Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Novel targeted therapies in chordoma: an update
Authors Di Maio S, Yip S, Al Zhrani GA, Alotaibi FE, Al Turki A, Kong E, Rostomily R
Received 12 June 2014
Accepted for publication 14 April 2015
Published 26 May 2015 Volume 2015:11 Pages 873—883
DOI https://doi.org/10.2147/TCRM.S50526
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Salvatore Di Maio,1 Stephen Yip,2 Gmaan A Al Zhrani,3,4 Fahad E Alotaibi,3,4 Abdulrahman Al Turki,3,4 Esther Kong,2 Robert C Rostomily5
1Division of Neurosurgery, Jewish General Hospital, McGill University, Montreal, QC, 2Department of Pathology and Laboratory Medicine, Vancouver General Hospital, University of British Columbia, Vancouver, BC, Canada; 3National Neuroscience Institute, Department of Neurosurgery, King Fahad Medical City, Riyadh, Saudi Arabia; 4Department of Neurology and Neurosurgery, The Montreal Neurological Institute and Hospital, McGill University Health Centre, Montreal, QC, Canada; 5Department of Neurological Surgery, University of Washington, University of Washington Medical Center, Seattle, WA, USA
Abstract: Chordomas are rare, locally aggressive skull base neoplasms known for local recurrence and not-infrequent treatment failure. Current evidence supports the role of maximal safe surgical resection. In addition to open skull-base approaches, the endoscopic endonasal approach to clival chordomas has been reported with favorable albeit early results. Adjuvant radiation is prescribed following complete resection, alternatively for gross residual disease or at the time of recurrence. The modalities of adjuvant radiation therapy reported vary widely and include proton-beam, carbon-ion, fractionated photon radiotherapy, and photon and gamma-knife radiosurgery. As of now, no direct comparison is available, and high-level evidence demonstrating superiority of one modality over another is lacking. While systemic therapies have yet to form part of any first-line therapy for chordomas, a number of targeted agents have been evaluated to date that inhibit specific molecules and their respective pathways known to be implicated in chordomas. These include EGFR (erlotinib, gefitinib, lapatinib), PDGFR (imatinib), mTOR (rapamycin), and VEGF (bevacizumab). This article provides an update of the current multimodality treatment of cranial base chordomas, with an emphasis on how current understanding of molecular pathogenesis provides a framework for the development of novel targeted approaches.
Keywords: chordomas, cell lines, radiation therapy, skull-base neoplasms, surgery, molecular genetics
Introduction
Chordomas are rare primary bone neoplasms that typically originate from the spine or skull base. Conventional practice generally involves maximal safe surgical removal, often followed by focused radiation therapy. Despite this, many patients ultimately succumb to local treatment failure and recurrence. To date, no chemotherapeutic agent has demonstrated sufficient efficacy to constitute part of the first-line therapy for these locally aggressive neoplasms. Progress in the understanding of chordomas’ molecular pathogenesis has given rise to efforts to treat chordomas with targeted therapies. This article provides an essential review on cranial base chordomas, outlines current surgery and radiation-treatment paradigms, and further examines how the evolving understanding of molecular pathogenesis has informed potential new targeted chemotherapeutic approaches.
Description and epidemiology
Chordomas are rare neoplasms, occurring with an annual age-adjusted incidence of 0.02 per 100,000 person-years, and account for 1%–4% of all primary malignant bone tumors.1,2 Arising from embryonic notochord remnants, chordomas occur at any point along the skeletal neuraxis,2,3 with the sacrum and cranial base being the most frequently affected areas.4 Cranial base chordomas account for 35%–49% of all chordomas.3 Consistent with their site of origin, chordomas predominantly present in the extradural space and produce symptoms secondary to local growth and bony destruction. Of note, chordomas can rarely present as a solitary intradural lesion in the retroclival space, and while they behave in similar fashion to their classic extradural counterparts,5 intradural lesions must be distinguished from a more benign variant of intradural notochordal tumor termed ecchordosis physaliphora, which can have very subtle radiologic and pathologic differences from more malignant chordomas.6,7
The median age at presentation for cranial chordomas is in the sixth decade, slightly younger for sacral chordomas,2 and with rare occurrences in the pediatric population.2,8 Given the predilection for growth in the clivus and cavernous sinus regions, the most frequent clinical presentations of skull-base chordomas are cranial nerve deficits (eg, abducens or oculomotor nerve palsy).9
The median survival of cranial base chordomas is estimated at 6.29 years,4,10 with 5-year overall survival (OS) and progression-free survival (PFS) rates of 78.4% and 50.8%, respectively.11 The lethality of skull-base chordoma is largely due to local progression, although systemic metastasis has been reported in 12.5% of skull base/craniocervical tumors.12 A large retrospective study of cranial chordomas recently suggested a trend toward improvement in survival over time,3 with 5-year OS for the 1975–1984, 1985–1994, and 1995–2004 epochs of 48.5%, 73.0%, and 80.7%, respectively. The reasons for such improvements are unclear, but may reflect earlier detection and treatment (lead-time bias); better treatment modalities, including the addition of endoscopic approaches both as an adjunct to open procedures and as a standalone procedure, especially for midline clival tumors; refinements in the safety of open skull-base approaches; and use of greater sophistication in radiation therapy-delivery techniques, allowing for safer prescription of the requisite higher dose for chordomas.
Current treatment paradigm for skull-base chordomas
There is no uniform consensus regarding the optimal standard treatment for skull-base chordomas, and evidence regarding various paradigms stem from retrospective series over often long and inconsistent treatment eras. While it is generally agreed that a maximal safe removal of the tumor should be done first,13 a number of other practice differences exist, including the type of radiation therapy, the indications for radiation therapy (eg, after complete vs only incomplete resection; upfront or at the time of recurrence), and the management of metastases. To date, no chemotherapeutic agent have been approved for the first-line treatment of skull-base chordomas.14 At the time of recurrence, the respective roles of repeat resection, radiation therapy, and chemotherapy remain unclear.
Surgery
The principal goals of surgery beyond histologic confirmation of the lesion are to achieve a maximal safe resection, provide symptomatic improvement, and to facilitate adjuvant treatment, such as radiotherapy, by minimizing the treatment volume and maximizing the distance between the target volume and critical surrounding neurovascular structures. In the skull base, surgical resection of chordomas remains a challenge, due to anatomic constraints in accessing the skull base, as well as their locally aggressive growth pattern and involvement of surrounding neurological structures, such as the brain stem, cranial nerves, and internal carotid and/or vertebral arteries. These factors often exceed the ability of any single surgical approach to achieve maximal resection, thus necessitating complex combined or multiple approaches. Understandably, the majority of surgical series describe a practice of piecemeal intralesional resection to normal dural and/or bony margins, as opposed to en bloc resection with margins as described for their sacral counterparts.15
Despite these limitations, the literature supports the benefit of surgical resection on survival for chordomas. In a large review of 962 patients with spinal, sacral, or cranial base chordomas identified from the Surveillance, Epidemiology, and End Results (SEER) database, Jawad and Scully found surgical resection to be an independent predictor of OS.16 In addition, in a meta-analysis of 23 retrospective studies, 807 patients with cranial base chordomas were analyzed together, with a mean follow-up of 53.6 months.11 The rate of complete resection ranged widely across studies, from 0% to 73.7%. Those patients with a complete resection had a 5-year OS of 95% (vs 71% without) and 5-year PFS of 87% (vs 50% without). Despite a seemingly clear benefit of complete resection in this study, a few stipulations should be mentioned. The majority of included studies reported the routine use of adjuvant radiotherapy, including after complete resection. Most outcomes reported were for patients who had undergone open resection, with only a small minority of cases undergoing endoscopic endonasal resection. The careful assessment of a complete resection can be difficult, and may have differed across studies. For example, a complete resection may refer to the soft-tissue component of the tumor, versus a resection of the tumor and all affected surrounding bone and dura.
