neurons. Similarly, different layers of epidermis express different combinations of keratins. The many tissue-specific IFs enable a further diversity of function from their specific binding partners.
Nucleoskeleton
Tubulin is absent from the interphase nucleus. Though actin enhances the function of some helicases in yeast,26 there is no defined pathway for its regulated import and it remains controversial whether this nuclear actin assembles filaments.27 Although filaments can be forced by overexpressing actin carrying nuclear localization signals, it is noteworthy that wild-type actin has two conserved nuclear export signals to keep the nuclear levels low.28 Moreover, this minor actin population is dwarfed by lamins, which are typically present at ~3 million copies in a typical mammalian nucleus.29 Thus, actin and tubulin are not likely to contribute to the nucleoskeleton, which comprises principally the type-V IF lamins. Lamins differ from cytoplasmic IFs principally by having a longer coiled-coil region (+6 heptads),30,31 a nuclear localization sequence,32,33 and a C-terminal CaaX box that is farnesylated.34,35 All three of these differences contribute to the translocation of lamins into the nucleus.36,37
There are three genes encoding lamins: LMNA, LMNB1, and LMNB2. At least one B-type lamin is expressed in all cells, while lamin A is expressed later in differentiation. A combination of tissue differences in relative expression levels and several splice variants enable tissue specificity for the nucleoskeleton.38–40 The predominant LMNA splice variants in order are lamin A, C, AΔ10,41 and C2 that is unique to sperm cells.42 LMNB1 also has a tissue-specific splice variant, lamin B3,43 and others are yet to be identified. All splice variants share the coiled-coil domain, which is the most conserved region by sequence. Nonetheless, the coiled coils may differ structurally, as prediction algorithms suggest that lamin B2 might have two linkers fused into one long one, thus losing one of the four coiled coils. This should, in theory, increase flexibility and, though point mutations in the linkers did not yield notable assembly phenotypes in keratins,44 the lamin B2 was less stable in vitro45,46 and in vivo.47,48 Thus, differences in the relative expression levels of the different lamin subtypes in different tissues can yield distinct mechanical properties to the polymer.
As the lack of chromatin connections in Xenopus oocyte nuclei enables visualization of the inner membrane surface,7 different lamin subtypes were exogenously expressed in this system, yielding distinct assemblies.49 This suggests that different lamin subtypes form distinct networks and is further supported by immunogold-labeling electron microscopy and superresolution studies in certain cell types,50 while other studies observed Förster resonbance energy transfer (FRET) between different lamin subtypes and these subtypes interacted in vitro.45,51,52 Thus, this issue remains unresolved.
As mentioned above, lamins also differ from cytoplasmic IFs in that they are posttranslationally modified with the addition of a C-terminal farnesyl lipid moiety. As lamins are highly insoluble, association with the membrane through this modification could help prevent off-target aggregation and thus support proper assembly. At the same time, as both nuclear localization sequence and CaaX box contributed to lamin targeting,36 lamin association with the membrane might enable a backup system for transit through the peripheral channels of the NPC, though this has never been tested. Finally, this lipid modification may directly link the lamin polymer to the membrane to anchor it at the inner nuclear surface.53,54 This idea is supported by observations in the Xenopus oocyte system where B-type lamins appeared to assemble closer to the membrane than lamin A.49 B-type lamins keep their farnesyl moiety, while it is only transient in lamin A due to subsequent cleavage of several C-terminal residues.55 Mutations that prevent the subsequent removal of the farnesyl group perturb cells,56 but the functional reason for its transient addition in the case of lamin A remains obscure.
Network connectivity
The three central filament systems of the cytoplasm are interconnected through cytoskeletal-associated proteins. They must also be connected to the nucleoskeleton because the nucleus was “pulled” in the same direction as cytoplasmic filaments in experiments where a pipette tip was applied outside the nucleus in the cytoplasm.57 These and other observations led to the cellular tensegrity model proposed by Donald Ingber58 that explains the cytoskeleton as a 3D assembly comprises compression-resistant struts (microtubules) connected by stretchable linkers under tension (actin filaments). Thus, the cell is more like a tent with poles (compression) and anchored ropes (tension) as opposed to a brick and mortar structure just under compression stress.
As the nucleoskeleton comprises just the lamin IFs, there are apparently no countering forces in the isolated nucleus to generate tensegrity, but at the same time, the filaments are elastic, deformable, and capable of withstanding strong compression or stretch/tension forces. However, in intact cells this elastic nucleoskeleton is connected to the cytoplasmic filament network with its mixed properties principally through the LINC complex.8 The core components of LINC are SUN-domain NETs embedded in the inner nuclear membrane that bind lamins in the nucleoplasm and Klarsicht/ANC-1/Syne homology (KASH)-domain NETs in the nuclear envelope lumen that in turn connect to cytoplasmic filaments from the outer nuclear membrane. The 120-residue SUN domain defines a family with at least five members in mammals, all of which have been found in the inner nuclear membrane, though all but SUN1 and SUN2 are tissue-specific.59 The combination of a KASH domain that binds SUN domains and spectrin repeats defines the nesprin family.8,60,61 In mammalian cells, there are at least four SYNE genes that give rise to multiple nesprin isoforms through alternative splicing and transcription initiation.62 Nesprin-1 and -2 interact directly with the actin cytoskeleton.63–65 Nesprin-3 binds plectin through which it is suggested to connect to cytoplasmic IFs,66 though plectin interfaces with all three cytoplasmic filament systems. Nesprin-4 and the related KASH5 (that lacks spectrin repeats), respectively, bind kinesin and dynein, plus- and minus-end-directed microtubule-dependent motors.67,68 These interactions may support a connection of the nucleoskeleton to microtubules, though the tissue-specific expression of nesprin-4 in hair cells of the inner ear and of KASH5 in germ cells means that either microtubule connections are tissue-specific or other NETs mediate connections to the microtubules. An interesting study suggested that some nesprins bind each other to generate a protein scaffold on the outer nuclear membrane,69 thus providing a more stable surface for connections to cytoplasmic filaments.
In the context of filament network connections or cellular “tensegrity,” LINC acts as a local force projection site for the nucleus. Disruption of LINC should therefore not only affect the connection itself but would have an effect on the architecture and function of both the nucleus and cytoplasm. This has been investigated by several laboratories, each using different specially designed tools to deform the nucleus/cytoplasm and measure the effects of this strain on the system. The Wirtz laboratory found that disruption of LINC by overexpression of dominant-negative SUN or KASH domains leads to abnormal actin and vimentin organization and causes the nucleus to deform less under applied stress.70 More tellingly, disruption of LINC profoundly reduces 3D cell migration in a soft collagen matrix.71 As cell migration is principally directed by lammelipodia at the leading edge of the cell, the ability of this process to be affected by disrupting the connection of the actin cytoskeleton to the nucleus at the far end of the entire actin network strongly supports mechanical aspects predicted by tensegrity. As expected for an interconnected network, nucleoskeletal disruption also affects these properties, with the Lammerding laboratory finding that Lmna-/- mouse cells72 have more fragile and easily deformable nuclei and exhibit migration defects in 2D assays compared to wild-type.73 Although cytoskeleton architecture was visually intact, an overall increase in stiffness was measured by microrheology.73 The Broers and Ramaekers laboratories obtained similar results using a different approach, with Lmna-/- nuclei exhibiting an isotropic deformation upon indentation, though the rest of the cell had an anisotropic deformation.74 Thus, nucleoskeletal weakening by disruption of lamin A or nucleo-cytoplasmic connectivity makes the nucleus more deformable, but at the same time it impairs force transmission so that it is irresponsive to external mechanical stimuli.
