Back to Journals » International Journal of Nanomedicine » Volume 11
Novel walnut peptide–selenium hybrids with enhanced anticancer synergism: facile synthesis and mechanistic investigation of anticancer activity
Authors Liao W ![]() , Zhang R, Dong C, Yu Z, Ren J
, Zhang R, Dong C, Yu Z, Ren J
Received 12 July 2015
Accepted for publication 5 October 2015
Published 11 April 2016 Volume 2016:11 Pages 1305—1321
DOI https://doi.org/10.2147/IJN.S92257
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Carlos Rinaldi
Wenzhen Liao,1 Rong Zhang,1 Chenbo Dong,2 Zhiqiang Yu,3 Jiaoyan Ren1
1College of Light Industry and Food Sciences, South China University of Technology, Guangzhou, Guangdong, People’s Republic of China; 2Civil and Environmental Engineering, Rice University, Houston, TX, USA; 3School of Pharmaceutical Science, Guangdong Provincial Key Laboratory of New Drug Screening, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China
Abstract: This contribution reports a facile synthesis of degreased walnut peptides (WP1)-functionalized selenium nanoparticles (SeNPs) hybrids with enhanced anticancer activity and a detailed mechanistic evaluation of its superior anticancer activity. Structural and chemical characterizations proved that SeNPs are effectively capped with WP1 via physical absorption, resulting in a stable hybrid structure with an average diameter of 89.22 nm. A panel of selected human cancer cell lines demonstrated high susceptibility toward WP1-SeNPs and displayed significantly reduced proliferative behavior. The as-synthesized WP1-SeNPs exhibited excellent selectivity between cancer cells and normal cells. The targeted induction of apoptosis in human breast adenocarcinoma cells (MCF-7) was confirmed by the accumulation of arrested S-phase cells, nuclear condensation, and DNA breakage. Careful investigations revealed that an extrinsic apoptotic pathway can be attributed to the cell apoptosis and the same was confirmed by activation of the Fas-associated with death domain protein and caspases 3, 8, and 9. In addition, it was also understood that intrinsic apoptotic pathways including reactive oxygen species generation, as well as the reduction in mitochondrial membrane potential, are also involved in the WP1-SeNP-induced apoptosis. This suggested the involvement of multiple apoptosis pathways in the anticancer activity. Our results indicated that WP1-SeNP hybrids with Se core encapsulated in a WP1 shell could be a highly effective method to achieve anticancer synergism. Moreover, the great potential exhibited by WP1-SeNPs could make them an ideal candidate as a chemotherapeutic agent for human cancers, especially for breast cancer.
Keywords: selenium nanoparticles, walnut peptides, human cancer cell lines, antiproliferative activity, apoptosis
Introduction
Cancer, one of the most significant health concerns,1 and its related global apprehension are on the increase despite the appreciable recent advances in cancer therapeutics. This is primarily due to the inadequacies of the state-of-the-art remedial procedures. For example, chemotherapy drugs tend to show non-specific cytotoxicity and will kill not only cancerous tumor cells, but also damage normal tissues.2,3 In addition, the non-specific distribution, poor water solubility, instability, and the development of multidrug resistance over prolonged usage limit the bioavailability and clinical applications of many anticancer drugs.4,5
Nanotechnology and nanomaterials owing to their unique optical, electronic, magnetic, photoresponsive, and structural properties have demonstrated immense promise for employing them in tumor-targeted therapy, bio-imaging, and diagnosis.6–10 It is well documented that nanoparticle-based drugs have many advantages including targeted action, reduction of toxicity, and resistance to degradation.11 However, precisely controlling the grain/particle size, modulating the surface properties, increasing the stability, and neutralizing the toxicity/hazards are the major challenges in the synthesis as well as in application of drug-loaded nanomaterials for therapeutic applications.12,13
Selenium (Se) is an essential trace element with unique physiological and pharmacological properties.14,15 Many studies have demonstrated that supplemental Se uptake can reduce several disease risks including cancers, muscle disorders, diabetes mellitus, and cardiovascular diseases.16,17 Moreover, Se plays a critical role in the activation of seleno-enzymes as well.18 However, it is demonstrated that the chemical form of seleno-compounds can have determinant effects on its biological activities. For example, though elemental Se is insoluble and biologically inert, an intimate correlation was observed between the particle size of elemental Se powder and the bioavailability, where particles with 5–200 nm diameter exhibited high efficiency as free radical scavengers compared with bigger particulates.19
Due to the abundant bioavailability and miniscule toxicity compared with organic or inorganic seleno-compounds, Se nanoparticles (SeNPs) are subjected to increasing research.20–24 However, before employing them as an anticancer agent, its cellular uptake, stability, biosafety, and the mechanism of its curative actions need to be better understood.
Several methods including laser ablation, γ-irradiation, lithography, electro-deposition, pyrolysis, and chemical vapor deposition have been applied to synthesize SeNPs. However, the comparatively expensive, complex, and hazardous nature of these techniques limit their practical utility. Biosynthesis of SeNPs using biomolecules as templates to stabilize nanoparticles, on the other hand, might have an advantage over other physical and chemical methods, as it is inexpensive, eco-friendly, and can be done at room temperature without advanced tools or special conditions. A large volume of reports utilizing various biomolecules, including polysaccharides, sucrose, glucose, chitosan, and polysaccharide-protein complexes, illustrate the possibility as well as the promise of biomolecule-regulated synthesis of SeNPs.25 Nevertheless, the use of bioactive peptides for the size regulation, stabilization, and functionalization-based surface modification during the synthesis of SeNPs has never been explored in great detail. Bioactive peptides are defined as specific protein fragments that have beneficial effects on living beings. It is established that bioactive peptides can exhibit positive effects such as immunomodulatory, antioxidative, antihypertensive, antimicrobial, and antithrombotic activities when used at optimal dosage.26 In the present study, we use a bioactive peptide extracted from degreased walnut meal for the first time for the facile synthesis of size-selected SeNPs. Here, the peptide efficiently caps the in-situ generated SeNPs and efficiently controls their size. The structure and chemical encapsulation in the walnut peptides-selenium nanoparticles (WP1-SeNPs) was characterized by Fourier transform infrared spectroscopy (FTIR), energy dispersive X-ray (EDX) analysis, Zetasizer particle size analysis, and scanning electron microscopy (SEM). Furthermore, the in vitro antiproliferative activity of WP1-SeNPs on selected human cancer cells was investigated. The underlying mechanism of the apoptosis-inducing activities of WP1-SeNPs was also studied in human breast carcinoma MCF-7 cells.
Materials and methods
Materials
Degreased walnut meal was purchased from Huizhi Resource Co., Ltd. (Yunnan, People’s Republic of China). Trypsin (46 million U/g protein 150 USP [United States Pharmacopoeia] units/mg) was obtained from Shisheng Technology Co., Ltd. (Hangzhou, People’s Republic of China) and Fisher Scientific Co. (Fair Lawn, NJ, USA). Sodium selenite (Na2SeO3), bicinchoninic acid (BCA) kits, 5,5′,6,6′-tetraethyl-imidacarbocyanine iodide (JC-1), 4′,6-diamidino-2-phenyindole (DAPI), 2′,7′-dichlorofluorescein diacetate (DCFH-DA), diethyl pyrocarbonate (DEPC), and thiazolyl blue tetrazolium bromide (MTT) were all of analytical grade and purchased from Sigma Company (St Louis, MO, USA). Five cell lines, including the human breast cancer MCF-7 cell line, the human pulmonary carcinoma A549 cell line, the human prostate carcinoma PC3 cell line, and the human cervical carcinoma HeLa cell line, were supplied from American Type Culture Collection (ATCC, Rockville, MD, USA). Another two cell lines, the human normal liver HL-7702 (L02) cell line and the human gastric carcinoma SGC-7901 cell line, were obtained from the Medical College of Sun Yat-Sen University (Guangzhou, Guangdong, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium (DMEM), Hank’s balanced salt solution, heat-inactivated fetal bovine serum (FBS), phosphate-buffered saline (PBS, pH 7.4), penicillin, and streptomycin were obtained from Gibco Life Technologies (Grand Island, NY, USA). DNA ladder kits were purchased from the Omega Bio-Tek Company (Norcross, GA, USA). The model substrates for caspase-3 and -7 (Ac-DEVD-AMC), caspase-8 (Ac-IETD-AFC), and caspase-9 (Ac-LEHD-AFC) were obtained from Cal Biochem (La Jolla, CA, USA). The ultrapure water was obtained from a Milli-Q water purification system (Millipore, Bedford, MA, USA). All the other chemical reagents used in this study were of analytical grade. The study was approved by the institutional review board (IRB) of Centers for Disease Control and Prevention (CDCP) in Guangdong, China. All the experiments related to human cell lines were granted ethical approval.
Preparation of WP1
Degreased walnut meal was mixed with distilled water (1:7, w/v) and homogenized at 4,000 rpm using a IKA T25 homogenizer system (IKA Works, Guangzhou, Guangdong, People’s Republic of China). The mixture was preheated to 55°C and trypsin was added. The enzyme–to-substrate ratio was fixed at 1:100 (w/v). The temperature was maintained at 55°C for 22 hours to facilitate hydrolysis. Subsequently, the mixture was heated to boil and was boiled for 10 minutes to inactivate the protease. The hydrolysate was centrifuged using a GL-21M refrigerated high-speed centrifuge (Xiang Yi Instrument Co., Ltd., Changsha, Hunan, People’s Republic of China) at 8,000 rpm for 20 minutes at room temperature. The supernatant was collected and fractionated through ultrafiltration membranes in a bioreactor system (Vivaflow 200, Vivascience, Sartorius, Goettingen, Germany) with molecular weight cutoffs of 3, 5, and 10 kDa, respectively. The WP1 with molecular weight distributions of >10 kDa were classified as WP1. The samples were then lyophilized (R2L-100KPS, Kyowa Vacuum Engineering, Tokyo, Japan) for further use.