To achieve maximal resection, traditional open skull-base approaches (summarized elsewhere)17,18 are most commonly used, but the less invasive endoscopic approaches have been increasingly adopted. The endoscope has been employed as an adjunct to open resection or as a standalone endonasal approach to resect midline skull-base chordomas.13,19–27 Advances in such techniques as pituitary gland mobilization, posterior clinoidectomy, and skeletonization of the petrous carotid arteries have permitted access to the entire rostrocaudal extent of the clivus via this approach.28 However, comparisons of endoscopic versus microscopic resection of cranial base chordomas are invariably prone to selection bias, and understandably long-term comparisons of the rate of complete resection, recurrence, and OS have to date not been published. In one systematic review,29 26 open-surgery studies published from 1987 to 2010 were compared with 12 studies of either endoscope-assisted or fully endoscopic resection of cranial base chordomas from 2002 to 2010. No survival data were available for the endoscopic cohort, and follow-up for this latter group was limited to 18.5 months. The reported rate of complete resection was 61.0%, although the rate of petrous involvement and dural invasion was lower in the endoscopic group. The main limitation of this technique remains the risk of cerebrospinal fluid leakage and meningitis, largely mitigated by meticulous layered closure and vascularized mucosal flaps. Nevertheless, the risk in all recently published larger (n>10) series with an intradural tumor component remains significant: 8%–33% developed cerebrospinal fluid leakage and 0%–14% meningitis.19,20,23,30
Radiotherapy
Although radiotherapy is an important therapeutic adjunct for cranial base chordomas, issues regarding both the timing of adjuvant radiotherapy, specifically after complete resection or only for residual/recurrent disease, and optimal type of radiotherapy are largely unresolved in the literature. Two recent meta-analyses based on the SEER database failed to demonstrate a benefit in survival in patients who received radiotherapy;31,32 however, this was possibly due to a number of sources of error, including but not limited to not controlling for the presence or absence of postoperative residual disease, histologic subtype, tumor size, patient age, type and dose of radiotherapy, era of treatment, and other factors. The difficulty of answering these questions is evident, and the following discussion attempts to interpret the available literature as much as possible.
There is significant heterogeneity in the literature regarding the use of adjuvant radiotherapy following surgical resections described as “complete” or “gross total”. In one meta-analysis, among patients reported to have gross total resection of a skull-base chordoma, only 37.9% of patients were referred for adjuvant radiation therapy.11 Despite gross total resection, however, the recurrence rate following surgical removal of skull-base chordomas can be high. The results of large surgical series show a 5-year PFS of 22.5%–74.2% with complete upfront resection, and pooled numbers are too small to assess the benefit of radiotherapy in this subgroup.11 The concept of gross total resection in cranial base chordoma is difficult to define or prove, given the challenge surgeons have in identifying and removing microscopic invasion adjacent to the tumor, and of assessing microscopic disease on postoperative imaging. Histopathologic studies of chordoma-growth patterns point to an infiltrative propensity to invade bone and submucous tissue and along the loose connective tissue among local vessels, nerves, and muscles in multilayers or multilobular fashion.33 Chordomas lack a fibrous capsule,34 and tumor cells clearly invade intralesional fibrous septa, particularly in advanced stages of the disease. The rationale for adjuvant radiation therapy in light of these features is thus to improve local control.13
Despite this, the evidence supporting the use of radiation therapy following complete surgical removal is mixed.35 In one large series of cranial base chordomas in which aggressive microsurgical resection was performed,36 53 of 74 (71.6%) patients had complete resection either after single- or multistage resection. Only patients with gross residual disease received adjuvant radiation therapy. Five-year recurrence-free survival was 47%. In a larger updated series by the same authors,37 a trend toward routine adjuvant radiation therapy was observed, despite a similar rate of complete resection in early (1988–1999) and late (2000–2011) treatment eras. Five-year recurrence-free survival in the early and late-treatment eras was not statistically different: 55% and 59%, respectively. Five-year OS, however, was significantly better in the latter treatment era (93% vs 64%). Although there was a modest reduction in perioperative complications in the latter era, it is also conceivable that the improvement in survival may also at least in part be attributable to more routine use of adjuvant radiotherapy. This remains to be proven, however, in a comparative prospective fashion.
In addition to the timing of adjuvant radiotherapy, the optimal type of contemporary radiotherapy is controversial and as-yet unproven in the literature. Chordomas exhibit an increased dose–response relationship relative to other tumors.38 Therefore, the principal challenge of delivering safe and effective radiation therapy is to achieve clinically relevant doses while mitigating toxicity to adjacent neurovascular structures, such as the optic and other cranial nerves, pituitary gland, and brain stem. Proton-beam therapy has classically been considered to be well-suited for chordomas, due to its unique radiobiological properties,39 although evidence regarding its clinical superiority over other modern delivery techniques (eg, radiosurgery and fractionated stereotactic radiotherapy), radiation sources, and dose schedules has been questioned.40 To date, in addition to proton-beam radiotherapy, fractionated photon radiotherapy, CyberKnife and gamma-knife radiosurgery, and carbon-ion radiotherapy have all been reported for cranial base chordomas. Superiority of one radiotherapy modality over another has yet to be demonstrated, although large cohort comparisons are lacking. A discussion of each form of radiotherapy used currently for cranial base chordomas follows.
Charged heavy-particle therapy, such as with protons, takes advantage of a number of physical properties to deliver a high dose of radiation to tumors, while sparing falloff exposure to surrounding tissues. For cranial base chordomas, 5-year recurrence-free survival using proton-based therapy ranges from 59% to 73%.39,41,42 Amichetti et al performed a systematic review of the literature comparing proton-based radiotherapy with other available forms of radiation therapy for skull-base chordomas.43 There were no direct comparative studies, and the results of seven proton-beam therapy series including 416 patients were reviewed along with ten studies and 191 patients who underwent conventional forms of photon-based treatment. Mean dose delivery for proton-beam therapy was 66–83 cobalt gray equivalent. The average 5-year local control and OS were 69.2% and 79.8%, respectively. Only one study reported 10-year recurrence free survival and OS, which was 54%.39 This was found to be superior to conventional photon radiotherapy, which had 33.5% and 53.5% average local control and OS at 5 years, respectively. The years of treatment for the photon group were from 1938 to 2005, and significantly earlier than included proton-beam studies.