For all the distinct characteristics that different lamin subtypes can confer to the polymers they form, much more variation can be contributed by the many tissue-specific nucleoskeletal components. For example, the LINC-component splice variant nesprin-2ε1 is specific to early embryonic cells while short nesprins 1α2 and 2α1 are restricted to heart and muscle tissues75 and several nesprin and SUN genes are tissue-specifically expressed.67,68,76 Several widely expressed and tissue-specific NETs may influence LINC or support separate interactions with cytoplasmic filaments. NET5 (Samp1) interacts with the LINC component SUN177 and also contributes to more specialized TAN lines that serve as tracks for nuclear migration within the cell.78 NET5 also binds lamin A and is observed at spindle poles during mitosis, while its knockdown affects centrosome positioning,79 suggesting an additional role mediating nucleoskeletal associations with microtubules. Interestingly, two NETs found in proteomics of muscle nuclear envelopes, Tmem214 and WFS1, similarly accumulate at spindle poles during mitosis, and Tmem214 along with a third muscle NET KLHL31 partially tracks with microtubules on the nuclear surface in interphase.12 Future work will likely reveal additional NETs that participate in specific nucleoskeletal connections with cytoplasmic filament systems.
Chromatin, a component of the nucleoskeleton?
Thus far, we have focused on the side of the lamin polymer facing outward, but the nucleoplasmic face is not less active. Many labs have shown that lamins can bind specific types of DNA/chromatin including beta-heterochromatin in Drosophila, MARs, SARs, telomeres, centromeres, and core histones – specifically H2A and H2B.76 There appear to be both weak binding sites in the lamin coiled-coil domain80,81 and high-affinity binding sites in the C-terminal domain.82,83 Furthermore, lamins can bind a variety of transcriptional regulators including pRb, cFos, and Mok2.84 This is thought to sequester these factors away from their target genes that tend to be more internal in the nucleus, but if a gene is proximal to lamins, it could have an activating effect. Indeed, ChIP-sequencing of lamin A-associated chromatin revealed a mixture of active and repressed genes.85 However, this study could not distinguish between contributions from a lesser nucleoplasmic pool of lamins and the membrane-associated polymer. It is also unclear whether lamin contributions to spatial genome organization are directed by lamin–chromatin interactions, lamin–transcriptional regulator interactions, or lamin–NET interactions. Nonetheless, defective lamin B1 results in release of chromosome 18 from the periphery,86 and lamin A mutations associated with human disease (see below) cause changes in the positioning of certain chromosomes.87
Several NETs also interact directly with chromatin and/or transcriptional regulators. Lamin B receptor (LBR) binds H3/H4 histones,88 favoring histone H3 with the silencing lysine 9 tri-methylation mark,89 and also binds heterochromatin protein 1 (HP1).90 LAP2β binds barrier-to-autointegration factor (BAF),76 the transcriptional repressor germ cell-less (gcl),91 and the chromatin-remodeling factor histone deacetylase 3.92 Interestingly, emerin was subsequently shown to bind these same factors,93,94 but it also has additional specific interactions with the transcriptional repressor Btf95 and the transcription factor Lmo7.96 These NETs are all widely expressed, and so could only contribute tissue-specific genome regulatory functions through the medium of tissue-specific chromatin or chromatin/DNA regulating binding partners. In contrast, there are several highly tissue-specific NETs that direct tissue-specific patterns of genome organization. TAPBPL, STT3A, NET5, NET29, NET39, NET45, and NET47 all can direct particular subsets of chromosomes to the nuclear periphery.11,97
All of the genome-linked NETs tested thus far also bind lamins, and so are also parts of the nucleoskeleton. This should enable lamins and NETs to work in concert to achieve their functions in genome organization and regulation. Indeed, LBR and lamin A together achieve a general pattern of heterochromatin distribution at the nuclear periphery that is disrupted when these components are removed.98 More specific gene positioning can be achieved with a different nexus of LAP2β with lamin B1 and histone deacetylase 3 that directs the IgH and Cyp3a loci to the nuclear periphery in lymphocytes.99 Lamin and interacting NET effects on the genome also include RNA splicing,100 DNA replication,101,102 and DNA damage repair responses.103
All of the above examples focus on the role of the genome as the repository of the genetic material and the regulation of that material. Chromosomes, however, can also be viewed as the largest individual molecules of the cell. In fact, a chromosome makes a megadalton molecule like titin, the largest protein in the cell and a contributor to the organization of the actin cytoskeleton, seem tiny in comparison. Such large molecules could easily contribute to the tensegrity network of the nucleoskeleton and through this to that of the whole cytoskeleton. While this is a rather novel way to view the genome, it is consistent with observations that during mitosis the attachment of a large chromosome to individual microtubules actually stabilize the microtubules and eventually the whole microtubule spindle.104 We propose that in interphase the decondensed chromosomes could act as a buffer for forces generated by cytoskeletal mechanics similar to how a cushion absorbs force when sat on. This idea also may shed light on the logic of evolution in making the protein nucleoskeleton almost exclusively from IFs and associated NETs. The extreme elasticity and tensile strength of the IF lamin polymer enables it to stretch under considerable force without breaking. As it sits between the powerful forces of the cytoplasmic filaments in their tensegrity network on one side and the powerful forces of the chromosomes on the other side, the lamin polymer endures probably the strongest pushing and pulling forces in the cell. That most chromatin binding NETs also bind to lamins and lamins also bind chromatin supports a multiplicity of docking sites for the genome, while having many different, mostly multispanning transmembrane proteins as part of this nexus provides for strong interactions with the membrane.16 Having multiple NETs engage in multiple connections to multiple cytoplasmic filament networks further distributes the load-bearing in the greater genome–nucleoskeleton–cytoplasmic filament tensegrity system. At this stage, however, this is just speculation.
