Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 12
NSAID associated bilateral renal infarctions: a case report
Authors Jeon Y, Lis JB, Chang WG
Received 12 April 2019
Accepted for publication 4 July 2019
Published 1 August 2019 Volume 2019:12 Pages 177—181
DOI https://doi.org/10.2147/IJNRD.S212010
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Yejoo Jeon,* Jonathan B Lis,* William G Chang
Yale School of Medicine, Department of Internal Medicine, Yale University, New Haven, CT, USA
*These authors contributed equally to this work
Abstract: Renal infarctions (RIs) are caused by interruptions in the renal arterial blood flow. RIs are generally considered to be rare, however we present the case of a 37 year old woman whose renal infarction was likely due to the vasoconstrictive effects of non-steroidal anti-inflammatory drugs. Although high-dose non-steroidal anti-inflammatory drugs (NSAIDs) are known to cause a decrease in renal perfusion, they have not been accepted as causative agents in renal infarction. Theoretically, patients in prostaglandin dependent states should be more vulnerable to renovascular vasoconstriction and resulting hypoperfusion in the presence of NSAIDs. Given the high prevalence of NSAID use, we suspect that this mechanism of renal injury may be more prevalent than previously thought.
Keywords: renovascular, acute kidney injury, vasoconstriction, prostaglandins
Introduction
Renal infarctions (RIs) are induced by disruption of arterial flow to the renal parenchyma that could lead to persistent kidney dysfunction or herald underlying cardiovascular or hematological pathology. RI is relatively rare, as an emergency department (ED) study reported RI in 0.004% of visits.1 In retrospective analyses, RI was defined as the result of an “acute disruption of renal blood flow in the renal arteries or their branches … ”, and diagnosed by radiological findings of wedge-shaped parenchymal deficits. From 438 patients diagnosed with RIs, 55.7% had a cardiogenic cause, 7.5% had renal artery injury, 6.6% were hypercoagulable, and 30.1% were idiopathic.2 We argue that NSAIDs should be added as a potential cause of RI.
NSAIDs are commonly used drugs that are also associated with a wide range of adverse effects.3 Pharmacologically, NSAIDs inhibit cyclooxygenases necessary for the synthesis of prostaglandins (PGs). Human and animal studies suggest that NSAIDs can induce renal vasoconstriction in clinical states where vasodilatory renal PGs maintain renal blood flow.4,5 However, the role for NSAIDs in RI has not been widely reported. We present a case of a patient with bilateral renal segmental hypoperfusion and acute kidney injury (AKI) in the setting of significant NSAID use.
Case presentation
A 37-year-old Hispanic woman with a history of cholestasis during pregnancy presented with acute on chronic abdominal pain. She had been previously evaluated for chronic right upper quadrant (RUQ) pain and chronically mildly elevated liver function tests (LFTs) but had no sonographic evidence of cholelithiasis or choledocolithiasis. She experienced an acute worsening of her right-sided abdominal pain, prompting a visit to the ED. She was given a diagnosis of pelvic congestion syndrome after prominent variceal veins were noted within the pelvis on transvaginal ultrasound. She was given 60 mg of intravenous ketorolac tromethamine and discharged with ibuprofen 600 mg every 6 hrs as needed. She took ibuprofen three times per day for six days, but developed epigastric pain that diminished her appetite. She decreased her ibuprofen intake to 2 doses per day with acetaminophen. She was taking no other medications and denied herbal supplements. Her right-sided abdominal pain worsened and she developed new left-sided abdominal pain, prompting a second ED visit 7 days after the first.