Carbon-ion radiotherapy exerts similar physical characteristics as proton-based treatment, but with higher relative biological effectiveness, which is of interest in relatively radio-resistant tumors, such as chordomas.44–50 Early series from carbon-ion facilities in Germany and Japan have reported 3-year recurrence-free survival of 70%–80.6% for skull-base chordomas.44,47 A single-center randomized clinical trial of proton- versus carbon-radiation therapy in patients with cranial base chordomas is ongoing,48 although other preliminary data for sacral chordomas suggest a lack of difference in clinical efficacy between ion type.49 The primary disadvantages of proton and carbon ion-based radiotherapy are global availability and investment cost.50
The results of more modern studies of fractionated photon-based radiotherapy for skull-base chordomas appears closer to proton-based therapy compared to earlier studies. Techniques are nevertheless variable from one study to another, including whether photon therapy is administered alone or in combination with particle therapy.51,52 In a recent small series of fractionated photon-based radiotherapy,53 5-year OS was 76.4%, and 37.5% of patients were free of progression at 5 years. All patients either had gross residual disease after surgery or were being treated for symptomatic recurrences, however. Similar results were obtained by Debus et al54 who treated 37 patients with chordomas of the skull base or upper cervical spine. Local control was 50% and OS 82% at 5 years. In places where proton-beam or carbon-ion radiotherapy centers are not available, photon-based radiotherapy appears to confer grossly similar local control rates, although true comparative studies would be of great interest.
Smaller volume tumors have also been amenable to stereotactic radiosurgery (SRS), and early results have suggested comparable outcomes to other radiation modalities for residual or recurrent cases.55 SRS has been shown to be valuable in treating small residual postoperative and recurrent chordomas.56 Krishnan et al and Kano et al reported that using an SRS boost (15.0 Gy median margin dose) with or without fractionated radiation therapy (50.4 Gy median margin dose), the 5-year tumor-control rate was 55% and 65%, respectively.55,57 Liu et al and Hasegawa et al had a 5-year treated tumor-control rate of 66% and 72% when using 12.7 Gy and 15 Gy median margin dose, respectively. In both series, OS was 80% and 84% at 5 years, respectively.58,59 In a multicenter study of 71 patients who underwent gamma-knife SRS for chordomas, an 80% 5-year actuarial OS and 66% treated tumor-control rate after SRS was reported for the combined cohort. In this study, the median patient age was 45 years, the median SRS target volume was 7.1 cm3, and the median tumor-margin dose was 15.0 Gy.56
Irrespective of the type of radiotherapy administered, a number of factors have been identified that may influence the response rate of radiotherapy. These include age, sex, tumor heterogeneity, extent of resection, presence of necrosis in the pretreatment biopsy, elevated tumor volume, and radiotherapy dose delivered.60,61 Among cases with subtotal resection, a residual tumor volume under 25–30 cm3 appears associated with better local control using adjunctive radiation therapy.29,41,42,62 In one study of 42 patients with skull-base chordomas, a gross tumor volume ≤25 mL was associated with excellent local control using spot-scanning-based proton radiotherapy.63 The typical at-risk structures during dosimetric planning include the optic apparatus, pituitary gland, and brain stem, and need to be carefully mapped pretreatment.