Signaling through the nucleoskeleton
The connectivity of these various networks could support mechanotransduction of signals directly from the plasma membrane/extracellular matrix to the nucleus. Direct mechanical signaling should transmit a force applied at one end of the network to its other end in a matter of microseconds in contrast to the several seconds it would take for protein interactions and trafficking through the cell and nuclear pore complexes to get a normal protein–chemical signaling pathway from the plasma membrane to the nucleus.105 Consistent with the idea that mechanotransduction does indeed occur through cytoplasmic filament–nucleoskeleton connections, two different types of high-tension and low-tension actin fibers have been observed at the nuclear membrane. The high-tension fibers presumably contribute to tensegrity for the overall cell shape and stability. The low-tension fibers are focused at the actin cap on the nuclear surface and connect to the nucleoskeleton via the LINC complex.106 If high-tension fibers were involved in mechanotransduction, it is likely that the cell would become overstimulated due to the considerable dynamics of the cytoskeleton; therefore, it makes sense for the cell to have an independent low-tension network for the separate function of signal transduction.
An underlying contribution of mechanotransduction in nuclear membrane signaling is further indicated by impaired nuclear localization and signaling function of the mechanosensitive transcription factor megakaryoblastic leukemia 1 (MKL1) in cells from Lmna-/- mice.107 This impaired function appears to be a consequence of disrupted actin dynamics due to emerin mislocalization with its binding partner, lamin A, gone because exogenous overexpression of emerin rescued both the actin dynamics and the MKL1 function.107 Other protein signaling cascades that depend on nucleoskeletal NETs include emerin impacting on β-catenin signaling108 and MAN1 on Smad and TGF-β signaling.109,110 Additionally, the mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) pathways are affected, respectively, by emerin111 and lamin depletion,112 though there is no evidence at this point to indicate any links to mechanotransduction or tensegrity.
Mutations in nucleoskeletal proteins cause human disease
Mutations in lamins and several NETs of the nucleoskeleton cause a wide range of human diseases that include several muscular dystrophies, cardiomyopathies, dermopathy, neuropathies, bone disorders, lipodystrophies, and premature aging syndromes (Table 1).113 There are three core mechanisms proposed to explain how a mutation in a nuclear envelope protein could yield pathology. The first is that the cell cycle and/or stem cell maintenance is affected, resulting in a failure to regenerate damaged tissues from satellite cells. The second is that regulation of gene expression is altered, thus interfering with normal tissue function and metabolism. The third is that a weakening of the nucleoskeletal connections to the cytoplasmic filament systems results in increased susceptibility to mechanical stress – especially in cells suffering an intense mechanical strain such as muscle.114,115 It is likely that all three mechanisms contribute to disease pathology: for example, in muscle, increased susceptibility to mechanical stress could result in altered chromatin organization, which causes altered gene expression that finally results in satellite cells undergoing premature differentiation, thus depleting the pool of cells that can regenerate damaged tissue.
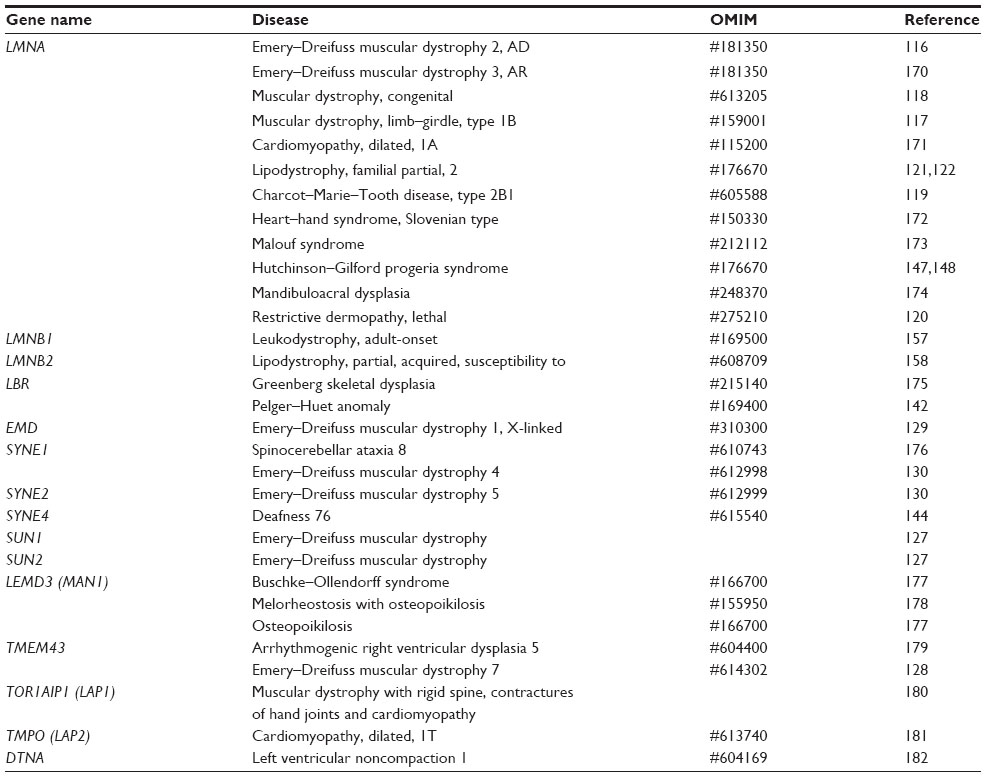 | Table 1 Diseases linked to nucleoskeletal proteins |
The tissue specificity of nuclear-envelope-linked diseases is curious, as different mutations in the widely expressed lamin A protein can cause distinct diseases such as muscular dystrophies,116–118 neuropathies,119 dermopathies,120 and lipodystrophies.121,122 Even more strikingly, each of the lamin A-linked muscular dystrophies affects a separate or partly overlapping set of specific muscle groups – shoulder, upper arm, calf muscles, and heart in Emery–Dreifuss muscular dystrophy (EDMD),123 proximal muscles (shoulder girdle and pelvic girdle) in limb–girdle muscular dystrophy (LGMD),124 and heart in dilated cardiomyopathy (DCM).125 Some of this tissue specificity of pathology could be due to the postulated tissue-specific partner proteins,126 but particularly in the muscular dystrophies the mechanical instability may well be the driving force.