Preparation of WP1-SeNPs
The lyophilized WP1 was dissolved in distilled water at a concentration of 3 mg/mL, and mixed with freshly prepared sodium selenite solution (4 mM/L) under constant stirring. The ascorbic acid solution (1 mM/L) was added to achieve a ratio of WP1:ascorbic acid:sodium selenite solutions 4:4:1 (v/v). The mixture was placed in an electronic temperature-controlled shaker (Model THZ-82A, Yuejin Medical Instrument Co. Ltd., Shanghai, People’s Republic of China) and shaken (150 rpm) in the absence of light at 45°C for 24 hours. The mixture was then dialyzed with 500 Da molecular weight cutoff dialysis bag as per the manufacturer’s specifications (Spectrum Medical Industries, Inc., Los Angeles, CA, USA) against deionized water until no Se component was detected in inductively coupled plasma atomic emission spectroscopy analysis. The WP1-SeNPs solution was lyophilized for further use.
Characterization of WP1-SeNPs
Scanning electron microscope
The surface morphology of the synthesized WP1-SeNPs was examined using SEM (Model 1530VP, LEO, Oberkochen, Germany). A drop of as-prepared WP1-SeNPs solutions (3 mg/mL) was placed on a glass slide and then spin coated to obtain a thin film. The glass slides were mounted on a stub with double stick adhesive tape and sputter-coated with a thin layer of gold-palladium. The metal-coated samples were characterized using the SEM at an accelerating potential of 15 kV.
EDX system
To determine the elemental composition of the WP1-SeNPs, an energy dispersive EX-250 system (HORIBA Ltd., Kyoto, Japan) X-ray spectrometer (EDX, Voyager ш NORAN instruments, Inc., Middletown WI) equipped with a cold field emission system was utilized at 5 kV with an analysis time of 70 live seconds.
Size distribution and zeta potential analysis
Zetasizer Nano ZS particle analyzer (Malvern Instruments Ltd., Malvern, UK) was employed for the analysis of the size distribution and zeta potential of the WP1-SeNPs. A series of WP1-SeNP solutions with different concentrations of WP1 range from 0 to 5 mg/mL were prepared, and the aqueous suspensions were put into the glass cuvette, and the average diameter and zeta potential of the particles were measured.
FTIR assay
The infrared spectra of WP1-SeNPs were analyzed using an FTIR spectrophotometer (Bruker, Ettlingen, Germany) equipped with OPUS 3.1 software. The WP1-SeNP samples (1 mg) were blended with KBr powder and then pressed into the pellets and scanned in the range of 4,000–400 cm−1.
Cell culture
The selected human cell lines were maintained in DMEM supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and 10% of FBS at 37°C in a 5% CO2 atmosphere in a humidified incubator (100% relative humidity).
Cell viability determination using the MTT assay
Cell viability was analyzed using the MTT assay based on the ability of live cells to convert the yellow MTT to a purple formazan dye.27 Briefly, both normal cells and cancer cells were placed into 96-well culture plates at a density of 5×103 cells/well at 37°C. After 24 hours, different concentrations of 200 μL WP1 or WP1-SeNPs were added and were incubated for 48 hours, each concentration group was tested in triplicate. Then, 20 μL of MTT solution (5 mg/mL) was added to each well and incubated for another 4 hours at 37°C. Afterward, the medium containing MTT was removed from each well, and it was replaced with 100 μL of DMSO to dissolve the dark blue formazan formed in surviving cells with intact mitochondria. Later, the absorbance at 570 nm was read using a microplate spectrophotometer (Versamax, Molecular Devices, Sunnyvale, CA, USA). Results were expressed as the percentage of MTT reduction assuming that the absorbance of the negative control (cells group without treatment by WP1-SeNPs) was 100%.
Cell morphology assay by polarized optical microscope
The morphological changes of MCF-7 cells treated with WP1-SeNPs were characterized using polarized optical microscope (POM). The cells were placed on 6-well culture plates with a density of 5×103 cells/well at 37°C. After 24 hours, cells were treated with WP1-SeNPs at different concentrations (125, 250, and 500 μg/mL) and was kept under the same condition for another 48 hours. Later, the cell morphology was captured using a POM (YYP-230, Shanghai Optical Instrument Factory, Shanghai, People’s Republic of China). The untreated cells were used as controls in the study.
Cell morphology assay by SEM
Cell imaging was carried out using a SEM (Hitachi TM3000, Hitachi Ltd., Tokyo, Japan). A coverslip was placed in each well of six-well culture plates. MCF-7 cells with a density of 5×103 cells/well were seeded on the coverslips and incubated at 37°C for 24 hours to create cell monolayer. After incubation, the cells were treated with WP1-SeNPs for different time intervals (12, 36, and 48 hours). Then, the treated cell monolayer was washed twice with PBS (pH 7.4) and fixed with 2.5% glutaraldehyde. After that, the cell film was placed on the specimen holder, and the images were acquired at an acceleration voltage of 15 kV.
Nuclear morphology analysis by Hoechst 33258
MCF-7 cells at a density of 5×103 cells/well were seeded in a six-well plate and treated with different concentrations (125, 250, and 500 μg/mL) of WP1-SeNPs for 48 hours. After treatment, the cells were washed with PBS for three times and detached by introducing trypsin into a microcentrifuge tube. Then, the cells were fixed with 1 mL of methanol (4%) at 4°C for 10 minutes. After fixation, the cells were collected via centrifugation at 2,000 rpm for 3 minutes followed by resuspension in 50 μL of PBS. Then, the cell suspension was transferred onto a glass slide. Next, the samples were allowed to dry naturally, and the dry cells were stained with 100 μL of Hoechst 33258 for 10 minutes at room temperature. The cells were then twice washed using PBS and examined under a fluorescence microscope (Nikon Eclipse 80i, Nikon, Tokyo, Japan) with the excitation and emission wavelengths of 350 nm and 460 nm, respectively.
Apoptosis measurement and cell-cycle analysis by flow cytometry
The apoptosis of MCF-7 was determined by flow cytometry through an Annexin V-FITC/PI apoptosis detection kit (Bipec Biopharma Corporation, Cambridge, MA, USA). Briefly, WP1-SeNPs treated or untreated cells were harvested by trypsinization and washed three times with PBS. Then, the treated cells were suspended with binding buffer in the kit and divided into two replicates. Annexin V and FITC were incubated in one of the replicates of the cell suspension in the dark for 15 minutes at 37°C. The other replicate of the cell suspension was used as a control. Then, the stained cells were analyzed using flow cytometry after the addition of propidium iodide (PI) to each sample. The samples were excited at wavelength 488 nm. The signal from FITC was recorded at 516 nm, whereas the signal from PI was recorded at 560 nm. The data were collected and analyzed with the Cell Quest Research Software. Cell-cycle distribution was analyzed using flow cytometry as well. For this, MCF-7 cells were treated with different concentrations (125, 250, and 500 μg/mL) of WP1-SeNPs solution for 48 hours and then harvested and washed with PBS. The treated cells were stained with PI-containing RNase in the dark followed by fixation with 70% alcohol solution at −20°C overnight. Labeled cells were washed with PBS (pH 7.4) twice for flow-cytometry analysis. For each sample, 10,000 events were recorded. Cell-cycle distribution was determined by the amount of PI intercalated to DNA and analyzed by Cell Quest Research Software.
Determination of mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential was detected using a lipophilic cationic dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide). For the assessment, MCF-7 cells were treated with WP1-SeNPs for 48 hours in six-well plates. A sample without WP1-SeNPs treatment was maintained as a control. The cells were harvested by trypsinization and washed with PBS (pH 7.4), then incubated with 10 mg/mL of JC-1 at 37°C for 10 minutes in the dark. The stained cells were washed and suspended in PBS (pH 7.4) before subjecting to flow-cytometric analysis. The excitation wavelength was 488 nm, with the emissions wavelength at 525 nm for green fluorescence detection and at 590 nm for red fluorescence. The fluorescence intensity was detected using the fluoroskan ascent microplate fluorometer (Thermo Electron Corporation, Vantaa, Finland) at Ex/Em =590/525 nm.
Measurement of intracellular reactive oxygen species production
The effects of WP1-SeNPs on the intracellular reactive oxygen species (ROS) production in MCF-7 cells were evaluated using a fluorescence probe DCFH-DA. Briefly, MCF-7 cells were incubated with 10 μM of DCFH-DA at 37°C, in a non-CO2 incubator for 30 minutes. Then, the cells were washed three times with PBS (pH 7.4) to remove extra DCFH-DA. Then, the cells were incubated with 500 μg/mL of WP1-SeNPs at 37°C for 45 minutes. Cells were collected in the suspension, and the intracellular ROS level was measured using a fluorescence microscope (Nikon Eclipse 80i, Nikon). The fluorescence intensity of MCF-7 cells was analyzed using the fluoroskan ascent microplate fluorometer (Thermo Electron Corporation) at Ex/Em =488/525 nm.
DNA fragmentation assay
Initially, MCF-7 cells were treated with specific concentration of WP1-SeNPs at 37°C for 48 hours. Afterward, the treated cells were washed with PBS and lysed in 500 μL lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM EDTA, and 0.5% Triton X-100) for 20 minutes. The lysates were dissolved in RNase (0.03 mg/mL) and proteinase K (0.25 mg/mL), and then incubated at 37°C for 30 minutes followed by further incubation at 55°C for another 1 hour. The nuclear DNA was then extracted with phenol-chloroform, isolated in isopropyl alcohol, and stored at −20°C for 15 minutes. The samples were resuspended in DEPC water followed by washing with 70% ethanol. The DNA was precipitated and electrophoresed in 1.5% agarose gels for fragmentation analysis. Later, the gels were stained with ethidium bromide (1 mg/mL) and were visualized by UV transilluminator (Tanon 1600; Tanon, Shanghai, People’s Republic of China).