Investigation of novel therapeutic targets
Despite the challenges posed by the rarity of chordoma, advances in the molecular understanding of chordomas have led to the identification of promising targetable pathways and prognostic markers. These include brachyury, receptor tyrosine kinases (RTKs), and downstream pathways. Figure 1 lists some of the known pathways implicated in the pathogenesis of chordoma and the accompanying table (Table 1) lists the matched targeted therapeutics. Loss of chromosome 1p36, 9p loss of heterozygosity, and an elevated Ki67 proliferative index all correlate with aggressive behavior and shorter OS in skull-base chordomas.64,65 Additionally, loss of chromosome 1q, gain of 2p, and aberrant brachyury copy-number changes and subsequent protein expression are associated with disease recurrence.
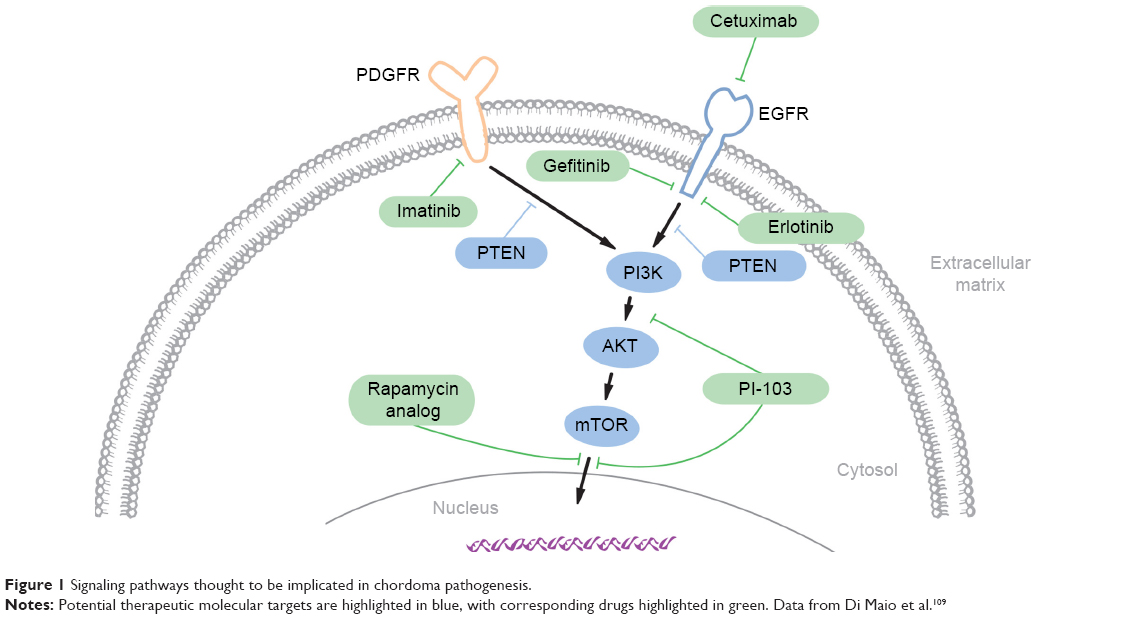 | Figure 1 Signaling pathways thought to be implicated in chordoma pathogenesis. |
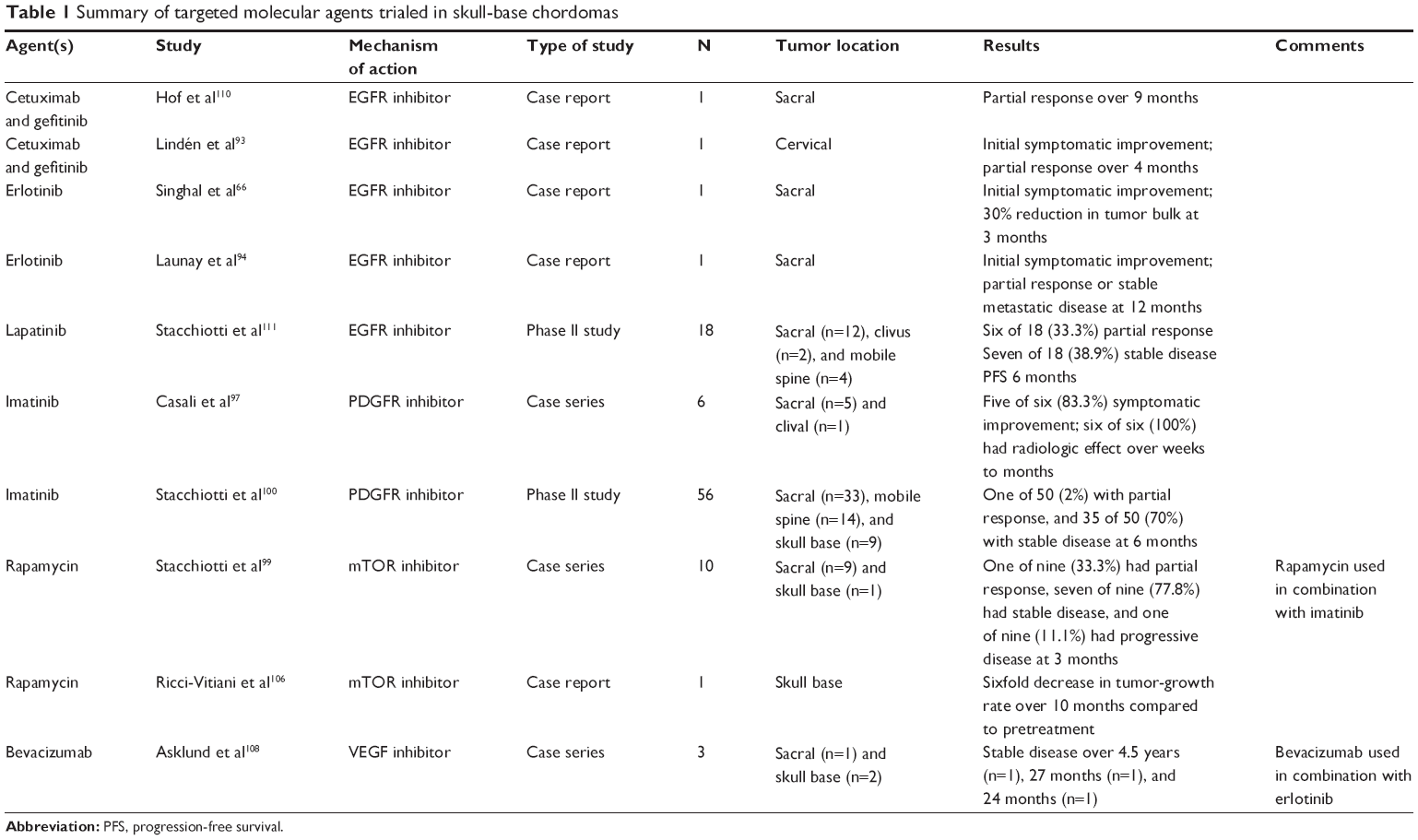 | Table 1 Summary of targeted molecular agents trialed in skull-base chordomas |
The brachyury or T gene located on chromosome 6q27 encodes a developmentally regulated transcription factor essential for notochordal development and formation of posterior mesodermal elements.65,66 Normally silenced in postdevelopmental tissue, brachyury is aberrantly reexpressed at high levels in chordoma cells,67 providing a diagnostic adjunct in differentiating chordoma from other tumors with similar histology and geographical location.68,69 The impact of brachyury experimental gain and loss of function on increased and decreased cell proliferation, respectively, highlights its biological and functional importance for chordoma growth and progression.70–73
Several lines of evidence also suggest a causative role of brachyury overexpression in chordoma formation.74 A study of four familial chordoma cohorts identified recurrent germ-line duplication within chromosome 6q27, which contains the brachyury gene.75 However, the vast majority of chordoma patients lack a family history, and in these more common sporadic tumors, only 6.8% of patients (16 of 236) exhibited genomic amplification of brachyury.70,76,77 Therefore, aberrant overexpression of brachyury in the latter group is potentially related to epigenetic changes rather than genomic amplification.76 Also, a nonsynonymous single-nucleotide polymorphism or SNP (rs2305089) in brachyury is strongly associated with development of chordoma,78 as well as worse OS in spinal chordoma patients.79 Lastly, genomic copy-number status and subsequent protein expression of brachyury is associated with shorter PFS on a cohort of 37 skull-base chordomas.65 However, a separate study published in the same year found no prognostic value of brachyury expression in a cohort of spinal chordomas.80 Whether this represents inherent biological differences from tumors arising from the spine and skull base remains to be tested in additional studies.