EDMD has thus far been linked not only to lamin A but also to the NETs emerin, nesprin-1, nesprin-2, SUN1, SUN2, and Tmem43 (Luma).116,127–130 Four of these NETs are components of the LINC complex8 while emerin connects to both lamins131 and the postulated tensegrity chromosomal component93–96 and Tmem43 binds lamins, SUN2, and emerin.128 Some degree of tissue specificity can be observed in the proteins in this system, as EDMD disease-causing mutations have been found in the more muscle-specific nesprin isoforms 1α and 2β and myotubes generated in vitro from biopsy of patients with these mutations had disrupted the sarcomere structure.130 While all these connections support the mechanical instability hypothesis, it is notable that, though some lamin A mutations resulted in filament assembly defects in vitro, others did not,132 indicating that this is not a general characteristic of the disease. At the same time, a potential chromatin disruption role is supported by electron microscopy observations of disrupted peripheral dense chromatin in EDMD patient biopsy sections. Furthermore, defects in gene expression were observed in biopsy from other EDMD patients, particularly of the muscle myoD gene and the cell cycle regulator pRb that plays a role in both satellite cell maintenance and the cell cycle withdrawal necessary for myotube differentiation.133 More telling for the myoD defects, lamin A was required for myoD repositioning away from the nuclear envelope associated with its activation in Caenorhabditis elegans, and this repositioning failed when lamin A carrying specific EDMD lamin point mutations was expressed.134
The finding that several interacting components of the nucleoskeleton can each, when mutated, cause the same EDMD disease pathology supports the idea of tensegrity playing a role in the nuclear envelope system. Even more compelling to the idea of tensegrity is the finding that the combination of multiple nucleoskeletal protein mutations leads to more severe disease than the individual component mutations (Figure 3). For example, emerin mutations, which alone cause mild clinical presentation with the patients fully ambulatory, when combined with SUN1 mutations yield wheelchair dependency.127 Also lamin A mutations, when combined with emerin, SUN1, or SUN2 mutations, result in a more severe presentation.18,127,135,136 The tensegrity model is even more strongly supported by a similar increase in disease severity when a nucleoskeleton mutation (emerin) is combined with a mutation in cytoplasmic filaments (desmin).137 Although such modifier mutations are yet to be found in LGMD, it is compelling in light of this model that, apart from mutations in lamin A, this disease can also be caused by mutations in the cytoplasmic filament system proteins plectin,138 desmin,139 and titin140 as well as the connected exoskeleton protein integrin alpha-7.141 Thus, weakening of any part of the nucleoskeletal–cytoskeletal–exoskeletal system can generate a similar pathology consistent with mechanical instability.
 | Figure 3 Combined mutations at different points of nucleoskeletal–cytoplasmic filament contacts could yield more severe disease pathologies. |
Another nucleoskeletal NET evidently linked to mechanical instability is the LBR. In contrast to most of the NETs above that are involved in LINC connections to cytoplasmic filaments, LBR functions in connecting the nucleoskeleton to chromatin. LBR mutations cause a blood granulocyte disorder characterized by abnormal nuclear morphology and chromatin organization named the Pelger–Huet anomaly.142 Granulocyte nuclei normally undergo extensive cytoskeletal reorganization to become highly lobulated when they further differentiate to neutrophils; however, the nuclei remain spherical in Pelger–Huet anomaly.142 Interestingly, granulocyte nuclei have high levels of lamin B2, the “weakest” lamin, compared to other subtypes.45,143 Thus, LBR connections to chromatin likely provide a counter force to support lobulation in a weak lamin background while disruption of this tensegrity element by loss of LBR results in failure to lobulate.
Two other diseases are likely linked to nucleoskeletal mechanotransduction. In one, a mutation in tissue-specific nesprin-4 affects positioning of the nucleus specifically in the mechanosensory hair cells leading to autosomal recessive deafness.144 The other is familial cardiomyopathy with conduction defect, where mice expressing a specific lamin A cardiomyopathy mutation (N195K/N195K) exhibit the same defects in the MKL1 mechanosensitive transcription factor as the Lmna-/- mice.107 Mouse models of lamin A-linked cardiomyopathy also affect the MAPK and ERK kinase pathways.145,146
The progeroid syndromes appear to be quite different, exhibiting defects in the processing and assembly of lamins. The normal transient lamin A farnesylation is disrupted in slightly different ways to yield the Hutchinson–Gilford progeria syndrome (HGPS),147,148 mandibuloacral dysplasia,149 and restrictive dermopathy (RD),120 but all these disorders are characterized by the failure to remove the farnesyl group. HGPS is most commonly caused by a heterozygous mutation resulting in a 50-amino acid deletion in lamin A, which includes the C-terminal cleavage site for the protease ZMPSTE24 needed to remove the farnesyl group.144,148 The resulting mutated protein is commonly referred to as progerin. In contrast, RD is caused by mutations resulting in a loss of ZMPSTE24 that cause an accumulation of farnesylated, unprocessed full-length prelamin A. Thus, both remain farnesylated, but progerin has an additional deletion and, being heterozygous 50% of the lamin A, is wild-type. These differences account for an enormous distinction in pathologies. HGPS patients appear healthy at birth but develop a progeroid phenotype including extreme short stature, low body weight, early hair loss, lipodystrophy, scleroderma, decreased joint mobility, and osteolysis within the course of 1–2 years. In most cases, cardiovascular problems cause death in the second decade.150 RD invariably yields neonatal death or death in early infancy. Clinical features include tightly adherent thin skin, generalized joint contractures, dysplasia of clavicles, increased subcutaneous fat, and respiratory insufficiency.56 Mandibuloacral dysplasia, caused by partial loss of ZMPSTE24 function due to combinations of missense and nonsense mutations, is characterized by postnatal growth retardation, craniofacial anomalies, skin changes such as atrophy and speckled hyperpigmentation, as well as accumulation of fat in the neck and moderate lipodystrophy of the limbs with a median age of death of 30 years.56
As the permanently farnesylated B-type lamins formed a different type of network from lamin A when exogenously expressed in Xenopus, it is likely that the maintained association with the membrane alters the polymer structure. However, the principal findings to date favor the genome face of lamins in the pathology of HGPS, with chromatin disorganization, DNA damage, and genome instability as the primary culprits.151–153 This is further supported by the involvement of the NET-interacting chromatin protein BAF in another progeroid disorder, the Nestor–Guillermo progeria syndrome (OMIM #614008).154 But progeria is also linked to LMNA mutations not associated with a failure of lipid processing. Patients with these mutations often have phenotypes overlapping between different progeroid syndromes or other laminopathies.155,156
Finally, it is worth noting that lamin B1 has been linked to leukodystrophy157 and lamin B2 to partial lipodystrophy,158 though little is known about the pathogenic mechanism, and lamins have also been linked to cancer. In fact, loss of lamin A was one of the first historical biomarkers associated with increased metastasis;159 however, the focus on this was dropped because such trends differed in different tumor types.160 Nonetheless, recent work has indicated that nucleoskeleton involvement in basic cellular processes such as maintenance of nuclear shape and size, centrosomal positioning, cell migration and signal transduction, and DNA damage repair all likely contribute to the formation of a wide range of cancers.161,162
Therapeutic target identification and screening approaches
Thus far, there are no treatments to “cure” any of the nucleoskeleton-linked muscular dystrophies. Nonetheless, some approaches are currently being investigated.163 One such approach is exon skipping, because the exon–intron structure of lamin A enables the removal of some entire exons within the coiled-coil domain without disrupting the heptad structure of the coiled coil.164
Another promising approach is the targeting of signaling pathways affected by the nucleoskeleton. Inhibitors of ERK and MAPK have yielded positive effects on preventing cardiomyopathy in mouse models expressing lamin A mutations causative of the disease;145,165 also MAPK inhibition in an EDMD mouse model improved the muscular phenotype.166
In the case of HGPS, due to the accumulation of the farnesylated prelamin A, the farnesyl transferase was targeted. However, while this yielded considerable improvements in the tissue culture aspects of nuclear morphology and chromosome organization,55 there were only minor benefits in aspects such as the overall body weight in the patients.167 Potential therapies for HGPS are now targeting its links to genome organization/stability, as chemical inhibition of NAT10, a lysine acetyletransferase, was also found to restore normal nuclear morphology in HGPS cultured cells.168
To the extent that tissue-specific NETs may contribute to particular nucleo/cytoskeletal structures, these also could be targeted. As lamin A and core LINC components are universally expressed, targeting these tissue-specific partners should have less damaging off-target effects. However, as these partners are only beginning to be identified, such therapies are not likely to be realized in the near future.