Caspase activity assay
Harvested cell pellets were suspended in cell lysis buffer and incubated on ice for 1 hour. Subsequently, it was centrifugated at 11,000× g for 30 minutes at 4°C. The supernatants were collected and immediately measured for protein concentration by a BCA kit. For caspase activity assay, the cell lysates were placed in 96-well plates and then the specific caspase substrates (Ac-DEVD-AMC for caspase-3/7, Ac-IETD-AMC for caspase-8, and Ac-LEHD-AMC for caspase-9) were added. Plates were then incubated at 37°C for 1 hour, and the caspase activity was determined by fluorescence intensity using the fluoroskan ascent microplate fluorometer (Thermo Electron Corporation) with the excitation and emission wavelengths at 380 and 440 nm, respectively.
Western blot analysis
MCF-7 cells treated with WP1-SeNPs for 48 hours were incubated with lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM EDTA, and 0.5% Triton X-100) (Beyotime Co., Shanghai, People’s Republic of China) to obtain total cellular proteins. The protein concentration was examined by BCA assay using a BCA kit. An equal amount of proteins was loaded to 12% tricine gels and electrophoresed. Then, it was transferred onto nitrocellulose membrane (Bio-Rad Laboratories Inc., Hercules, CA, USA) and blocked with 5% non-fat milk in Tris-buffered saline Tween 20 (TBST) buffer for 1 hour. After that, the membranes were incubated with primary antibodies at 1:1,000 dilution in 5% non-fat milk overnight at 4°C under continuous agitation. Then, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase at 1:2,000 dilution for 1 hour at room temperature, followed by three times washing with TBST. Protein bands were visible on X-ray film using enhanced chemiluminescence detection. The β-actin was used to confirm the comparable amount of proteins in each lane.
Statistical analysis
All experiments were carried out at least in triplicate and results were expressed as a mean ± standard deviation. Statistical analysis was done using SPSS 19.0 software (IBM Corporation, Armonk, NY, USA). The difference between the control and the experimental groups was analyzed using a two-tailed Student’s t-test. Differences with P<0.05 (*) were considered as statistically significant. The difference between three or more groups was analyzed by one-way analysis of variance multiple comparisons.
Results and discussion
Preparation and characterization of WP1-SeNPs
In the present work, we developed a facile method to synthesize WP1-functionalized SeNPs via anchoring WP1 to the surface of the SeNPs. Here, size controllable and highly stable WP1-SeNPs hybrids encapsulated in a coherent peptides capping layer were generated by a redox system in the presence of sodium selenite and ascorbic acid (Figure 1). When SeO32− and ascorbic acid reacted in the presence of peptides in aqueous solution, Se was reduced from SeO32− through oxidation-reduction reaction and soon polymerized/encapsulated with WP1 to form WP1-SeNPs. The strong conjugation and anchoring of peptide molecules on the surface of SeNPs facilitated the size-controlled formation of WP1-SeNPs. At optimum concentration of the WP1, the as-synthesized WP1-SeNPs were highly stable, homogeneous, and spherical in nature. Without surface embellishment by peptide molecules, the prepared SeNPs exhibited serious aggregation due to their high surface energy, leading to noticeable precipitation. Due to the effective interaction between SeNPs and WP1, the size of the SeNPs can be conveniently modulated by regulating the concentrations of peptide introduced into the reaction ensemble and optimal concentration leading to the smallest nanoparticle was identified. Zetasizer Nano ZS particle analyzer (Malvern Instruments Ltd.) was employed to investigate the size distribution of WP1-SeNPs and their stability under different conditions. As illustrated in Figure 2A, the study using a series of concentration-gradient peptide solutions revealed that the average particle size of WP1-SeNPs decreased with the increase in the peptide concentration. The minimum particle size (~89 nm) was achieved when the peptide solution concentration reached 3 mg/mL and the average particle size did not show any appreciable change (P>0.05) upon further increase in concentration of the peptide solution to 5 mg/mL. Also, as can be observed from Figure 2B and C, in the absence of the capping peptides, the size distribution of SeNPs was highly inhomogeneous and some large particles with sizes in the range of 1,000 nm or even 10,000 nm are present (Figure 2B). However, at the optimal concentration of peptide solutions (3 mg/mL), a highly homogeneous nanoparticle within 100 nm (average ~89 nm) and narrow size distribution was obtained (Figure 2C). Hence, uncapped SeNPs in the aqueous environment were seriously agglomerated giving rise to micro-particles because of their high surface energy and lead to significant precipitation. However, the strong capping efficiency of WP1 stabilizes smaller nanoparticles with an average diameter of ~89 nm. These results proved that the presence of peptides is a key factor for modifying and controlling size distribution of SeNPs.
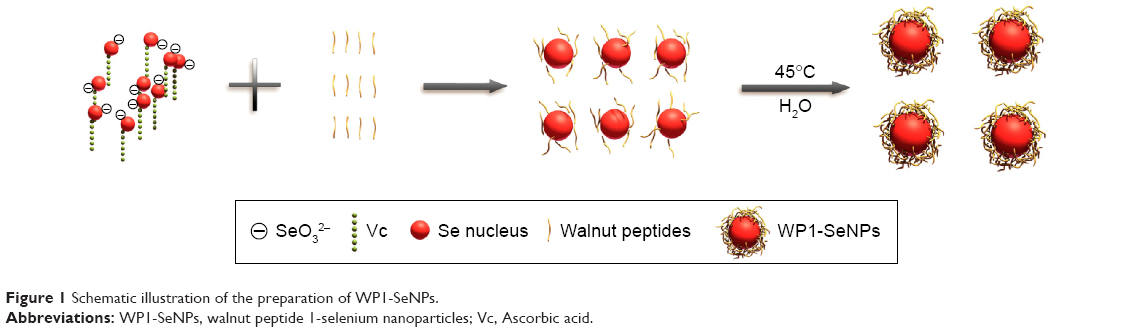 | Figure 1 Schematic illustration of the preparation of WP1-SeNPs. |
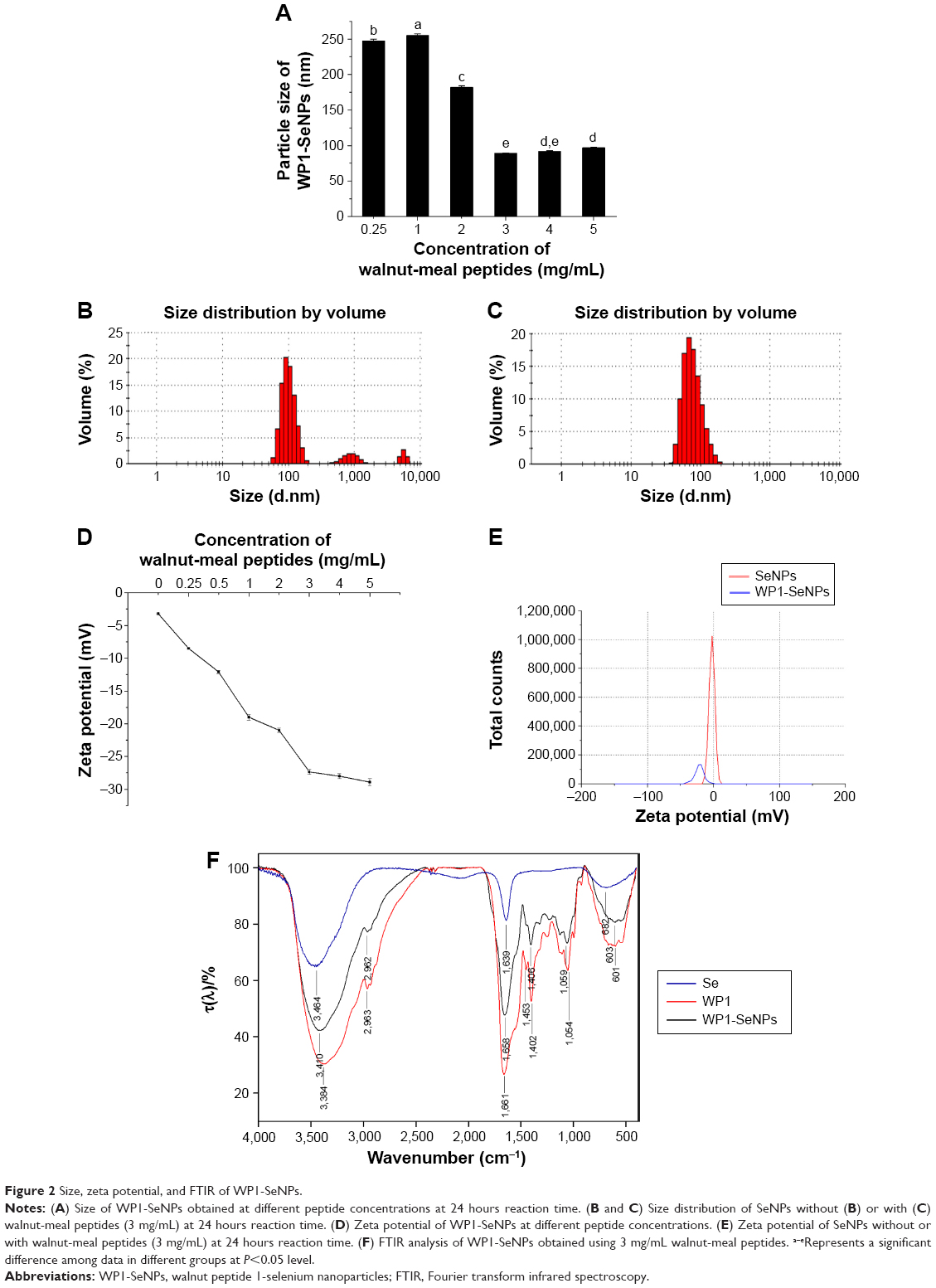 | Figure 2 Size, zeta potential, and FTIR of WP1-SeNPs. |
The morphology of the WP1-SeNPs was characterized using SEM. Figure 3 displays SEM image of SeNPs in the absence (A) and presence (B) of surface anchored WP1. It can be clearly observed that in the absence of peptides, SeNPs are seriously aggregated resulting in the formation of large particles chunks. However, the presence of peptide promoted the generation of homogeneous spherical NPs with high dispersibility as well as stability. Additionally, the SEM results clearly revealed that the average particles size of WP1-SeNPs was remarkably smaller compared with that of the SeNPs without WP1, which further confirmed that the size of SeNPs can be successfully regulated by WP1.