Given the tight correlation between genomic aberrations of brachyury leading to its temporally aberrant reexpression in chordoma, brachyury may be considered to be a driver oncogene in chordoma.65,70,71 As mentioned earlier, brachyury gain- and loss-of-function experiments have further established its putative role in the malignant growth of chordoma. Enforced silencing of brachyury in the JHC7 and UCH-1 chordoma cell lines result in growth cessation, senescence, and differentiation.70,71 Approximately 81.1% of 37 skull-base chordomas were found to be immunopositive for brachyury protein expression, indicating its potential central role in regulating growth in the majority of skull-base chordomas.65 A potential mechanism by which brachyury may contribute to chordoma’s malignant phenotype may involve an activating role in epithelial–mesenchymal transition (EMT). EMT represents a mechanism for normal developmental and tissue-repair mechanisms, but can also be co-opted in cancer development.81,82 EMT can mediate infiltrative and invasive behavior in tumor cells via the selective activation of “mesenchymal” genetic programs, such as expression of the SNAIL gene. This is relevant in chordomas, as recent evidence has highlighted brachyury-mediated SNAIL activation in these tumors.71,74
In the absence of active therapeutic approaches that directly target brachyury, inactivation of downstream or interacting signaling pathways provides an alternative means to short-circuit its function. For instance, the extensive cross talk between brachyury and other progrowth signaling pathways is of therapeutic significance for targeting brachyury in chordoma. FGFR/MEK/ERK signal-transduction pathways appear to mediate downstream signaling of brachyury in chordomas.83 Interestingly, these same pathways appear to effect positive feedback back on brachyury, which makes components of the FGFR/MEK/ERK pathways and brachyury potential therapeutic targets. A different study highlights the interaction between brachyury and the EGFR signaling pathway.73
Reexpression of brachyury exclusively in chordoma, and its role in driving cancer behavior, makes brachyury an attractive target for therapeutic intervention. Since the protein is not expressed in nonchordoma normal tissues, targeting of the brachyury protein, its function, and associated signal-transduction pathways proves a very attractive clinical option. Recent advancements in the field of tumor immunology have rekindled the interest in its incorporation in cancer treatment.84 The brachyury protein therefore represents a logical target for immunotherapy in chordoma, given its pivotal role in the initiation and progression of the disease.69,85
Protein tyrosine kinases (PTKs) mediate phosphorylation of selected tyrosine residues, which results in functional activation of many proteins and plays a crucial role in cancer development.86 RTKs are specialized, transmembrane PTKs that mediate signaling via sampling of the external environment. RTKs are composed of extracellular domains that bind cognate environmental ligands and an intracellular domain that mediates the signaling event via dimerization and binding to other signaling molecules. Mutations and overexpression of RTKs, such as PDGFα and -β, EGFR, MET, and HER2/NEU are central to the development of many cancers; therefore, they are attractive targets of therapeutic intervention. Hyperactive RTKs not only result in growth proliferation but also other procancer “hallmarks”, such as enhanced cell survival and angiogenesis, which together contribute to tumorigenesis.87
A large proportion of chordomas demonstrate EGFR overexpression, which is associated with aggressive clinical behavior. In a single study, 69% of chordoma samples expressed EGFR and 40% display EGFR amplification.88 This is recapitulated in three other studies with a cumulative number of 79.6% (43 of 54) chordoma cases demonstrating EGFR expression.89–91 Given these observations, and the efficacy of EGFR inhibitors gefitinib and erlotinib in large-cell lung adenocarcinoma patients with activating EGFR mutations,92 antibody-based blockade or pharmacologic inhibition of EGFR signaling in chordoma is a logical potential therapeutic option. Anecdotal reports of partial responses and clinical improvement in recurrent sacral and skull-base chordoma with adjuvant single-agent or combined EGFR antibody (cetuximab) and EGFR-specific (gefitinib or erlotinib) or broader-spectrum tyrosine-kinase inhibitors (lapatinib)66,93,94 suggest that inhibition of EGFR protein and the downstream signaling pathway might offer a reasonable therapeutic option for some chordoma patients. However, larger, more controlled studies are required to define more precisely the indications for treatment and efficacy.
The PDGF receptor (PDGFR) is another PTK expressed in chordoma. Although PDGFR promotes chordoma cell proliferation through activation of the PI3K/AKT, RAS/ERK, and STAT pathways,95 the PDGFRβ isoform is preferentially upregulated and localized in the stromal components of chordomas,89,96–98 suggesting a role in providing a supportive tumor microenvironment. Therefore, responses to PDGFR inhibition may reflect the relative abundance of tumor-associated stromata. In practice, the PDGFR preferential PTK inhibitor imatinib has demonstrated modest activity in small uncontrolled case series97,99 of chordoma patients with advanced disease, prompting a Phase II trial of 56 patients that realized overall clinical benefit in 64% and stable disease in 70% of patients.100 Despite some success with PDGFR inhibition in chordoma, a majority of treated patients do not show decline in tumor size, which may reflect the inability of targeting PDGFR expressing stromal elements to effect cytologic reduction of neoplastic chordoma cells. The presence of a prominent demineralized matrix in chordoma is another factor that may limit drug delivery and underlie cases that show suboptimal response to any systemic therapy.96 These observations indicate the critical need to consider the role of the tumor microenvironment as a barrier to drug delivery when designing future treatment strategies.
Targeting PTKs in chordoma can be challenging, due to their multiplicity and functional redundancy. Strategies to address this challenge could include simultaneous inhibition of multiple activated PTKs or targeting common signaling pathways downstream of the PTKs. For instance, signaling from multiple RTKs, including EGFR and PDGFR, converges on the PI3K/AKT/mTOR pathway, which is in turn negatively regulated by the PTEN tumor suppressor. Components of the PI3K/AKT/mTOR pathway are activated,95,101,102 and PTEN is suppressed in chordoma-tumor samples,103 suggesting that inhibition of this pathway alone or in combination with PTK inhibitors could have therapeutic activity against chordoma. Preclinical experimental studies using inhibitors of mTORC1 (rapamycin), mTOR (MLN0128), and PI3K/AKT/mTOR (PI-103) support this approach,101,103,104 while the addition of the mTOR inhibitor to imatinib showed additional clinical benefit in patients with advanced imatinib-resistant chordomas.99 These preliminary results indicate the therapeutic potential of targeting downstream effectors of activated PTKs and their potential effectiveness in combination with other targeted therapies.
The rarity of skull-base chordomas limits not only the number of treatments that can be practically tested in clinical trials but also the statistical power required to identify meaningful differences in outcome. Therefore, the application of precision or personalized oncology based on the molecular profiling of aberrant pathways in individual patients holds great promise for advancing the treatment of chordoma patients.105 For example, a chordoma patient treated with rapamycin on the basis of aberrant mTOR-pathway signaling identified in molecular profiling of patient-derived cells resulted in a sixfold reduction in tumor-growth rate.106 Additional targets for chordoma therapy will emerge from ongoing efforts at molecular profiling. For example, in addition to the targets discussed earlier, proangiogenic pathways mediated by VEGFR-2 and iNOS with relevance to other cancers have been implicated in chordoma.107 In a small clinical series, combined use of erlotinib and bevacizumab, a humanized anti-VEGF antibody, led disease stabilization in three chordoma patients.108 Ultimately, the establishment of large and well-annotated databases will be crucial to realize the benefits of personalized chordoma therapy. As proposed for other cancers, ongoing iterative analysis of individual patient molecular profiles and treatment responses generated from these databases is expected to better inform therapeutic choices in a prospective fashion.
Conclusion
Management of chordomas affecting the base of the skull remains challenging, although epidemiologic data suggest an improvement in survival and local control in more recent eras. Complete safe removal after initial diagnosis appears to improve survival relative to incomplete resection. Endoscopic endonasal approaches may be useful in achieving safe resection of those chordomas affecting the midline clivus. Multiple modalities of radiation therapy have been used for chordoma, and results in well-selected individuals are comparable. To date, no chemotherapeutic agent has formed part of the standard treatment of chordomas. Based on an increasing knowledge of the molecular pathogenesis of skull-base chordomas, a number of targeted therapies have been attempted, with modest results for recurrent cases. Future systemic treatments based on affected pathways and application of the principals of personalized oncology will hopefully reduce the burden of this rare tumor.
Disclosure
The authors report no conflicts of interest in this work.