Wider considerations and future directions
The observation that multiple components that interact in the nucleoskeleton and cytoplasmic filament systems can all yield variants of the same disease is a powerful argument for the idea of a tensegrity-type network connectedness in the function of the nuclear envelope and nucleoskeleton. While the idea that the genome could itself be a component of the overall cellular tensegrity system may be viewed by some as radical, it nonetheless comports with much of the existing data on the nucleoskeleton. This idea of the mechanical connectedness of these systems has recently gained traction with recent findings that lamin A levels scale with tissue elasticity. Low lamin A levels better supported differentiation of fat, while higher lamin levels improved differentiation of the much stiffer bone tissue.169 Matrix stiffness directly influenced lamin A levels, suggesting that its expression is increased to compensate/normalize the overall force applied on the nucleus in these tissues. Thus, the mechanics of the nucleo–cytoskeleton nexus are important for many aspects of nuclear function in both normal human health and disease.
Acknowledgments
Work in the Schirmer lab is supported by Wellcome Trust Senior Research Fellowship 095209 and Centre Grant 092076. PLT is supported by an MRC Studentship. AM is supported by a Principal’s Scholarships from the University of Edinburgh.
Disclosure
The authors declare no conflicts of interest in this work.
References
Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology. 4th ed. New York: WH Freeman; 2000. | |
Sperry AO. The dynamic cytoskeleton of the developing male germ cell. Biol Cell. 2012;104(5):297–305. | |
Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A. 1986;83(17):6450–6454. | |
Krohne G, Debus E, Osborn M, Weber K, Franke WW. A monoclonal antibody against nuclear lamina proteins reveals cell type-specificity in Xenopus laevis. Exp Cell Res. 1984;150(1):47–59. | |
Zackroff RV, Goldman AE, Jones JC, Steinert PM, Goldman RD. Isolation and characterization of keratin-like proteins from cultured cells with fibroblastic morphology. J Cell Biol. 1984;98(4):1231–1237. | |
Gerace L, Comeau C, Benson M. Organization and modulation of nuclear lamina structure. J Cell Sci Suppl. 1984;1:137–160. | |
Aebi U, Cohn J, Buhle L, Gerace L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323(6088):560–564. | |
Crisp M, Liu Q, Roux K, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172(1):41–53. | |
Dreger M, Bengtsson L, Schoneberg T, Otto H, Hucho F. Nuclear envelope proteomics: novel integral membrane proteins of the inner nuclear membrane. Proc Natl Acad Sci U S A. 2001;98(21):11943–11948. | |
Schirmer EC, Florens L, Guan TL, Yates JR, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301(5638):1380–1382. | |
Korfali N, Wilkie GS, Swanson SK, et al. The leukocyte nuclear envelope proteome varies with cell activation and contains novel transmembrane proteins that affect genome architecture. Mol Cell Proteomics. 2010;9(12):2571–2585. | |
Wilkie GS, Korfali N, Swanson SK, et al. Several novel nuclear envelope transmembrane proteins identified in skeletal muscle have cytoskeletal associations. Mol Cell Proteomics. 2011;10(1):M110.003129. | |
Korfali N, Wilkie GS, Swanson SK, et al. The nuclear envelope proteome differs notably between tissues. Nucleus. 2012;3(6):552–564. | |
Dittmer TA, Sahni N, Kubben N, et al. Systematic identification of pathological lamin A interactors. Mol Biol Cell. 2014;25(9):1493–1510. | |
Zastrow MS, Vlcek S, Wilson KL. Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci. 2004;117(pt 7):979–987. | |
Zuleger N, Robson MI, Schirmer EC. The nuclear envelope as a chromatin organizer. Nucleus. 2011;2(5):339–349. | |
Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7(12):940–952. | |
Meinke P, Nguyen TD, Wehnert MS. The LINC complex and human disease. Biochem Soc Trans. 2011;39:1693–1697. | |
de Las Heras JI, Meinke P, Batrakou DG, et al. Tissue specificity in the nuclear envelope supports its functional complexity. Nucleus. 2013;4(6):460–477. | |
Wilkie GS, Schirmer EC. Guilt by association: the nuclear envelope proteome and disease. Mol Cell Proteomics. 2006;5(10):1865–1875. | |
Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem. 1994;63:345–382. | |
Strelkov SV, Herrmann H, Aebi U. Molecular architecture of intermediate filaments. Bioessays. 2003;25(3):243–251. | |
Vikstrom KL, Lim SS, Goldman RD, Borisy GG. Steady-state dynamics of intermediate filament networks. J Cell Biol. 1992;118(1):121–129. | |
Okabe S, Hirokawa N. Dynamics of the neuronal microtubules and intermediate filaments studied by laser fluorescence photobleach recovery method and caged fluorescence UV photoactivation method. Mol Biol Cell. 1992;3:A2–A2. | |
Janmey PA, Euteneuer U, Traub P, Schliwa M. Viscoelastic properties of vimentin compared with other filamentous biopolymer networks. J Cell Biol. 1991;113(1):155–160. | |
Shen X, Ranallo R, Choi E, Wu C. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol Cell. 2003;12(1):147–155. | |
Belin BJ, Mullins RD. What we talk about when we talk about nuclear actin. Nucleus. 2013;4(4):291–297. | |
Wada A, Fukuda M, Mishima M, Nishida E. Nuclear export of actin: a novel mechanism regulating the subcellular localization of a major cytoskeletal protein. EMBO J. 1998;17(6):1635–1641. | |
Gerace L, Burke B. Functional organization of the nuclear envelope. Annu Rev Cell Biol. 1988;4:335–374. | |
Peter M, Kitten GT, Lehner CF, et al. Cloning and sequencing of cDNA clones encoding chicken lamins A and B1 and comparison of the primary structures of vertebrate A- and B-type lamins. J Mol Biol. 1989;208(3):393–404. | |
Weber K, Plessmann U, Ulrich W. Cytoplasmic intermediate filament proteins of invertebrates are closer to nuclear lamins than are vertebrate intermediate filament proteins; sequence characterization of two muscle proteins of a nematode. EMBO J. 1989;8(11):3221–3227. | |
Loewinger L, McKeon F. Mutations in the nuclear lamin proteins resulting in their aberrant assembly in the cytoplasm. EMBO J. 1988;7(8):2301–2309. | |
Lazebnik YA, Takahashi A, Moir RD, et al. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci U S A. 1995;92(20):9042–9046. | |
Holtz D, Tanaka RA, Hartwig J, McKeon F. The CaaX motif of lamin A functions in conjunction with the nuclear localization signal to target assembly to the nuclear envelope. Cell. 1989;59(6):969–977. | |
Nigg EA, Kitten GT, Vorburger K. Targeting lamin proteins to the nuclear envelope: the role of CaaX box modifications. Biochem Soc Trans. 1992;20(2):500–504. | |