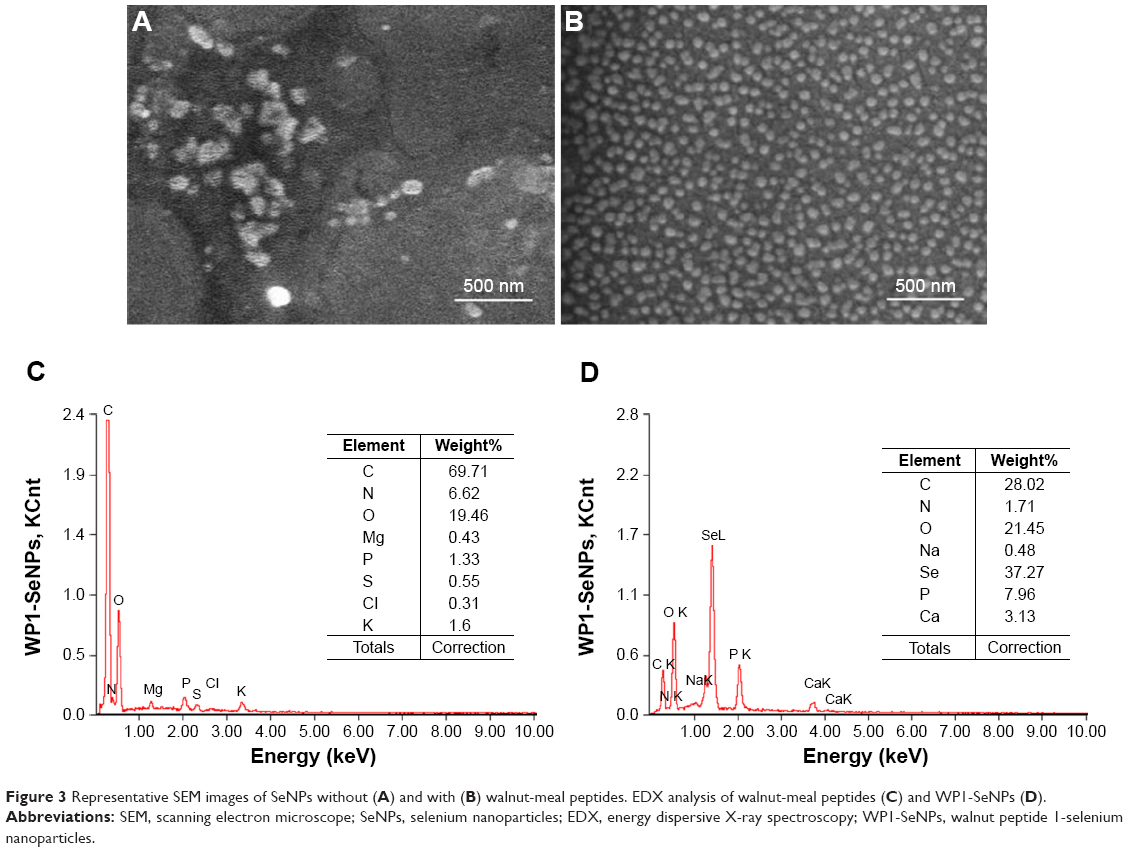 | Figure 3 Representative SEM images of SeNPs without (A) and with (B) walnut-meal peptides. EDX analysis of walnut-meal peptides (C) and WP1-SeNPs (D). |
The stability of nanoparticles in aqueous solutions is considered as a key factor for their potential industrial applications. Thus, zeta potential measurement was further performed to investigate the effects of incorporation of different concentrations of the peptide on the stability of WP1-SeNPs. It was found that zeta potential value increases (negative values) upon increasing the peptide concentration (Figure 2D). Similar to the size evolution study, the increase in concentration of WP1 above the optimal value (3 mg/mL) did not show any significant changes in zeta potential. At the optimal concentration, theWP1-SeNPs exhibited a zeta potential value of −27.35 mV, with the average diameter of 89.22 nm. As can be observed in Figure 2E, the zeta potential of bare SeNPs was −3.2 mV, which increased to −27.35 mV after surface decoration by peptide molecules with the concentration of 3 mg/mL, pointing to the increased surface charge and resultant increase in stability of WP1-SeNPs compared with bare SeNPs. Based on the average diameter and the stability of obtained nanoparticles, 3 mg/mL concentration of peptides as a capping agent was fixed as the optimal concentration and was applied in following experiments.
FTIR analysis (Equinox 55, Bruker, Ettlingen, Germany) was conducted to elucidate the mechanism or mode of chemical binding between the peptide molecules and the SeNPs in WP1-SeNPs. Compared with the FTIR spectrum of pristine WP1, no new absorbance peaks were observed in the WP1-SeNPs sample (Figure 2F), indicating that the surface anchoring does not involve any new covalent bond formation. However, considerable shifts toward higher wave number region were observed for the absorption peaks corresponding to the stretching vibrations of hydroxyl (3,384–3,410 cm−1) and C–H bonds (1,406–1,453 cm−1) in WP1-SeNPs sample compared with WP1, illustrating that Se was captured by peptide molecules that could be attributed to the physical absorption and the formation of Se–O and Se–N bonds of peptide.
To determine elemental compositions of WP1-SeNPs samples, EDX (EX-250 system, HORIBA Ltd.) was used. EDX investigation of WP1 alone revealed the presence of C (69.71%), O (19.46%), and N (6.62%), which makes up to 95.79% of its total elemental compositions (Figure 3C). Further, EDX analysis for WP1-SeNPs (Figure 3D) exhibited the strong characteristic signal of Se atom with element weight appeared to be 37.27%, together with C (28.02%), O (21.45%), and P (7.96%). These four main elements were approximately 94.70% of the total element weight in WP1-SeNPs. This result confirmed that Se was successfully conjugated with WP1.
Cell viability tests by MTT assay
The in vitro cytotoxicity effects of WP1 and WP1-SeNPs on HL-7702 (L02) cell, MCF-7 cell, SGC-7901 cell, A549 cell, PC3 cell, and HeLa cell were evaluated using MTT assay. As shown in Figure 4A, WP1 did not exhibit any obvious cytotoxicity on most of the tested cell lines within a concentration of 500 μg/mL. Moreover, WP1 demonstrated potential to promote the growth of L02, MCF-7, A549, and PC3 cells, which could be ascribed to the ability of small peptides to serve as nutritional resources for cell growth. As for MCF-7, there was no apparent dose-dependence between peptide concentration and cell survival rate because the results showed no significant difference. Only at 1,000 μg/mL, WP1 showed mildly toxicity to HeLa cells (P<0.05). Generally, it can be concluded that WP1 are not toxic to most of the cell lines and they might even serve as nutritional factors for cell growth, including cancer cells. However, once WP1 were conjugated with Se elements to form nanosized particles, the WP1-SeNPs displayed significant cytotoxicity on specific cancer cells. Figure 4B illustrates the significant dose-dependent inhibitory capacity of WP1-SeNPs on the growth of all cancer cells, among which MCF-7 was the most sensitive cell line. When incubated with 500 μg/mL WP1-SeNPs, the cell viability of MCF-7 cells was greatly reduced to 33.3%, which was almost three times lower extent of viable cells compared with treatment using pristine WP1 (107.2%). Here, the combination of peptide and Se element to form nanoparticles exhibited lowered cytotoxicity on the normal human cell line (human normal hepatocyte L02), but heightened cytotoxicity against specific cancer cell lines. Even at a relatively lower concentration (125 μg/mL), WP1-SeNPs were highly effective to decrease the cell viability for most tested cells lines. Especially, as the concentration was increased to 250 μg/mL, the survival rate of MCF-7 cells reduced more than half compared with that at 125 μg/mL. At a concentration of 500 μg/mL, MCF-7 cells reached the lowest survival rate (14.93%). Therefore, it was concluded that MCF-7 is the most sensitive cell line to WP1-SeNPs treatment, and thus it was selected as an ideal cell line for the following anticancer studies.