References
Central Brain Tumor Registry of the United States. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2007. Hinsdale (IL): CBTRUS; 2011. | ||
Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12(11):1344–1350. | ||
Chambers KJ, Lin DT, Meier J, Remenschneider A, Herr M, Gray ST. Incidence and survival patterns of cranial chordoma in the United States. Laryngoscope. 2014;124(5):1097–1102. | ||
McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12(1):1–11. | ||
AlOtaibi F, Guiot MC, Muanza T, Di Maio S. Giant petroclival primary intradural chordoma: case report and systematic review of the literature. J Neurol Surg Rep. 2014;75(1):e160–e169. | ||
Golden LD, Small JE. Benign notochordal lesions of the posterior clivus: retrospective review of prevalence and imaging characteristics. J Neuroimaging. 2014;24(3):245–249. | ||
George B, Bresson D, Bouazza S, et al. [Chordoma]. Neuro-Chirurgie. 2014;60(3):63–140. French. | ||
Mirra JM, Nelson SD, Della Rocca C, Mertens F. Chordoma. In: Fletcher DM, Unni K, Mertens F, editors. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002:316–317. | ||
Walcott BP, Nahed BV, Mohyeldin A, Coumans JV, Kahle KT, Ferreira MJ. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13(2):e69–e76. | ||
McMaster M. Update on the epidemiology of chordoma: SEER registry data 1973–2007. Poster presented at: Third International Chordoma Research Workshop; March 17–19, 2011; Bethesda, MD. | ||
Di Maio S, Temkin N, Ramanathan D, Sekhar LN. Current comprehensive management of cranial base chordomas: 10-year meta-analysis of observational studies. J Neurosurg. 2011;115(6):1094–1105. | ||
Yasuda M, Bresson D, Chibbaro S, et al. Chordomas of the skull base and cervical spine: clinical outcomes associated with a multimodal surgical resection combined with proton-beam radiation in 40 patients. Neurosurg Rev. 2012;35(2):171–182; discussion 182–183. | ||
Fernandez-Miranda JC, Gardner PA, Snyderman CH, et al. Clival chordomas: a pathological, surgical, and radiotherapeutic review. Head Neck. 2014;36(6):892–906. | ||
Diaz RJ, Cusimano MD. The biological basis for modern treatment of chordoma. J Neurooncol. 2011;104(2):411–422. | ||
Boriani S, Bandiera S, Biagini R, et al. Chordoma of the mobile spine: fifty years of experience. Spine (Phila Pa 1976). 2006;31(4):493–503. | ||
Jawad MU, Scully SP. Surgery significantly improves survival in patients with chordoma. Spine (Phila Pa 1976). 2010;35(1):117–123. | ||
Di Maio S, Sekhar LN. Skull base approaches. In: Ellenbogen RG, Abdulrauf SI, editors. Principles of Neurological Surgery: Expert Consult – Online and Print. Philadelphia: Elsevier; 2012. | ||
Rostomily RC, Sekhar LN, Elahi F. Chordomas and chondrosarcomas. In: Sekhar LN, Fessler R, editors. Atlas of Neurosurgical Techniques. Brain. New York: Thieme; 2006. | ||
Tan NC, Naidoo Y, Oue S, et al. Endoscopic surgery of skull base chordomas. J Neurol Surg B Skull Base. 2012;73(6):379–386. | ||
Chibbaro S, Cornelius JF, Froelich S, et al. Endoscopic endonasal approach in the management of skull base chordomas – clinical experience on a large series, technique, outcome, and pitfalls. Neurosurg Rev. 2014;37(2):217–224; discussion 224–215. | ||
Saito K, Toda M, Tomita T, Ogawa K, Yoshida K. Surgical results of an endoscopic endonasal approach for clival chordomas. Acta Neurochir (Wien). 2012;154(5):879–886. | ||
Fraser JF, Nyquist GG, Moore N, Anand VK, Schwartz TH. Endoscopic endonasal transclival resection of chordomas: operative technique, clinical outcome, and review of the literature. J Neurosurg. 2010;112(5):1061–1069. | ||
Dehdashti AR, Karabatsou K, Ganna A, Witterick I, Gentili F. Expanded endoscopic endonasal approach for treatment of clival chordomas: early results in 12 patients. Neurosurgery. 2008;63(2):299–307; discussion 307–309. | ||
Jiang WH, Zhao SP, Xie ZH, Zhang H, Zhang J, Xiao JY. Endoscopic resection of chordomas in different clival regions. Acta Otolaryngol. 2009;129(1):71–83. | ||
Zhang Q, Kong F, Yan B, Ni Z, Liu H. Endoscopic endonasal surgery for clival chordoma and chondrosarcoma. ORL J Otorhinolaryngol Relat Spec. 2008;70(2):124–129. | ||
Frank G, Sciarretta V, Calbucci F, Farneti G, Mazzatenta D, Pasquini E. The endoscopic transnasal transsphenoidal approach for the treatment of cranial base chordomas and chondrosarcomas. Neurosurgery. 2006;59(1 Suppl 1):ONS50–ONS57. | ||
Fatemi N, Dusick JR, Gorgulho AA, et al. Endonasal microscopic removal of clival chordomas. Surg Neurol. 2008;69(4):331–338. | ||
Fernandez-Miranda JC, Gardner PA, Rastelli MM Jr, et al. Endoscopic endonasal transcavernous posterior clinoidectomy with interdural pituitary transposition. J Neurosurg. 2014;121(1):91–99. | ||
Komotar RJ, Starke RM, Raper DM, Anand VK, Schwartz TH. The endoscope-assisted ventral approach compared with open microscope-assisted surgery for clival chordomas. World Neurosurg. 2011;76(3–4):318–327; discussion 259–362. | ||
Koutourousiou M, Gardner PA, Tormenti MJ, et al. Endoscopic endonasal approach for resection of cranial base chordomas: outcomes and learning curve. Neurosurgery. 2012;71(3):614–624; discussion 624–625. | ||
Jones PS, Aghi MK, Muzikansky A, Shih HA, Barker FG 2nd, Curry WT Jr. Outcomes and patterns of care in adult skull base chordomas from the Surveillance, Epidemiology, and End Results (SEER) database. J Clin Neurosci. 2014;21(9):1490–1496. | ||
Bohman LE, Koch M, Bailey RL, Alonso-Basanta M, Lee JY. Skull base chordoma and chondrosarcoma: influence of clinical and demographic factors on prognosis; a SEER analysis. World Neurosurg. 2014;82(5):806–814. | ||
Oikawa S, Kyoshima K, Goto T, et al. Histological study on local invasiveness of clival chordoma. Case report of autopsy. Acta Neurochir (Wien). 2001;143(10):1065–1069. | ||
Naka T, Boltze C, Kuester D, et al. Intralesional fibrous septum in chordoma: a clinicopathologic and immunohistochemical study of 122 lesions. Am J Clin Pathol. 2005;124(2):288–294. | ||
Jian BJ, Bloch OG, Yang I, Han SJ, Aranda D, Parsa AT. A comprehensive analysis of intracranial chordoma and survival: a systematic review. Br J Neurosurg. 2011;25(4):446–453. | ||