Mical TI, Monteiro MJ. The role of sequences unique to nuclear intermediate filaments in the targeting and assembly of human lamin B: evidence for lack of interaction of lamin B with its putative receptor. J Cell Sci. 1998;111:3471–3485. | |
Monteiro MJ, Hicks C, Gu L, Janicki S. Determinants for intracellular sorting of cytoplasmic and nuclear intermediate filaments. J Cell Biol. 1994;127(5):1327–1343. | |
Broers JL, de Leij L, Rot MK, et al. Expression of intermediate filament proteins in fetal and adult human lung tissues. Differentiation. 1989;40(2):119–128. | |
Jansen MP, Machiels BM, Hopman AH, et al. Comparison of A and B-type lamin expression in reactive lymph nodes and nodular sclerosing Hodgkin’s disease. Histopathology. 1997;31(4):304–312. | |
Broers JL, Ramaekers FC. Dynamics of nuclear lamina assembly and disassembly. Symp Soc Exp Biol. 2004;56:177–192. | |
Machiels BM, Zorenc AH, Endert JM, et al. An alternative splicing product of the lamin A/C gene lacks exon 10. J Biol Chem. 1996;271(16):9249–9253. | |
Alsheimer M, Benavente R. Change of karyoskeleton during mammalian spermatogenesis: expression pattern of nuclear lamin C2 and its regulation. Exp Cell Res. 1996;228(2):181–188. | |
Furukawa K, Hotta Y. cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J. 1993;12(1):97–106. | |
Letai A, Fuchs E. The importance of intramolecular ion pairing in intermediate filaments. Proc Natl Acad Sci U S A. 1995;92(1):92–96. | |
Schirmer EC, Gerace L. The stability of the nuclear lamina polymer changes with the composition of lamin subtypes according to their individual binding strengths. J Biol Chem. 2004;279(41):42811–42817. | |
Panorchan P, Wirtz D, Tseng Y. Structure-function relationship of biological gels revealed by multiple-particle tracking and differential interference contrast microscopy: the case of human lamin networks. Phys Rev E Stat Nonlin Soft Matter Phys. 2004;70(4 pt 1):27. | |
Lammerding J. Mechanics of the nucleus. Compr Physiol. 2011;1(2):783–807. | |
Isermann P, Lammerding J. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol. 2013;23(24):009. | |
von Moeller F, Barendziak T, Apte K, Goldberg MW, Stick R. Molecular characterization of Xenopus lamin LIV reveals differences in the lamin composition of sperms in amphibians and mammals. Nucleus. 2010;1(1):85–95. | |
Shimi T, Pfleghaar K, Kojima S, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22(24):3409–3421. | |
Delbarre E, Tramier M, Coppey-Moisan M, Gaillard C, Courvalin JC, Buendia B. The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum Mol Genet. 2006;15(7):1113–1122. | |
Schirmer EC, Guan T, Gerace L. Involvement of the lamin rod domain in heterotypic lamin interactions important for nuclear organization. J Cell Biol. 2001;153(3):479–489. | |
Lutz RJ, Trujillo MA, Denham KS, Wenger L, Sinensky M. Nucleoplasmic localization of prelamin A: implications for prenylation-dependent lamin A assembly into the nuclear lamina. Proc Natl Acad Sci U S A. 1992;89(7):3000–3004. | |
Firmbach-Kraft I, Stick R. The role of CaaX-dependent modifications in membrane association of Xenopus nuclear lamin B3 during meiosis and the fate of B3 in transfected mitotic cells. J Cell Biol. 1993;123(6 pt 2):1661–1670. | |
Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria – new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46(12):2531–2558. | |
Navarro CL, Esteves-Vieira V, Courrier S, et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur J Hum Genet. 2014;22(8):1002–1011. | |
Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A. 1997;94(3):849–854. | |
Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol. 1997;59:575–599. | |
Mejat A, Misteli T. LINC complexes in health and disease. Nucleus. 2010;1(1):40–52. | |
Padmakumar VC, Libotte T, Lu W, et al. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci. 2005;118(pt 15):3419–3430. | |
Stewart-Hutchinson PJ, Hale CM, Wirtz D, Hodzic D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp Cell Res. 2008;314(8):1892–1905. | |
Burke B, Roux KJ. Nuclei take a position: managing nuclear location. Dev Cell. 2009;17(5):587–597. | |
Padmakumar VC, Abraham S, Braune S, et al. Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp Cell Res. 2004;295(2):330–339. | |
Zhang Q, Ragnauth C, Greener MJ, Shanahan CM, Roberts RG. The nesprins are giant actin-binding proteins, orthologous to Drosophila melanogaster muscle protein MSP-300. Genomics. 2002;80(5):473–481. | |
Zhen YY, Libotte T, Munck M, Noegel AA, Korenbaum E. NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J Cell Sci. 2002;115(pt 15):3207–3222. | |
Wilhelmsen K, Litjens SH, Kuikman I, et al. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol. 2005;171(5):799–810. | |
Roux KJ, Crisp ML, Liu Q, et al. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc Natl Acad Sci U S A. 2009;106(7):2194–2199. | |
Morimoto A, Shibuya H, Zhu X, et al. A conserved KASH domain protein associates with telomeres, SUN1, and dynactin during mammalian meiosis. J Cell Biol. 2012;198(2):165–172. | |
Lu W, Schneider M, Neumann S, et al. Nesprin interchain associations control nuclear size. Cell Mol Life Sci. 2012;69(20):3493–3509. | |
Lombardi ML, Zwerger M, Lammerding J. Biophysical assays to probe the mechanical properties of the interphase cell nucleus: substrate strain application and microneedle manipulation. J Vis Exp. 2011;(55):ii:3087. | |
Khatau SB, Bloom RJ, Bajpai S, et al. The distinct roles of the nucleus and nucleus-cytoskeleton connections in three-dimensional cell migration. Sci Rep. 2012;2(488):3. | |
Sullivan T, Escalante-Alcalde D, Bhatt H, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147(5):913–920. | |
Lammerding J, Schulze PC, Takahashi T, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113(3):370–378. | |
Broers JL, Peeters EA, Kuijpers HJ, et al. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet. 2004;13(21):2567–2580. | |
Duong NT, Morris GE, Lam le T, et al. Nesprins: tissue-specific expression of epsilon and other short isoforms. PLoS One. 2014;9(4):e94380. | |
Mattout-Drubezki A, Gruenbaum Y. Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol Life Sci. 2003;60(10):2053–2063. | |
Gudise S, Figueroa RA, Lindberg R, Larsson V, Hallberg E. Samp1 is functionally associated with the LINC complex and A-type lamina networks. J Cell Sci. 2011;124(pt 12):2077–2085. | |
Borrego-Pinto J, Jegou T, Osorio DS, et al. Samp1 is a component of TAN lines and is required for nuclear movement. J Cell Sci. 2012;125(pt 5):1099–1105. | |