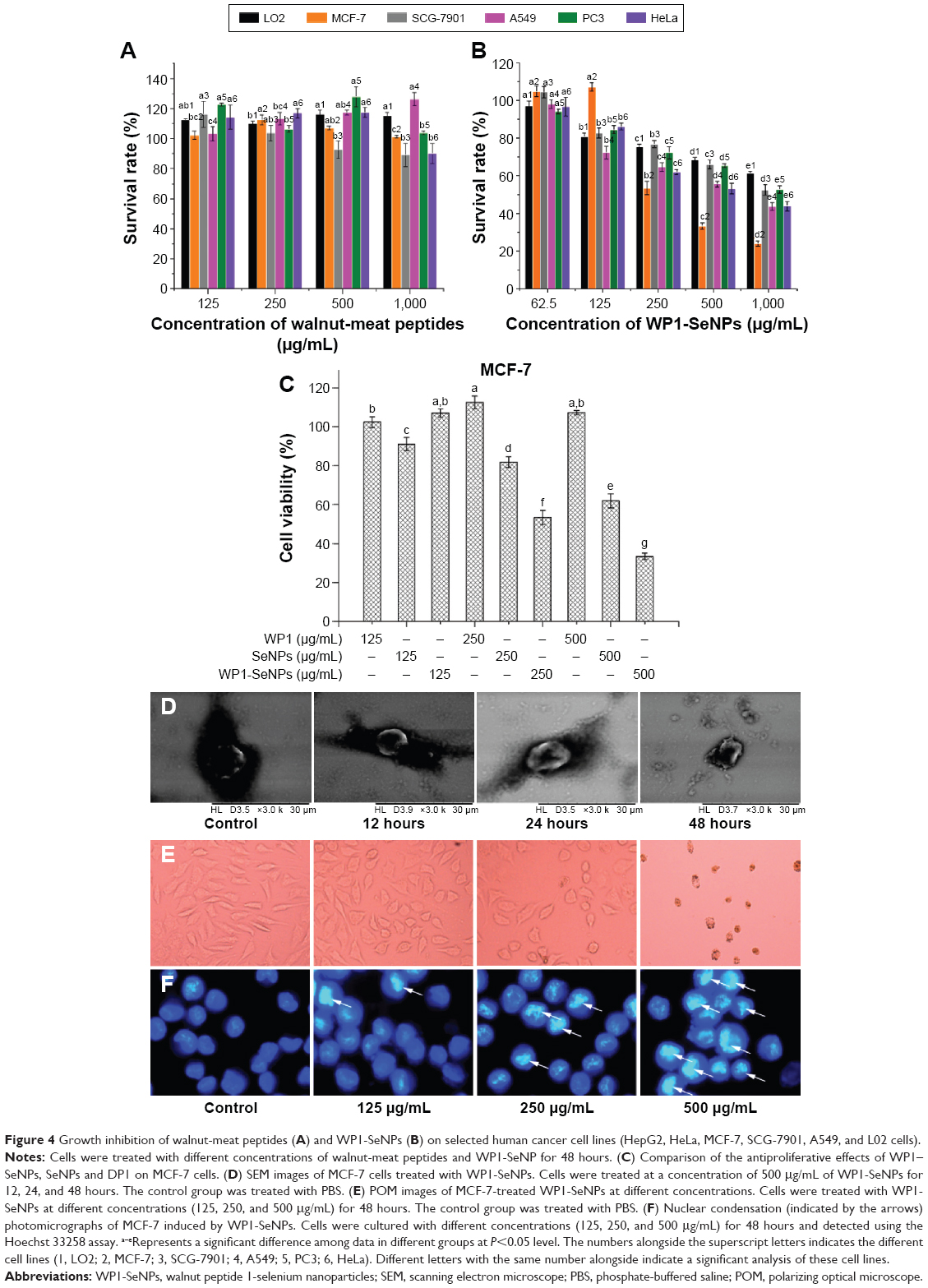 | Figure 4 Growth inhibition of walnut-meat peptides (A) and WP1-SeNPs (B) on selected human cancer cell lines (HepG2, HeLa, MCF-7, SCG-7901, A549, and L02 cells). |
The antiproliferative effects of WP1, SeNPs, and WP1-SeNPs against MCF-7 cells were examined to compare the effect of WP1 surface decoration on antiproliferative effect of SeNPs. As shown in Figure 4C, the bare SeNPs exhibited certain antiproliferative effect on MCF-7 cells. While decorated by WP1, the antiproliferative effect of WP1-SeNPs was significantly enhanced as compared with SeNPs. The main reason for this result might be that WP1-SeNPs possessed the better cellular uptake and stability than SeNPs. These results indicated that WP1 surface decoration is crucial to enhance the antiproliferative action of SeNPs on MCF-7 cells.
SEM observation
Apoptosis have been regarded as one of the most important mechanism behind the anticancer activity of Se. The significant biophysical characteristic of cell apoptosis is cellular morphology changes. Thus, we performed SEM imaging of WP1-SeNP-exposed MCF-7 cells to identify their morphological changes and develop a deep understanding of the WP1-SeNP-induced cell apoptosis and anticancer performance. Morphologic changes of MCF-7 cells treated with 500 μg/mL WP1-SeNPs for different time intervals (12, 24, and 48 hours, respectively) were investigated by SEM. As shown in Figure 4D, cancer cells without WP1-SeNPs treatment maintained its complete oblate structure and kept as regular shape even after several steps in fixation processing. After the first 12 hours of WP1-SeNPs treatment, the cell morphology did not change much, and after 24 hours, the cell borders started to change and demonstrated an obscure boundary. After 48 hours, the WP1-SeNP-treated cells displayed a rough and shrunken morphology with truncated lamellipodia, which indicated the apoptosis of MCF-7 cells. The results substantiated our proposition that WP1-SeNPs can exerted intense cytotoxicity on MCF-7 cells.
POM inspection
To further understand how the MCF-7 morphology changes when incubated with WP1-SeNPs, POM characterization was conducted. The POM images of MCF-7 cells are given in Figure 4E and F. It can be clearly observed that the MCF-7 cells exhibit a dose-dependent cytotoxicity when treated with WP1-SeNPs. We can clearly see that as the dose of WP1-SeNPs is increased, a large number of the cells transformed round shape and shrunken in size and the number of adhering cells evidently decreased. The results clearly indicated that the WP1-SeNPs could induce the morphologic changes of MCF-7 cells and inhibit the growth of the cancer cells.
Hoechst 33258 staining detection
To obtain further insights into the apoptotic mechanisms of WP1-SeNP-induced MCF-7 cells, WP1-SeNP-treated cells were stained with Hoechst 33258 and measured using fluorescence microscope. As demonstrated in Figure 4E, treatment of MCF-7 cells with WP1-SeNPs resulted in a dose-dependent increase of fluorescent intensity. It is reasonable to assume that WP1-SeNPs caused typical apoptotic changes in the nuclear morphology leading to the condensation of cell nuclei in MCF-7 cell. The apoptotic changes were the most obvious at a concentration of 500 μg/mL, which agree well with the POM results. At this concentration, most of the cell nuclei were fragmentized in spherical particles.
Determination of DNA fragmentation
To further investigate apoptosis-mediated cell death caused by WP1-SeNPs in the most susceptible MCF-7 cell lines, DNA fragmentation analysis was performed. The formation of “ladder” or oligo nucleosomal DNA fragments owing to the fragmentation of DNA is considered as a biochemical marker of late apoptosis.28 In this study, the induction of apoptosis by WP1-SeNPs was confirmed using the agarose gel electrophoresis of fragmented DNA. DNA extracted from MCF-7 cells treated with different concentration (125, 250, and 500 μg/mL) of WP1-SeNPs was analyzed. Figure 5 depicts the ladder patterns of discontinuous DNA fragments from the samples. It is worth noting that while single-stranded DNA was observed in the control group, incubation with WP1-SeNPs triggered the progressive, concentration-dependent accumulation of fragmented DNA. Due to apoptosis-related inter nucleosomal cleavage, the pattern appeared as a specific laddering distribution of DNA fragmentation.
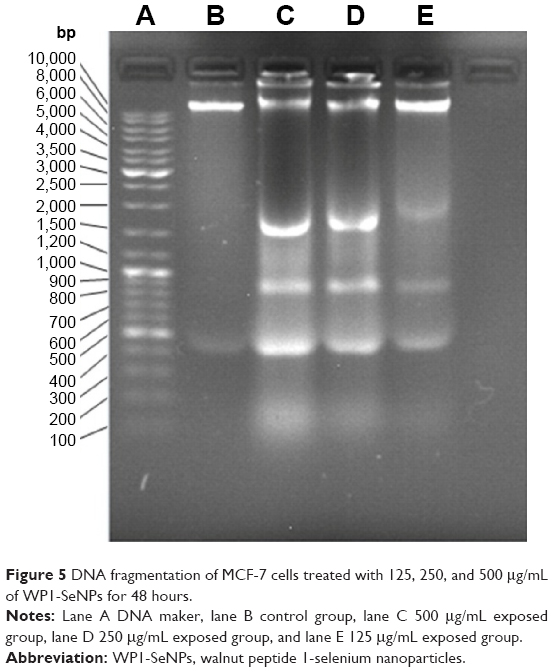 | Figure 5 DNA fragmentation of MCF-7 cells treated with 125, 250, and 500 μg/mL of WP1-SeNPs for 48 hours. |
Induction of apoptosis in MCF-7 by WP1-SeNPs
To assess the apoptosis period and apoptosis rates of MCF-7 cells induced by WP1-SeNPs, flow-cytometric analysis was carried out. Figure 6Ai–Aiv demonstrates the results of Annexin V-FITC/PI staining assay. The study revealed that early apoptosis occurred in a dose-dependent manner. MCF-7 cells respectively exposed to 125, 250, and 500 μg/mL of WP1-SeNPs showed a significant increase in early-stage apoptosis (12.07%, Figure 6Aii; 35.85%, Figure 6Aiii; 64.52%, Figure 6Aiv) while the ratio of control group was 1.30% (Figure 6Ai). Additionally, the ratio of late apoptosis and necrosis in the latter stages demonstrated that there was no distinct change in treated and untreated cells, indicating that WP1-SeNPs inhibited MCF-7 cell proliferation mainly in early-stage apoptosis, showing great potential of using WP1-SeNPs as anticancer drugs.