She denied urinary symptoms, fevers, joint pains, rashes, history of miscarriages, or smoking history. Her blood pressure was 141/90, heart rate 76, and temperature 99.6°F. She had right costovertebral angle tenderness, diffuse abdominal tenderness (mostly RUQ) with voluntary guarding, but no rebound tenderness. ED lab work included urinalysis (UA), LFTs, basic metabolic panel, and complete blood count (CBC). UA was normal (specific gravity 1.003, negative protein, blood, leukocytes, and nitrite). LFTs showed stably elevated alkaline phosphatase (126 U/L; normal 9–122 U/L) and alanine aminotransferase (42 U/L; normal 6–34 U/L). Her serum creatinine (Cr) was elevated compared to her baseline (1.73 mg/dL from 0.61 mg/dL). CBC was normal. CT abdominal pelvis with IV contrast (Figure 1) revealed multiple segmental wedge shaped areas of non-enhancement suggesting pyelonephritis or infarction. Patient was admitted for further assessment and monitoring. A kidney ultrasound with Doppler flows revealed patent renal arteries but bilaterally decreased vascular flow in the upper and lower poles of kidneys corresponding with areas of CT hypoperfusion (Figure 2 and Table 1). Urine culture and examination of urine sediment by microscopy were unremarkable, suggesting neither infection nor tubular necrosis. Hepatitis panel was negative and gallbladder ultrasound was normal. Telemetry and echocardiogram were normal. C-reactive protein (CRP) and the erythrocyte sedimentation rate (ESR) were elevated at 16.1mg/L (normal <3.0 mg/L) and 22 mm/hr (normal 0–20 mm/hr) respectively. An extensive rheumatologic lab work up was non-revealing. Lactate dehydrogenase (LDH) was normal at 219 U/L (normal 122–241 U/L).
 |
Figure 1 CT Scan of bilateral renal infarctions. CT scan of abdomen and pelvis with intravenous contrast (80cc of Omnipaque 350) showing wedge-shaped infarcts (arrows) of the bilateral kidneys. |
 |
Figure 2 Kidney ultrasounds with doppler flow on admission and on follow-up. |
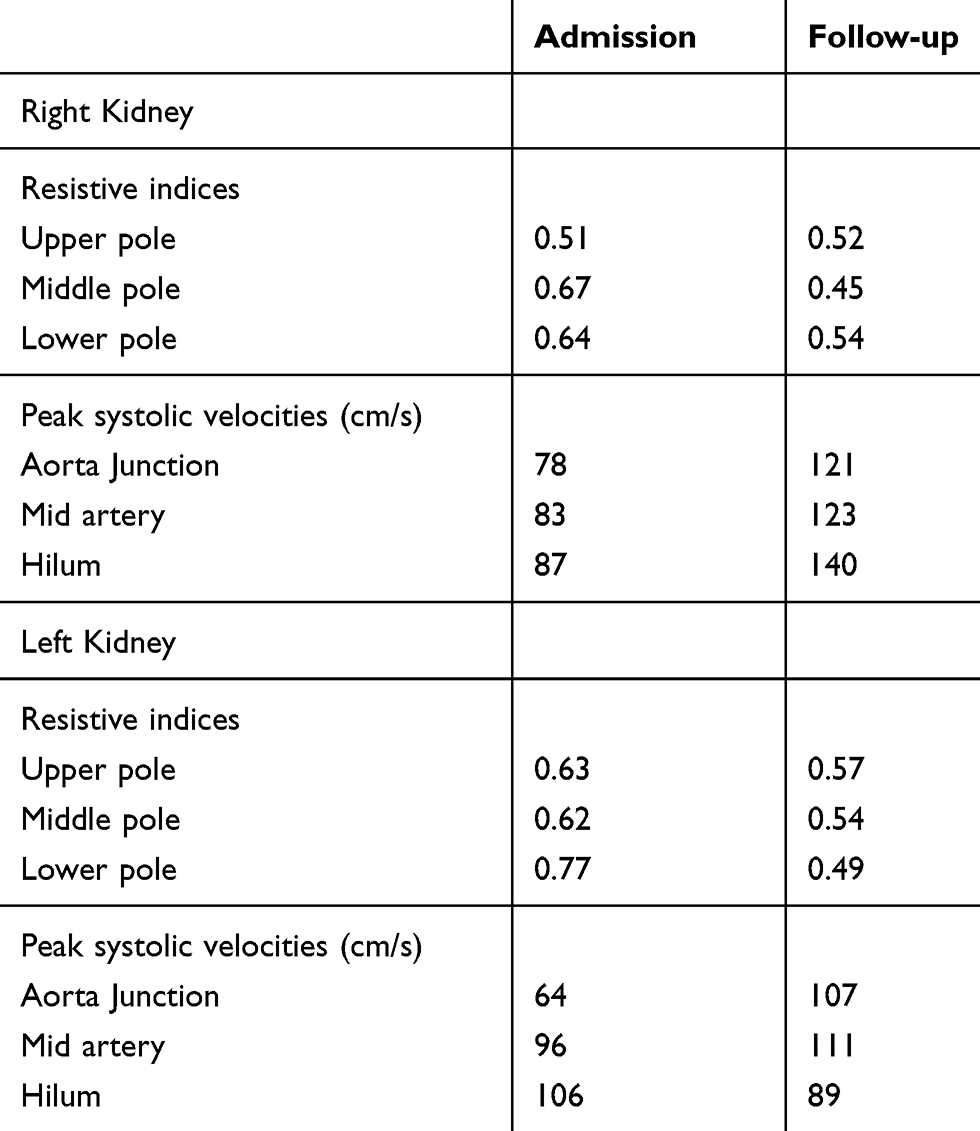 |
Table 1 Renal ultrasound measurements |
The patient remained clinically stable. She received 2 liters of normal saline in the ED. Cr started to downtrend by the next day and patient’s pain improved (Figure 3). The patient was not anti-coagulated and was discharged 3 days after admission. Follow-up 14 days after discharge showed normalization of Cr, and a renal ultrasound with improved vascularity (Figure 2 and Table 1). Patient has been advised to avoid future NSAID use.