Tzortzidis F, Elahi F, Wright D, Natarajan SK, Sekhar LN. Patient outcome at long-term follow-up after aggressive microsurgical resection of cranial base chordomas. Neurosurgery. 2006;59(2):230–237. | ||
Di Maio S, Rostomily R, Sekhar LN. Current surgical outcomes for cranial base chordomas: cohort study of 95 patients. Neurosurgery. 2012;70(6):1355–1360. | ||
Pearlman AW, Friedman M. Radical radiation therapy of chordoma. Am J Roentgenol Radium Ther Nucl Med. 1970;108(2):332–341. | ||
Munzenrider JE, Liebsch NJ. Proton therapy for tumors of the skull base. Strahlenther Onkol. 1999;175 Suppl 2:57–63. | ||
Brada M, Pijls-Johannesma M, De Ruysscher D. Current clinical evidence for proton therapy. Cancer J. 2009;15(4):319–324. | ||
Hug EB, Loredo LN, Slater JD, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91(3):432–439. | ||
Castro JR, Linstadt DE, Bahary JP, et al. Experience in charged particle irradiation of tumors of the skull base: 1977–1992. Int J Radiat Oncol Biol Phys. 1994;29(4):647–655. | ||
Amichetti M, Cianchetti M, Amelio D, Enrici RM, Minniti G. Proton therapy in chordoma of the base of the skull: a systematic review. Neurosurg Rev. 2009;32(4):403–416. | ||
Schulz-Ertner D, Karger CP, Feuerhake A, et al. Effectiveness of carbon ion radiotherapy in the treatment of skull-base chordomas. Int J Radiat Oncol Biol Phys. 2007;68(2):449–457. | ||
Ostroumov E, Hunter CJ. The role of extracellular factors in human metastatic chordoma cell growth in vitro. Spine (Phila Pa 1976). 2007;32(26):2957–2964. | ||
Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol. 2010;7(1):37–43. | ||
Takahashi S, Kawase T, Yoshida K, Hasegawa A, Mizoe JE. Skull base chordomas: efficacy of surgery followed by carbon ion radiotherapy. Acta Neurochir (Wien). 2009;151(7):759–769. | ||
Nikoghosyan AV, Karapanagiotou-Schenkel I, Münter MW, Jensen AD, Combs SE, Debus J. Randomised trial of proton vs carbon ion radiation therapy in patients with chordoma of the skull base, clinical phase III study HIT-1-Study. BMC Cancer. 2010;10:607. | ||
Mima M, Demizu Y, Jin D, et al. Particle therapy using carbon ions or protons as a definitive therapy for patients with primary sacral chordoma. Br J Radiol. 2014;87(1033):20130512. | ||
Peeters A, Grutters JP, Pijls-Johannesma M, et al. How costly is particle therapy? Cost analysis of external beam radiotherapy with carbon-ions, protons and photons. Radiother Oncol. 2010;95(1):45–53. | ||
Torres MA, Chang EL, Mahajan A, et al. Optimal treatment planning for skull base chordoma: photons, protons, or a combination of both? Int J Radiat Oncol Biol Phys. 2009;74(4):1033–1039. | ||
Feuvret L, Noel G, Weber DC, et al. A treatment planning comparison of combined photon-proton beams versus proton beams-only for the treatment of skull base tumors. Int J Radiat Oncol Biol Phys. 2007;69(3):944–954. | ||
Bugoci DM, Girvigian MR, Chen JC, Miller MM, Rahimian J. Photon-based fractionated stereotactic radiotherapy for postoperative treatment of skull base chordomas. Am J Clin Oncol. 2013;36(4):404–410. | ||
Debus J, Schulz-Ertner D, Schad L, et al. Stereotactic fractionated radiotherapy for chordomas and chondrosarcomas of the skull base. Int J Radiat Oncol Biol Phys. 2000;47(3):591–596. | ||
Kano H, Iqbal FO, Sheehan J, et al. Stereotactic radiosurgery for chordoma: a report from the North American Gamma Knife Consortium. Neurosurgery. 2011;68(2):379–389. | ||
Kano H, Lunsford LD. Stereotactic radiosurgery of intracranial chordomas, chondrosarcomas, and glomus tumors. Neurosurg Clin N Am. 2013;24(4):553–560. | ||
Krishnan S, Foote RL, Brown PD, Pollock BE, Link MJ, Garces YI. Radiosurgery for cranial base chordomas and chondrosarcomas. Neurosurgery. 2005;56(4):777–784. | ||
Liu AL, Wang ZC, Sun SB, Wang MH, Luo B, Liu P. Gamma knife radiosurgery for residual skull base chordomas. Neurol Res. 2008;30(6):557–561. | ||
Hasegawa T, Ishii D, Kida Y, Yoshimoto M, Koike J, Iizuka H. Gamma knife surgery for skull base chordomas and chondrosarcomas. J Neurosurg. 2007;107(4):752–757. | ||
Terahara A, Niemierko A, Goitein M, et al. Analysis of the relationship between tumor dose inhomogeneity and local control in patients with skull base chordoma. Int J Radiat Oncol Biol Phys. 1999;45(2):351–358. | ||
O’Connell JX, Renard LG, Liebsch NJ, Efird JT, Munzenrider JE, Rosenberg AE. Base of skull chordoma. A correlative study of histologic and clinical features of 62 cases. Cancer. 1994;74(8):2261–2267. | ||
Potluri S, Jefferies SJ, Jena R, et al. Residual postoperative tumour volume predicts outcome after high-dose radiotherapy for chordoma and chondrosarcoma of the skull base and spine. Clin Oncol (R Coll Radiol). 2011;23(3):199–208. | ||
Ares C, Hug EB, Lomax AJ, et al. Effectiveness and safety of spot scanning proton radiation therapy for chordomas and chondrosarcomas of the skull base: first long-term report. Int J Radiat Oncol Biol Phys. 2009;75(4):1111–1118. | ||
Horbinski C, Oakley GJ, Cieply K, et al. The prognostic value of Ki-67, p53, epidermal growth factor receptor, 1p36, 9p21, 10q23, and 17p13 in skull base chordomas. Arch Pathol Lab Med. 2010;134(8):1170–1176. | ||
Kitamura Y, Sasaki H, Kimura T, et al. Molecular and clinical risk factors for recurrence of skull base chordomas: gain on chromosome 2p, expression of brachyury, and lack of irradiation negatively correlate with patient prognosis. J Neuropathol Exp Neurol. 2013;72(9):816–823. | ||
Singhal N, Kotasek D, Parnis FX. Response to erlotinib in a patient with treatment refractory chordoma. Anticancer Drugs. 2009;20(10):953–955. | ||
Vujovic S, Henderson S, Presneau N, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209(2):157–165. | ||
Romeo S, Hogendoorn PC. Brachyury and chordoma: the chondroid-chordoid dilemma resolved? J Pathol. 2006;209(2):143–146. | ||
Schwab JH, Boland PJ, Agaram NP, et al. Chordoma and chondrosarcoma gene profile: implications for immunotherapy. Cancer Immunol Immunother. 2009;58(3):339–349. | ||
Presneau N, Shalaby A, Ye H, et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. J Pathol. 2011;223(3):327–335. | ||