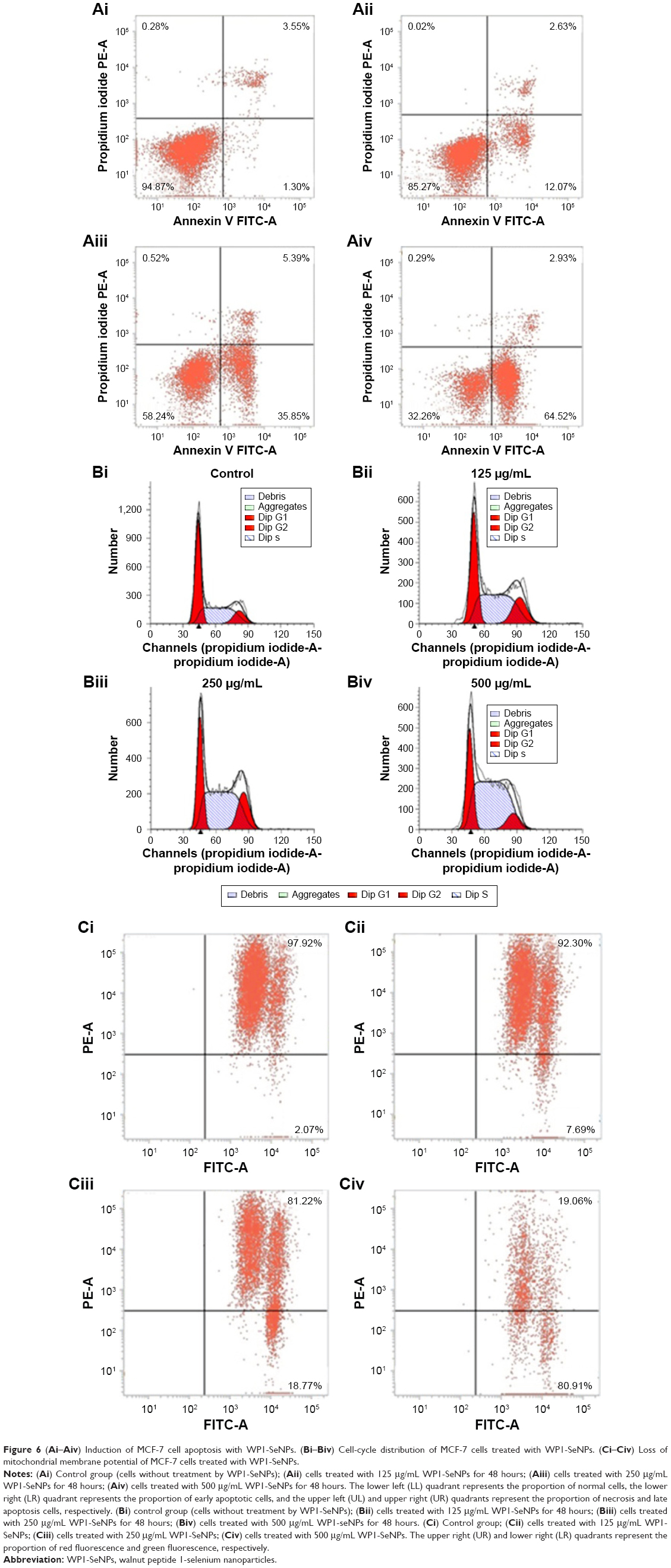 | Figure 6 (Ai–Aiv) Induction of MCF-7 cell apoptosis with WP1-SeNPs. (Bi–Biv) Cell-cycle distribution of MCF-7 cells treated with WP1-SeNPs. (Ci–Civ) Loss of mitochondrial membrane potential of MCF-7 cells treated with WP1-SeNPs. |
Effects of WP1-SeNPs on cell-cycle distribution
To comprehensively evaluate the effects of WP1-SeNPs on cell fate, the cell-cycle analysis was undertaken on cells treated with WP1-SeNPs. It should be noted that the inhibition of cancer cells proliferation can be caused by the induction of apoptosis, cell-cycle arrest, or a combination of these two processes.29 The mitosis and proliferation of cells get restrained if cells are arrested in the S-phase.30 To verify this hypothesis, PI flow-cytometric analysis was implemented to further identify the cell proliferation inhibition in MCF-7 cells treated with WP1-SeNPs. The DNA content and distribution histograms of MCF-7 cells exposed to WP1-SeNPs are displayed in Figure 6Bi–Biv and Table 1. The results suggested a dose-dependence on the proportion of S-phase arrest of MCF-7 cells. Specially, the S-phase arrest increased from 40.65% (control) to 65.66% (500 μg/mL) when treated with WP1-SeNPs. Besides, no significant change in G2/M phase was detected in the treated cells and the ratio of G0/G1-phase arrested cells was decreased in conjugation with the increase in S-phase arrested cells. Thus, it is reasonable to conclude that the inhibition of MCF-7 cells proliferation is mainly happening by S-phase cell arrest, confirming that WP1-SeNPs can restrain the proliferation of MCF-7 cells as well as induce the apoptosis of cancer cells.
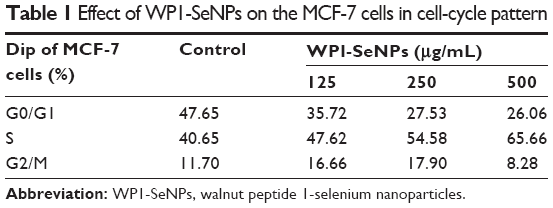 | Table 1 Effect of WP1-SeNPs on the MCF-7 cells in cell-cycle pattern |
WP1-SeNP-induced cell apoptosis via mitochondrial dysfunction
Cell apoptosis process induced by WP1-SeNPs was analyzed further by investigating the mitochondrial membrane potential of target cells. The change of mitochondrial membrane potential is an important indicator of activation of cell apoptosis.14,29 Thus, the mitochondrial membrane potentials of cells exposed to WP1-SeNPs were evaluated using flow cytometry via JC-1 staining. The loss or reduction of membrane potential in the WP1-SeNP-treated MCF-7 cells for 48 hours is manifested in Figure 6Ci–Civ. It revealed a dose-dependent increase in the reduction of mitochondrial membrane potential. It is evident that the proportion of the green fluorescence of cells treated with 250 μg/mL of WP1-SeNPs increased from 2.07% to 18.77% compared with the control group. An increase in green fluorescence (JC-1 monomer) is associated with the mitochondrial membrane depolarization, whereas the increase in red fluorescence (JC-1 aggregate) is related to the polarization of the mitochondrial membrane.31 The red/green fluorescence intensity ratio in the MCF-7 cells treated with different concentrations of WP1-SeNPs (125, 250, and 500 μg/mL) is displayed in Figure 7A. A clear concentration-dependent decrease in the ratio can be observed. Especially, the ratio decreased by 86%, 56%, and 35% after treatment with WP1-SeNPs with increasing concentration (from 0 to 500 μg/mL), respectively, indicating such WP1-SeNPs exposure could cause MCF-7 cell mitochondrial membrane potential depolarization and distinct mitochondrial damage. Also, as shown in Figure 7C, JC-1 staining data demonstrated that WP1-SeNP-treated (125, 250, and 500 μg/mL) cells exhibited a decline in red fluorescence and a dose-dependent enhancement of green fluorescence compared with the control group. These results indicated that treatment of WP1-SeNPs would induce the loss or reduction of mitochondrial membrane potential. Thus, it was concluded that WP1-SeNP-induced MCF-7 cells apoptosis has a contribution from mitochondrial dysfunction as well.
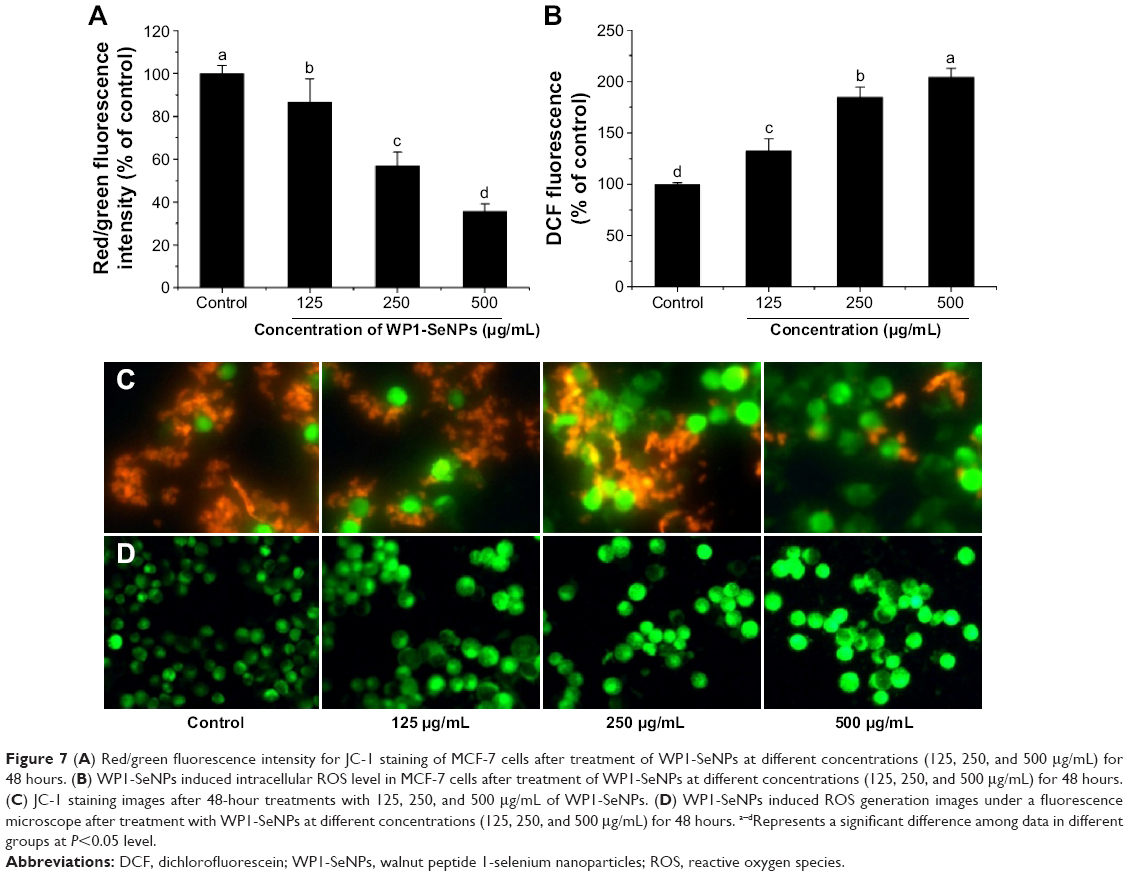 | Figure 7 (A) Red/green fluorescence intensity for JC-1 staining of MCF-7 cells after treatment of WP1-SeNPs at different concentrations (125, 250, and 500 μg/mL) for 48 hours. (B) WP1-SeNPs induced intracellular ROS level in MCF-7 cells after treatment of WP1-SeNPs at different concentrations (125, 250, and 500 μg/mL) for 48 hours. (C) JC-1 staining images after 48-hour treatments with 125, 250, and 500 μg/mL of WP1-SeNPs. (D) WP1-SeNPs induced ROS generation images under a fluorescence microscope after treatment with WP1-SeNPs at different concentrations (125, 250, and 500 μg/mL) for 48 hours. a–dRepresents a significant difference among data in different groups at P<0.05 level. |
WP1-SeNPs induced mitochondrial dysfunction via ROS overproduction
Mitochondrial respiratory chain is regard as the main source of intracellular ROS (ie, hydrogen peroxide and superoxide), which are produced during oxidative metabolism. The production of intracellular ROS can be correlated with mitochondrial dysfunction and the reaction of apoptosis.32–35 A number of apoptotic stimuli induce apoptosis through ROS generation. Previous reports indicated that several metal oxide nanoparticles had been demonstrated to trigger the generation of excess ROS leading to modification and damage of cellular DNA, proteins, and lipids, which can cause cell apoptosis.35–37 Thus, it is necessary to evaluate ROS generation of target cell line incubated with WP1-SeNPs via measuring dichlorofluorescein (DCF) fluorescence intensity. As demonstrated in Figure 7D, MCF-7 cells cultured in the presence of WP1-SeNPs (125, 250, and 500 μg/mL) exhibit a concentration-dependent increase in oxidized DCF fluorescence, suggesting an overproduction of intracellular ROS production. It should be noted that no significant change was observed in the fluorescence intensity of the control group. The DCF fluorescence intensity, indicative of degree of intracellular ROS, increased by 132%, 184%, and 204% after exposure to 125, 250, and 500 μg/mL of WP1-SeNPs, respectively (Figure 7B, P<0.05). These results indicated that ROS overproduction is also involved in theWP1-SeNP-induced apoptosis of MCF-7 cells.