 |
Figure 3 Graph of serum Cr including baseline, admission, hospital, and follow-up. |
Discussion
This clinical case raises interesting questions about RI. Radiologically, pyelonephritis could present similarly. However, the patient showed no signs of infection, and this alternative diagnosis was dismissed early in her hospitalization. Like most RI patients, our patient had abdominal/flank pain, but our patient was significantly younger than the 60-year-old mean age reported. In the largest study of RI patients, only 20.9% had AKI, and laboratory abnormalities typically consisted of elevated LDH and CRP.2 Although our patient had an elevated CRP, her LDH was normal. In our opinion, LDH is an important test in the evaluation of RI. LDH, which is present within all cells, is used as a marker for cell death. The lack of increased LDH and the lack of any signs of renal tubular damage in the urine sediment suggested that there was minimal renal damage associated with the hypoperfusion seen by CT. In addition, her Cr rapidly improved with some volume resuscitation and holding of NSAIDs. Workup failed to identify any cardiogenic, autoimmune, renovascular, or hypercoaguability etiology. It is our opinion that her prominent radiological findings were due to a vasoconstrictive effect of her NSAID use prior to admission. This is supported by the rapid improvement in her Cr after NSAIDs were discontinued. We propose that NSAIDs should be recognized as a distinct non-thromboembolic cause of RI.
There has been one similar case reported in which a previously healthy Korean man presented with hypertension, bilateral flank pain, AKI, and CT showing bilateral wedge shaped hypoattenuation days after taking high dose NSAIDs.6 His work-up was also negative for other etiologies. He was treated conservatively, and his kidney function returned to baseline. Follow-up CT demonstrated improved perfusion. It was concluded that his clinical syndrome was due to NSAID related renal ischemia.
Transient renal hypoperfusion has been described in other clinical scenarios as well. Acute renal failure with loin pain after anaerobic exercise (ALPE) has been reported.7,8 CT in these cases demonstrate bilateral patchy wedge shaped hypo-attenuation with delayed persistence of enhancement in these same areas from the gradual resolution of vasoconstriction.7,8 It has been suggested that vasospasm of intrarenal vessels is secondary to non-myoglobin nephrotoxins or reactive oxygen species secondary to muscle injury, leading to ischemia, acute tubular necrosis, and loin pain.7,8 Our patient denied any strenuous exercise prior to admission. Another syndrome of AKI related to NSAID intake is “syndrome of flank pain and acute renal failure after binge drinking and NSAIDs”.9,10 This syndrome has been reported in healthy patients who present with severe back pain and AKI after heavy ethanol consumption and NSAID intake. These patients saw resolution of pain and renal dysfunction with conservative therapy and intravenous fluids. Pathology was attributed to ethanol induced volume depletion creating a PG dependent state and NSAIDs induced loss of renal perfusion. Our patient denied significant alcohol intake.