Hsu W, Mohyeldin A, Shah SR, et al. Generation of chordoma cell line JHC7 and the identification of brachyury as a novel molecular target. J Neurosurg. 2011;115(4):760–769. | ||
Fernando RI, Litzinger M, Trono P, Hamilton DH, Schlom J, Palena C. The T-box transcription factor brachyury promotes epithelial-mesenchymal transition in human tumor cells. J Clin Invest. 2010;120(2):533–544. | ||
Nelson AC, Pillay N, Henderson S, et al. An integrated functional genomics approach identifies the regulatory network directed by brachyury (T) in chordoma. J Pathol. 2012;228(3):274–285. | ||
Nibu Y, José-Edwards DS, Di Gregorio A. From notochord formation to hereditary chordoma: the many roles of brachyury. Biomed Res Int. 2013;2013:826435. | ||
Yang C, Schwab JH, Schoenfeld AJ, et al. A novel target for treatment of chordoma: signal transducers and activators of transcription 3. Mol Cancer Ther. 2009;8(9):2597–2605. | ||
Le LP, Nielsen GP, Rosenberg AE, et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PloS One. 2011;6(5):e18846. | ||
Shalaby AA, Presneau N, Idowu BD, et al. Analysis of the fibroblastic growth factor receptor-RAS/RAF/MEK/ERK-ETS2/brachyury signalling pathway in chordomas. Mod Pathol. 2009;22(8):996–1005. | ||
Pillay N, Plagnol V, Tarpey PS, et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet. 2012;44(11):1185–1187. | ||
Varga P, Fisher C, Rhines L, et al. Prognostic significance of T gene SNP s2305089 in individuals with spinal column chordoma. Poster presented at: AACR Annual Meeting 2014; April 5–9, 2014; San Diego, CA. | ||
Zhang L, Guo S, Schwab JH, et al. Tissue microarray immunohistochemical detection of brachyury is not a prognostic indicator in chordoma. PloS One. 2013;8(9):e75851. | ||
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. | ||
Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–1449. | ||
Hu Y, Mintz A, Shah SR, Quinones-Hinojosa A, Hsu W. The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis. 2014;35(7):1491–1499. | ||
Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11(11):805–812. | ||
Palena C, Polev DE, Tsang KY, et al. The human T-box mesodermal transcription factor brachyury is a candidate target for T-cell-mediated cancer immunotherapy. Clin Cancer Res. 2007;13(8):2471–2478. | ||
Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–370. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Shalaby A, Presneau N, Ye H, et al. The role of epidermal growth factor receptor in chordoma pathogenesis: a potential therapeutic target. J Pathol. 2011;223(3):336–346. | ||
Fasig JH, Dupont WD, LaFleur BJ, Olson SJ, Cates JM. Immunohistochemical analysis of receptor tyrosine kinase signal transduction activity in chordoma. Neuropathol Appl Neurobiol. 2008;34(1):95–104. | ||
Weinberger PM, Yu Z, Kowalski D, et al. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch Otolaryngol Head Neck Surg. 2005;131(8):707–711. | ||
Ptaszyński K, Szumera-Ciećkiewicz A, Owczarek J, et al. Epidermal growth factor receptor (EGFR) status in chordoma. Pol J Pathol. 2009;60(2):81–87. | ||
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. | ||
Lindén O, Stenberg L, Kjellén E. Regression of cervical spinal cord compression in a patient with chordoma following treatment with cetuximab and gefitinib. Acta Oncol. 2009;48(1):158–159. | ||
Launay SG, Chetaille B, Medina F, et al. Efficacy of epidermal growth factor receptor targeting in advanced chordoma: case report and literature review. BMC Cancer. 2011;11:423. | ||
Tamborini E, Virdis E, Negri T, et al. Analysis of receptor tyrosine kinases (RTKs) and downstream pathways in chordomas. Neuro Oncol. 2010;12(8):776–789. | ||
Tamborini E, Miselli F, Negri T, et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res. 2006;12(23):6920–6928. | ||
Casali PG, Messina A, Stacchiotti S, et al. Imatinib mesylate in chordoma. Cancer. 2004;101(9):2086–2097. | ||
Orzan F, Terreni MR, Longoni M, et al. Expression study of the target receptor tyrosine kinase of Imatinib mesylate in skull base chordomas. Oncol Rep. 2007;18(1):249–252. | ||
Stacchiotti S, Marrari A, Tamborini E, et al. Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol. 2009;20(11):1886–1894. | ||
Stacchiotti S, Longhi A, Ferraresi V, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30(9):914–920. | ||
Schwab J, Antonescu C, Boland P, et al. Combination of PI3K/mTOR inhibition demonstrates efficacy in human chordoma. Anticancer Res. 2009;29(6):1867–1871. | ||
Presneau N, Shalaby A, Idowu B, et al. Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway. Br J Cancer. 2009;100(9):1406–1414. | ||
Han S, Polizzano C, Nielsen GP, Hornicek FJ, Rosenberg AE, Ramesh V. Aberrant hyperactivation of akt and mammalian target of rapamycin complex 1 signaling in sporadic chordomas. Clin Cancer Res. 2009;15(6):1940–1946. | ||
Davies JM, Robinson AE, Cowdrey C, et al. Generation of a patient-derived chordoma xenograft and characterization of the phosphoproteome in a recurrent chordoma. J Neurosurg. 2014;120(2):331–336. | ||
Shrager J, Tenenbaum JM. Rapid learning for precision oncology. Nat Rev Clin Oncol. 2014;11(2):109–118. | ||
Ricci-Vitiani L, Runci D, D’Alessandris QG, et al. Chemotherapy of skull base chordoma tailored on responsiveness of patient-derived tumor cells to rapamycin. Neoplasia. 2013;15(7):773–782. | ||
Akhavan-Sigari R, Gaab MR, Rohde V, et al. Expression of vascular endothelial growth factor receptor 2 (VEGFR-2), inducible nitric oxide synthase (iNOS), and Ki-M1P in skull base chordoma: a series of 145 tumors. Neurosurg Rev. 2014;37(1):79–88. | ||
Asklund T, Sandström M, Shahidi S, Riklund K, Henriksson R. Durable stabilization of three chordoma cases by bevacizumab and erlotinib. Acta Oncol. 2014;53(7):980–984. | ||
Di Maio S, Kong E, Yip S, Rostomily R. Converging paths to progress for skull base chordoma: review of current therapy and future molecular targets. Surg Neurol Int. 2013;4:72. | ||
Hof H, Welzel T, Debus J. Effectiveness of cetuximab/gefitinib in the therapy of a sacral chordoma. Onkologie. 2006;29(12):572–574. | ||
Stacchiotti S, Tamborini E, Lo Vullo S, et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann Oncol. 2013;24(7):1931–1936. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.