WP1-SeNPs induced caspase-dependent apoptosis in MCF-7 cells
Caspases, a family of cysteine proteases, serve as vital components in regulating apoptosis. To determine whether caspase activation is involved in the WP1-SeNP-induced cell apoptosis, the activities of initiator caspases (caspase-8 and caspase-9) and effector caspases-7 were examined using fluorometric assays. The analysis (Figure 8A) revealed that there is a dose-dependent triggering of two caspase initiators, caspase-8 (Fas/tumor necrosis factor (TNF)-mediated) and caspase-9 (mitochondrial-mediated), as well as a caspase effector (caspase-3), WP1-SeNPs incubation with MCF-7 cells. These results confirm that both intrinsic and extrinsic pathways contribute to WP1-SeNP-induced apoptosis. Furthermore, the activity of caspase-8 was significantly higher than those of caspase-3 and caspase-9 when treated with 500 μg/mL WP1-SeNPs, suggesting that extrinsic pathway contributed more to the MCF-7 cells apoptosis than the intrinsic one.
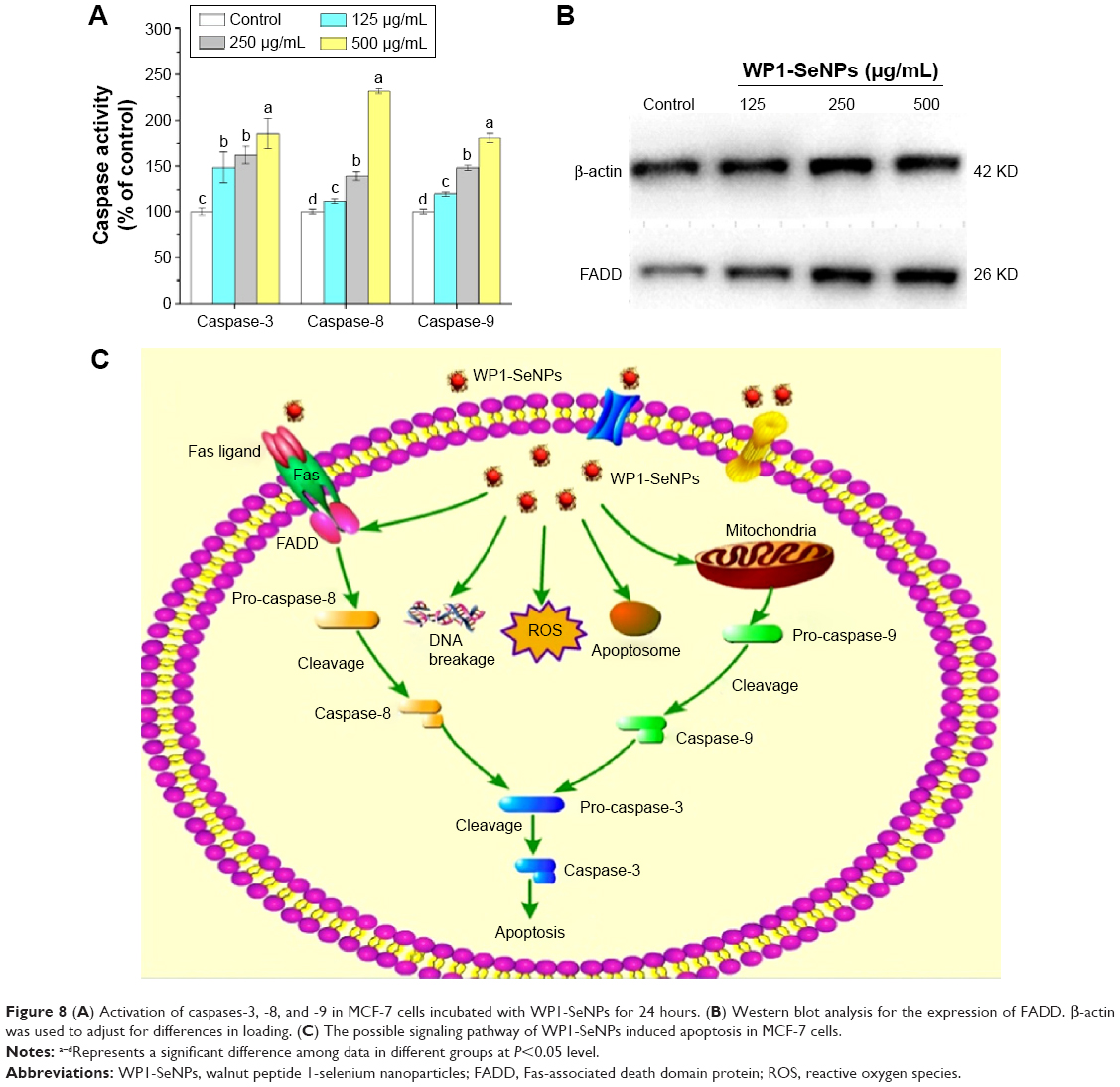 | Figure 8 (A) Activation of caspases-3, -8, and -9 in MCF-7 cells incubated with WP1-SeNPs for 24 hours. (B) Western blot analysis for the expression of FADD. β-actin was used to adjust for differences in loading. (C) The possible signaling pathway of WP1-SeNPs induced apoptosis in MCF-7 cells. |
Similarly, Huang et al reported that caspase-3, caspase-8, and caspase-9 were triggered by transferrin-conjugated SeNPs in a dose-dependent manner.38 Wu et al discovered that surface decoration of SeNPs by mushroom polysaccharide–protein complexes significantly increased the activities of caspase-3/7, -8, and -9 in a dose-dependent manner.39 Liu et al also demonstrated that 5-fluorouracil surface-functionalized SeNPs evoked activation of caspase-3, caspase-8, and caspase-9 in the MCF-7 cells in a dose-dependent manner.14 Our findings are consistent with previous reports where a similar dose-dependent triggering of caspases by surface-functionalized SeNPs were observed.
WP1-SeNPs induce Fas-associated death domain protein activation in MCF-7 cells
Apoptosis or programmed cell death is a key regulator of physiological growth control and regulation of tissue homeostasis.40 Fas-associated death domain protein (FADD) is a novel death domain-containing protein, which could interact with the death domain of Fas to initiate apoptosis.41 FADD is a component of the TNF receptor type 1 (TNF-R1) and Fas signaling complexes are involved in TNF-R1-induced and Fas-induced apoptosis.42 FADD acts as an adaptor protein used to connect both Fas receptor and caspase family, and this complex helps to activate other caspases. Therefore, we examined FADD to determine how such protein involved in cellular apoptosis that happens in the presence of WP1-SeNPs. The western blot analysis shown in Figure 8B illustrates a significant improvement in the expression levels of FADD in MCF-7 cells upon treatment with WP1-SeNPs. Our observation closely matches with that of Kumar et al who reported the anticancer activity of curcumin-loaded poly (2-hydroxyethyl methacrylate) nanoparticles in SKOV-3 cells and the notable increase in TRAIL (32 KDa), Fas (43 KDa), and FADD (26 KDa) proteins.43
Conclusion
The present study provides a facile method to design SeNP-based hybrid system with enhanced anticancer synergism by using walnut-meat peptides as the capping agent. The obtained WP1-SeNPs exhibited considerably high stability with controllable size distribution. Surface encapsulation by walnut-meat peptides significantly enhanced the anticancer activity of SeNPs. The five selected human cancer cell lines were found to be susceptible to WP1-SeNPs and MCF-7 cell was found to be most sensitive. The apoptosis-inducing activities of WP1-SeNPs on MCF-7 cells were further verified by the accumulation of S-phase cell arrest, nuclear condensation, and DNA breakage. The intrinsic signal pathway of WP1-SeNP-induced apoptosis in MCF-7 was confirmed to be mainly through the activation of FADD and caspases 3, 8, and 9, in combination with the mitochondrial membrane potential depletion as well as ROS generation. The possible signaling pathway of WP1-SeNPs induced apoptosis in MCF-7 cells is shown in Figure 8C. The results indicated that the synthesis of SeNPs using walnut-meat peptides as a modifier or stabilizer could be an innovative and effective way to achieve the bioavailability of SeNPs in anticancer treatment, especially for MCF-7 cell.