NSAIDs have long been implicated in several renal pathologies. NSAID related renal dysfunction may be seen in 1–5% of all NSAID users, which given its common use, represents a significant clinical problem.11 NSAIDs have been implicated in fluid and electrolyte imbalances, acute and chronic kidney injury, nephrotic syndrome, interstitial nephritis, and renal papillary necrosis.11 NSAIDs reduce PG synthesis by inhibiting cyclooxygenase 1 and 2. PGs are synthesized locally in the kidney by the glomerulus and medullary interstitial cells.11,12 PGI2, PGE2, and PGD2 mediate renal perfusion by decreasing vascular resistance.11 Interestingly, inhibition of PG synthesis has little impact on healthy individuals.12 However in PG dependent states, a patient may become vulnerable to NSAID related renal injury.5 In states of elevated adrenergic and renin-angiotensin-aldosterone stimulation, such as in hypovolemia, low cardiac output states, cirrhosis, or nephrotic syndrome, there is increased renal vasoconstriction which must be modulated by PG driven vasodilation to sustain normal renal blood flow.5 In these scenarios, blocking PG could lead to unopposed vasoconstriction and kidney ischemia. We suspect our patient was in a PG dependent state and developed NSAID induced renal vasoconstriction. She reported decreased oral intake of food due to epigastric pain that began after taking initiation of NSAIDs. This may have led to mild volume depletion. Alternatively, she may have underlying liver disease manifested as chronically elevated LFTs.
An interesting clinical question is whether such patients should be anticoagulated. In a case series, 81% of patients with RI were anticoagulated with heparin.2 Our patient and the case previously reported did not receive anticoagulation when NSAID induced vasoconstriction was suspected. Although anticoagulation would not benefit transient vasoconstriction, it would be reasonable to begin the hypercoaguability work-up and initiate anticoagulation while other etiologies for RI were investigated.
Clearly, more clinical experience is needed to make informed decisions. Given how commonly NSAIDs are used, we suspect that the prevalence of NSAID induced RI is more than we previously appreciated. We therefore invite readers to share their cases of suspected NSAID induced RI (http://bit.ly/NephNSAID) so that we may assess them and report the results in a future report. As part of this investigation, we will evaluate whether normal LDH in the setting of radiological signs of RI is a distinguishing characteristic of NSAID induced RI.
Ethics
Written informed consent has been provided by the patient to have case details and accompanying images published. Further institutional approval is not required to publish de-identified case information.
Acknowledgments
We would like to thank the Yale Radiology & Biomedical Imaging Department and the Yale Center for Clinical Investigation for useful discussions.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Huang CC, Lo HC, Huang HH, et al. ED presentations of acute renal infarction. Am J Emerg Med. 2007;25(2):164–169. doi:10.1016/j.ajem.2006.06.010
2. Oh YK, Yang CW, Kim YL, et al. Clinical characteristics and outcomes of renal infarction. Am J Kidney Dis. 2016;67(2):243–250. doi:10.1053/j.ajkd.2015.09.019
3. Conaghan PG. A turbulent decade for NSAIDs: update on current concepts of classification, epidemiology, comparative efficacy, and toxicity. Rheumatol Int. 2012;32(6):1491–1502. doi:10.1007/s00296-011-2263-6
4. Dunn MJ. Nonsteroidal antiinflammatory drugs and renal function. Annu Rev Med. 1984;35:411–428. doi:10.1146/annurev.me.35.020184.002211
5. Zambraski EJ. The effects of nonsteroidal anti-inflammatory drugs on renal function: experimental studies in animals. Semin Nephrol. 1995;15(3):205–213.
6. Yoon SH, Kim YL, Park SH, Kim CD, Choi JY, Yun SR. Renal infarction after NSAID treatment. Korean J Med. 2012;67:618–622. doi:10.3904/kjm.2012.82.5.618
7. Ishikawa I. Acute renal failure with severe loin pain and patchy renal ischemia after anaerobic exercise in patients with or without renal hypouricemia. Nephron. 2002;91(4):559–570. doi:10.1159/000065013
8. Maekawa M, Imaizumi T, Yamakawa T, Ito Y. Acute renal failure with severe loin pain and patchy renal vasoconstriction in a patient without hypouricemia, provoked by epileptic seizure. Intern Med. 2017;56(15):2001–2005. doi:10.2169/internalmedicine.56.8328
9. Calviño J, Bravo J, Millán B, Gonzalez-Tabares L. Flank pain and acute renal failure after binge drinking: a growing concern? Ren Fail. 2013;35(3):421–424. doi:10.3109/0886022X.2013.764253
10. Johnson GR, Wen SF. Syndrome of flank pain and acute renal failure after binge drinking and nonsteroidal anti-inflammatory drug ingestion. J Am Soc Nephrol. 1995;5(9):1647–1652.
11. Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med. 1999;106(5B):13S–24S. doi:10.1016/s0002-9343(99)00113-8
12. Clive DM, Stoff JS. Renal syndromes associated with nonsteroidal anti-inflammatory drugs. N Engl J Med. 1984;310(9):563–572. doi:10.1056/NEJM198403013100905
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.