Buch C, Lindberg R, Figueroa R, Gudise S, Onischenko E, Hallberg E. An integral protein of the inner nuclear membrane localizes to the mitotic spindle in mammalian cells. J Cell Sci. 2009;122(pt 12):2100–2107. | |
Shoeman RL, Traub P. The in vitro DNA-binding properties of purified nuclear lamin proteins and vimentin. J Biol Chem. 1990;265(16):9055–9061. | |
Glass JR, Gerace L. Lamins A and C bind and assemble at the surface of mitotic chromosomes. J Cell Biol. 1990;111(3):1047–1057. | |
Taniura H, Glass C, Gerace L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J Cell Biol. 1995;131(1):33–44. | |
Goldberg M, Harel A, Brandeis M, et al. The tail domain of lamin Dm0 binds histones H2A and H2B. Proc Natl Acad Sci U S A. 1999;96(6):2852–2857. | |
Dechat T, Adam SA, Taimen P, Shimi T, Goldman RD. Nuclear lamins. Cold Spring Harb Perspect Biol. 2010;2(11):8. | |
Lund E, Oldenburg AR, Delbarre E, et al. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013;23(10):1580–1589. | |
Malhas A, Lee CF, Sanders R, Saunders NJ, Vaux DJ. Defects in lamin B1 expression or processing affect interphase chromosome position and gene expression. J Cell Biol. 2007;176(5):593–603. | |
Puckelwartz MJ, Depreux FF, McNally EM. Gene expression, chromosome position and lamin A/C mutations. Nucleus. 2011;2(3):162–167. | |
Polioudaki H, Kourmouli N, Drosou V, et al. Histones H3/H4 form a tight complex with the inner nuclear membrane protein LBR and heterochromatin protein 1. EMBO Rep. 2001;2(10):920–925. | |
Makatsori D, Kourmouli N, Polioudaki H, et al. The inner nuclear membrane protein lamin B receptor forms distinct microdomains and links epigenetically marked chromatin to the nuclear envelope. J Biol Chem. 2004;279(24):25567–25573. | |
Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271(25):14653–14656. | |
Nili E, Cojocaru GS, Kalma Y, et al. Nuclear membrane protein LAP2beta mediates transcriptional repression alone and together with its binding partner GCL (germ-cell-less). J Cell Sci. 2001;114(pt 18):3297–3307. | |
Somech R, Shaklai S, Geller O, et al. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci. 2005;118(pt 17):4017–4025. | |
Demmerle J, Koch AJ, Holaska JM. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem. 2012;287(26):22080–22088. | |
Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci. 2001;114(pt 24):4567–4573. | |
Haraguchi T, Holaska JM, Yamane M, et al. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery-Dreifuss muscular dystrophy. Eur J Biochem. 2004;271(5):1035–1045. | |
Holaska JM, Rais-Bahrami S, Wilson KL. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum Mol Genet. 2006;15(23):3459–3472. | |
Zuleger N, Boyle S, Kelly DA, et al. Specific nuclear envelope transmembrane proteins can promote the location of chromosomes to and from the nuclear periphery. Genome Biol. 2013;14(2):2013–2014. | |
Solovei I, Wang AS, Thanisch K, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152(3):584–598. | |
Zullo JM, Demarco IA, Piqué-Regi R, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149(7):1474–1487. | |
Kumaran RI, Muralikrishna B, Parnaik VK. Lamin A/C speckles mediate spatial organization of splicing factor compartments and RNA polymerase II transcription. J Cell Biol. 2002;159(5):783–793. | |
Moir RD, Spann TP, Herrmann H, Goldman RD. Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J Cell Biol. 2000;149(6):1179–1192. | |
Singh M, Hunt CR, Pandita RK, et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol Cell Biol. 2013;33(6):1210–1222. | |
Lei K, Zhu X, Xu R, et al. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr Biol. 2012;22(17):1609–1615. | |
Alberts BJA, Lewis J, et al. Molecular Biology of the Cell. 4th ed. New York: Garland Science; 2002. | |
Wang N, Tytell JD, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009;10(1):75–82. | |
Chambliss AB, Khatau SB, Erdenberger N, et al. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci Rep. 2013;3(1087):18. | |
Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013;497(7450):507–511. | |
Markiewicz E, Tilgner K, Barker N, et al. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25(14):3275–3285. | |
Pan D, Estévez-Salmerón LD, Stroschein SL, et al. The integral inner nuclear membrane protein MAN1 physically interacts with the R-Smad proteins to repress signaling by the transforming growth factor-{beta} superfamily of cytokines. J Biol Chem. 2005;280(16):15992–16001. | |
Osada S, Ohmori SY, Taira M. XMAN1, an inner nuclear membrane protein, antagonizes BMP signaling by interacting with Smad1 in Xenopus embryos. Development. 2003;130(9):1783–1794. | |
Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet. 2007;16(15):1884–1895. | |
Muchir A, Wu W, Worman HJ. Reduced expression of A-type lamins and emerin activates extracellular signal-regulated kinase in cultured cells. Biochim Biophys Acta. 2009;1:75–81. | |
Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226(2):316–325. | |
Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Lamins in disease: why do ubiquitously expressed nuclear envelope proteins give rise to tissue-specific disease phenotypes? J Cell Sci. 2001;114(pt 1):9–19. | |
Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma? Nat Cell Biol. 2004;6(11):1062–1067. | |
Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21(3):285–288. | |
Muchir A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9(9):1453–1459. | |
Quijano-Roy S, Mbieleu B, Bönnemann CG, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. 2008;64(2):177–186. | |
De Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70(3):726–736. | |
Navarro CL, De Sandre-Giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13(20):2493–2503. | |
Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9(1):109–112. | |
Shackleton S, Lloyd DJ, Jackson SN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24(2):153–156. | |
Emery AE, Dreifuss FE. Unusual type of benign x-linked muscular dystrophy. J Neurol Neurosurg Psychiatry. 1966;29(4):338–342. | |
Bacon PA, Smith B. Familial muscular dystrophy of late onset. J Neurol Neurosurg Psychiatry. 1971;34(1):93–97. | |
Levitas A, Muhammad E, Harel G, et al. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet. 2010;18(10):1160–1165. | |
Schirmer EC, Foisner R. Proteins that associate with lamins: many faces, many functions. Exp Cell Res. 2007;313(10):2167–2179. | |
Meinke P, Mattioli E, Haque F, et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 2014;10(9):e1004605. | |