Acknowledgments
This study was supported by the Guangdong Natural Science Funds for Distinguished Young Scholars (No S2013050013954), Program for New Century Excellent Talents in University (NCET-13-0213), Guangdong Province Funded Research Projects (2013B010404001), and Key Laboratory of Aquatic Product Processing, Ministry of Agriculture, People’s Republic of China (NYJG201402). Also, we appreciate the kind help from Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, Guangdong, People’s Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Umar A, Dunn BK, Greenwald P. Future directions in cancer prevention. Nat Rev Cancer. 2012;12(12):835–848. | ||
Skilling KJ, Citossi F, Bradshaw TD, Ashford M, Kellam B, Marlow M. Insights into low molecular mass organic gelators: a focus on drug delivery and tissue engineering applications. Soft Matter. 2014;10(2):237–256. | ||
Hu X, Liu G, Li Y, Wang X, Liu S. Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals. J Am Chem Soc. 2015;137(1):362–368. | ||
Wang X, Yang L, Chen Z, Shin DM. Application of nanotechnology in cancer therapy and imaging. CA Cancer J Clin. 2008;58(2):97–110. | ||
Zhang H, Liu G, Zeng X, et al. Fabrication of genistein-loaded biodegradable TPGS-b-PCL nanoparticles for improved therapeutic effects in cervical cancer cells. Int J Nanomed. 2015;10:2461–2473. | ||
Shukla R, Hill E, Shi X, et al. Tumor microvasculature targeting with dendrimer-entrapped gold nanoparticles. Soft Matter. 2008;4(11):2160–2163. | ||
D’Arrigo G, Di Meo C, Gaucci E, et al. Self-assembled gellan-based nanohydrogels as a tool for prednisolone delivery. Soft Matter. 2012;8(45):11557–11564. | ||
Wen Y, Kolonich HR, Kruszewski KM, Giannoukakis N, Gawalt ES, Meng WS. Retaining antibodies in tumors with a self-assembling injectable system. Mol Pharm. 2013;10(3):1035–1044. | ||
Balducci A, Wen Y, Zhang Y, et al. A novel probe for the non-invasive detection of tumor-associated inflammation. Oncoimmunology. 2013;2(2):e23034. | ||
Hu X, Liu S. Recent advances towards the fabrication and biomedical applications of responsive polymeric assemblies and nanoparticle hybrid superstructures. Dalton Trans. 2015;44(9):3904–3922. | ||
Nalabotu SK, Kolli MB, Triest WE, et al. Intratracheal instillation of cerium oxide nanoparticles induces hepatic toxicity in male Sprague-Dawley rats. Int J Nanomed. 2011;6:2327–2335. | ||
Wen Y, Meng W. Recent in vivo evidences of particle-based delivery of small-interfering RNA (siRNA) into solid tumors. J Pharm Innov. 2014;9(2):158–173. | ||
Hu X, Hu J, Tian J, et al. Polyprodrug amphiphiles: hierarchical assemblies for shape-regulated cellular internalization, trafficking, and drug delivery. J Am Chem Soc. 2013;135(46):17617–17629. | ||
Liu J, Pang Y, Chen J, et al. Hyperbranched polydiselenide as a self assembling broad spectrum anticancer agent. Biomaterials. 2012;33(31):7765–7774. | ||
Yu S, Yin Y, Zhu J, et al. A modulatory bifunctional artificial enzyme with both SOD and GPx activities based on a smart star-shaped pseudo-block copolymer. Soft Matter. 2010;6(21):5342–5350. | ||
Navarro-Alarcon M, Lopez-Martinez MC. Essentiality of selenium in the human body: relationship with different diseases. Sci Total Environ. 2000;249(1–3):347–371. | ||
Rao L, Ma Y, Zhuang M, Luo T, Wang Y, Hong A. Chitosan-decorated selenium nanoparticles as protein carriers to improve the in vivo half-life of the peptide therapeutic BAY 55-9837 for type 2 diabetes mellitus. Int J Nanomed. 2014;9:4819–4828. | ||
Roy G, Sarma BK, Phadnis PP, Mugesh G. Selenium-containing enzymes in mammals: chemical perspectives. J Chem Sci. 2005;117(4):287–303. | ||
Zhang J, Wang X, Xu T. Elemental selenium at nano size (Nano-Se) as a potential chemopreventive agent with reduced risk of selenium toxicity: comparison with Se-methylselenocysteine in mice. Toxicol Sci. 2008;101(1):22–31. | ||
Loomis K, McNeeley K, Bellamkonda RV. Nanoparticles with targeting, triggered release, and imaging functionality for cancer applications. Soft Matter. 2011;7(3):839–856. | ||
Gao F, Yuan Q, Gao L, et al. Cytotoxicity and therapeutic effect of irinotecan combined with selenium nanoparticles. Biomaterials. 2014;35(31):8854–8866. | ||
Kong L, Yuan Q, Zhu H, et al. The suppression of prostate LNCaP cancer cells growth by selenium nanoparticles through Akt/Mdm2/AR controlled apoptosis. Biomaterials. 2011;32(27):6515–6522. | ||
Sun D, Liu Y, Yu Q, et al. The effects of luminescent ruthenium(II) polypyridyl functionalized selenium nanoparticles on bFGF-induced angiogenesis and AKT/ERK signaling. Biomaterials. 2013;34(1):171–180. | ||
Li Y, Li X, Wong Y-S, et al. The reversal of cisplatin-induced nephrotoxicity by selenium nanoparticles functionalized with 11-mercapto-1-undecanol by inhibition of ROS-mediated apoptosis. Biomaterials. 2011;32(34):9068–9076. | ||
Chen T, Wong Y-S, Zheng W, Bai Y, Huang L. Selenium nanoparticles fabricated in Undaria pinnatifida polysaccharide solutions induce mitochondria-mediated apoptosis in A375 human melanoma cells. Colloids Surf B Biointerfaces. 2008;67(1):26–31. | ||
Korhonen H. Milk-derived bioactive peptides: from science to applications. J Funct Foods. 2009;1(2):177–187. | ||
Zhang Y, Kong Q, Huang Y, Wang G, Chang KJ. Inhibition of c-FLIP by RNAi enhances sensitivity of the human osteogenic sarcoma cell line U2OS to TRAIL-induced apoptosis. Asian Pac J Cancer Prev. 2015;16(6):2251–2256. | ||
Sanpui P, Chattopadhyay A, Ghosh SS. Induction of apoptosis in cancer cells at low silver nanoparticle concentrations using chitosan nanocarrier. ACS Appl Mater Interfaces. 2011;3(2):218–228. | ||
Zheng S, Li X, Zhang Y, et al. PEG-nanolized ultrasmall selenium nanoparticles overcome drug resistance in hepatocellular carcinoma HepG2 cells through induction of mitochondria dysfunction. Int J Nanomed. 2012;7:3939. | ||
Luo H, Wang F, Bai Y, Chen T, Zheng W. Selenium nanoparticles inhibit the growth of HeLa and MDA-MB-231 cells through induction of S phase arrest. Colloids Surf B Biointerfaces. 2012;94:304–308. | ||
Danz EDB, Skramsted J, Henry N, Bennett JA, Keller RS. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med. 2009;46(12):1589–1597. | ||
Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. | ||
Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505–513. | ||
Liao W, Ning Z, Chen L, et al. Intracellular antioxidant detoxifying effects of diosmetin on 2,2-Azobis(2-amidinopropane) dihydrochloride (AAPH)-induced oxidative stress through inhibition of reactive oxygen species generation. J Agric Food Chem. 2014;62(34):8648–8654. | ||
Liao W, Luo Z, Liu D, Ning Z, Yang J, Ren J. Structure characterization of a novel polysaccharide from Dictyophora indusiata and its macrophage immunomodulatory activities. J Agric Food Chem. 2015;63:535–544. | ||
Foldbjerg R, Dang DA, Autrup H. Cytotoxicity and genotoxicity of silver nanoparticles in the human lung cancer cell line, A549. Arch Toxicol. 2011;85(7):743–750. | ||
Xia T, Kovochich M, Brant J, et al. Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm. Nano Lett. 2006;6(8):1794–1807. | ||
Huang Y, He L, Liu W, et al. Selective cellular uptake and induction of apoptosis of cancer-targeted selenium nanoparticles. Biomaterials. 2013;34(29):7106–7116. | ||
Wu H, Li X, Liu W, et al. Surface decoration of selenium nanoparticles by mushroom polysaccharides-protein complexes to achieve enhanced cellular uptake and antiproliferative activity. J Mater Chem. 2012;22(19):9602–9610. | ||
Fulda S, Debatin K. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25(34):4798–4811. | ||
Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81(4):505–512. | ||
Hu W-H, Johnson H, Shu H-B. Activation of NF-κB by FADD, Casper, and caspase-8. J Biol Chem. 2000;275(15):10838–10844. | ||
Kumar SSD, Surianarayanan M, Vijayaraghavan R, Mandal AB, MacFarlane DR. Curcumin loaded poly(2-hydroxyethyl methacrylate) nanoparticles from gelled ionic liquid – In vitro cytotoxicity and anti-cancer activity in SKOV-3 cells. Eur J Pharm Sci. 2014;51(23):34–44. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.