Liang WC, Mitsuhashi H, Keduka E, et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol. 2011;69(6):1005–1013. | |
Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8(4):323–327. | |
Zhang Q, Bethmann C, Worth NF, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16(23):2816–2833. | |
Clements L, Manilal S, Love DR, Morris GE. Direct interaction between emerin and lamin A. Biochem Biophys Res Commun. 2000;267(3):709–714. | |
Wiesel N, Mattout A, Melcer S, et al. Laminopathic mutations interfere with the assembly, localization, and dynamics of nuclear lamins. Proc Natl Acad Sci U S A. 2008;105(1):180–185. | |
Melcon G, Kozlov S, Cutler DA, et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15(4):637–651. | |
Mattout A, Pike BL, Towbin BD, et al. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr Biol. 2011;21(19):1603–1614. | |
Li P, Meinke P, Huong Le TT, Wehnert M, Noegel AA. Contribution of SUN1 mutations to the pathomechanism in muscular dystrophies. Hum Mutat. 2014;35(4):452–461. | |
Taranum S, Vaylann E, Meinke P, et al. LINC complex alterations in DMD and EDMD/CMT fibroblasts. Eur J Cell Biol. 2012;91(8):614–628. | |
Muntoni F, Bonne G, Goldfarb LG, et al. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain. 2006;129(pt 5):1260–1268. | |
Gundesli H, Talim B, Korkusuz P, et al. Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophy. Am J Hum Genet. 2010;87(6):834–841. | |
Cetin N, Balci-Hayta B, Gundesli H, et al. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: distinct histopathological outcomes compared with desminopathies. J Med Genet. 2013;50(7):437–443. | |
Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71(3):492–500. | |
Hayashi YK, Chou FL, Engvall E, et al. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet. 1998;19(1):94–97. | |
Hoffmann K, Dreger CK, Olins AL, et al. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly). Nat Genet. 2002;31(4):410–414. | |
Olins AL, Hoang TV, Zwerger M, et al. The LINC-less granulocyte nucleus. Eur J Cell Biol. 2009;88(4):203–214. | |
Horn HF, Brownstein Z, Lenz DR, et al. The LINC complex is essential for hearing. J Clin Invest. 2013;123(2):740–750. | |
Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18(2):241–247. | |
Muchir A, Pavlidis P, Decostre V, et al. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117(5):1282–1293. | |
Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298. | |
De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):17. | |
Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12(16):1995–2001. | |
Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140(23):2603–2624. | |
Liu B, Wang J, Chan KM, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11(7):780–785. | |
Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany N Y). 2009;1(1):28–37. | |
Gonzalez-Suarez I, Redwood AB, Perkins SM, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009;28(16):2414–2427. | |
Puente XS, Quesada V, Osorio FG, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88(5):650–656. | |
Madej-Pilarczyk A, Kmieć T, Fidziańska A, et al. Progeria caused by a rare LMNA mutation p.S143F associated with mild myopathy and atrial fibrillation. Eur J Paediatr Neurol. 2008;12(5):427–430. | |
Starke S, Meinke P, Camozzi D, et al. Progeroid laminopathy with restrictive dermopathy-like features caused by an isodisomic LMNA mutation p.R435C. Aging. 2013;5(6):445–459. | |
Padiath QS, Saigoh K, Schiffmann R, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38(10):1114–1123. | |
Hegele RA, Cao H, Liu DM, et al. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79(2):383–389. | |
Kuzmina SN, Buldyaeva TV, Akopov SB, Zbarsky IB. Protein patterns of the nuclear matrix in differently proliferating and malignant cells. Mol Cell Biochem. 1984;58(1–2):183–186. | |
Broers JL, Ramaekers FC. The role of the nuclear lamina in cancer and apoptosis. Adv Exp Med Biol. 2014;773:27–48. | |
Neumann S, Noegel AA. Nesprins in cell stability and migration. Adv Exp Med Biol. 2014;773:491–504. | |
Hutchison CJ. Do lamins influence disease progression in cancer? Adv Exp Med Biol. 2014;773:593–604. | |
Daniele N, Richard I, Bartoli M. Ins and outs of therapy in limb girdle muscular dystrophies. Int J Biochem Cell Biol. 2007;39(9):1608–1624. | |
Scharner J, Gnocchi VF, Ellis JA, Zammit PS. Genotype-phenotype correlations in laminopathies: how does fate translate? Biochem Soc Trans. 2010;38(pt 1):257–262. | |
Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation. 2011;123(1):53–61. | |
Muchir A, Kim YJ, Reilly SA, Wu W, Choi JC, Worman HJ. Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet Muscle. 2013;3(1):17. | |
Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109(41):16666–16671. | |
Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014;344(6183):527–532. | |
Swift J, Ivanovska IL, Buxboim A, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104. | |
Raffaele Di Barletta M, Ricci E, Galluzzi G, et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2000;66(4):1407–1412. | |
Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341(23):1715–1724. | |
Renou L, Stora S, Yaou RB, et al. Heart-hand syndrome of Slovenian type: a new kind of laminopathy. J Med Genet. 2008;45(10):666–671. | |
Chen L, Lee L, Kudlow BA, et al. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003;362(9382):440–445. | |
Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71(2):426–431. | |
Waterham HR, Koster J, Mooyer P, et al. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet. 2003;72(4):1013–1017. | |
Gros-Louis F, Dupré N, Dion P, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39(1):80–85. | |
Hellemans J, Preobrazhenska O, Willaert A, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet. 2004;36(11):1213–1218. | |
Debeer P, Pykels E, Lammens J, Devriendt K, Fryns JP. Melorheostosis in a family with autosomal dominant osteopoikilosis: report of a third family. Am J Med Genet A. 2003;1(2):188–193. | |
Merner ND, Hodgkinson KA, Haywood AF, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82(4):809–821. | |
Kayman-Kurekci G, Talim B, Korkusuz P, et al. Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: a novel gene related to nuclear envelopathies. Neuromuscul Disord. 2014;24(7):624–633. | |
Taylor MR, Slavov D, Gajewski A, et al; Familial Cardiomyopathy Registry Research Group. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26(6):566–574. | |
Ichida F, Tsubata S, Bowles KR, et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001;103(9):1256–1263. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.