Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Novel sulpiride-loaded solid lipid nanoparticles with enhanced intestinal permeability
Authors Ibrahim W, AlOmrani A, Yassin AE ![]()
Received 12 September 2013
Accepted for publication 3 November 2013
Published 19 December 2013 Volume 2014:9(1) Pages 129—144
DOI https://doi.org/10.2147/IJN.S54413
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Waheed M Ibrahim,1 Abdullah H AlOmrani,2 Alaa Eldeen B Yassin3
1Drug Sector, Saudi Food and Drug Authority, 2Department of Pharmaceutics, College of Pharmacy, King Saud University, 3Department of Pharmaceutical Sciences, College of Pharmacy, King Saud bin Abdulaziz University for Health Sciences, National Guard Health Affairs, Riyadh, Saudi Arabia
Background: Solid lipid nanoparticles (SLN), novel drug delivery carriers, can be utilized in enhancing both intestinal permeability and dissolution of poorly absorbed drugs. The aim of this work was to enhance the intestinal permeability of sulpiride by loading into SLN.
Methods: A unique ultrasonic melt-emulsification method with minimum stress conditions was used for the preparation of SLN. The mixture of the drug and the melted lipids was simply dispersed in an aqueous solution of a surfactant at a temperature that was 10°C higher than the melting points of the lipids using probe sonication, and was then simultaneously dispersed in cold water. Several formulation parameters were optimized, including the drug-to-lipid ratio, and the types of lipids and surfactants used. The produced SLN were evaluated for their particle size and shape, surface charge, entrapment efficiency, crystallinity of the drug and lipids, and the drug release profile. The rat everted sac intestine model was utilized to evaluate the change in intestinal permeability of sulpiride by loading into SLN.
Results: The method adopted allowed successful preparation of SLN with a monodispersed particle size of 147.8–298.8 nm. Both scanning electron microscopic and atomic force microscopic images showed uniform spherical particles and confirmed the sizes determined by the light scattering technique. Combination of triglycerides with stearic acid resulted in a marked increase in zeta potential, entrapment efficiency, and drug loading; however, the particle size was increased. The type of surfactant used was critical for particle size, charge, drug loading, and entrapment efficiency. Generally, the in vitro release profile demonstrated by all formulations showed the common biphasic mode with a varying degree of burst release. The everted sac model showed markedly enhanced sulpiride permeability in the case of the SLN-loaded formulation. The in situ results showed a very good correlation with the in vitro release data.
Conclusion: Incorporation of sulpiride into SLN results in enhanced intestinal permeability of sulpiride, that may in turn increase overall oral absorption of the drug. The superior attributes of the prepared SLN, specifically the high particle size uniformity and drug loading capacity, is considered novel, especially given the simplicity and modest nature of the sonication method used.
Keywords: ultrasonic melt-emulsification, everted sacs, atomic force microscopy, Dynasan®, stearic acid
Introduction
Sulpiride, a selective antagonist of central dopamine receptors and used as an antipsychotic, is classified under class IV in the biopharmaceutical classification system. This means that sulpiride has poor aqueous solubility and limited intestinal permeability.1 Consequently, high doses of the drug are needed to treat patients, resulting in problematic side effects, including sleeping disturbances, overstimulation, agitation, and mild extrapyramidal and cardiovascular effects.2
Solid lipid nanoparticles (SLN) have been attracting interest as a novel drug delivery carrier since their introduction by Müller et al for oral drug delivery in the mid 1990s.3 These particles are mainly formed by cooling dispersions of melted lipids in aqueous surfactant solution. The most commonly used lipids include triglyceride esters of hydrogenated fatty acids such as tristearin, partial glycerides such as Imwitor®, and fatty acids such as stearic acid or hydrogenated palm oil (Dynasan® P60 or Softisan® 154).4,5 Emulsifiers are used to stabilize the lipid dispersion. Use of a combination of emulsifiers can increase their efficiency by preventing particle agglomeration.4
SLN can be utilized in a number of applications, including improved drug solubility,6 controlled drug release, and drug targeting,4,7,8 with better permeability through the blood-brain barrier, a reduction in the therapeutic dose, enhanced bioavailability,9 and increased drug stability during long-term storage.4,8 SLN are amenable to both lyophilization and steam sterilization, so have fewer storage and drug leakage problems. SLN improve protein stability, prevent proteolytic degradation, and have been recommended for administration by a number of methods including the oral, parenteral, topical,4,7,10 and pulmonary routes.7,10
The adhesive properties of nanoparticles are reported to increase drug bioavailability and reduce or minimize erratic absorption.11 Several mechanisms have been suggested to account for the absorption of nanoparticles through the intestinal mucosa, ie, through the Peyer’s patches, intracellular uptake, or the paracellular pathway. The stability of SLN on contact with gastrointestinal fluids is a critical issue, because a particle size in the nano range maximizes the surface area available for enzymatic degradation.12
Dependence of the drug loading capacity on solubility of the drug in the lipid, the high burst release, and the relatively high water content of the dispersions (70%–99.9%) are considered shortcomings in the performance of SLN, so need improvement.10,13,14 Approaches to overcome these limitations include the incorporation of small amounts of oils within the solid lipid to increase the number of imperfections in the crystal lattice of the lipid, thereby avoiding expulsion of the loaded drug onto the surface. Combinations of spatially different lipids may be used to provide more room for accommodation of guest molecules and thus improve the loading capacity.
The aim of this work was to enhance the intestinal permeability of sulpiride by its incorporation into an optimized SLN formulation with a small particle size and high drug entrapment efficiency.
Materials and methods
The lipids Dynasan® 118, Softisan® 154, and Imwitor® 900K were acquired from Sasol Germany GmbH (Witten, Germany). Stearic acid, Tween 80, Span 80, and Poloxamer 188 were obtained from Sigma-Aldrich Chemical Company (St Louis, MO, USA). All other materials were of analytical grade and used as received.
Preparation of sulpiride-loaded SLN
Based on the ultrasonic melt-emulsification method, certain weights of lipids were melted by heating at approximately 10°C above the lipid melting point. Certain weights of sulpiride were dissolved in the melted lipids. An aqueous solution of a 1% surfactant (Tween 80, Span 80, or Poloxamer 188) and 0.5% sodium dioxycholate, as a cosurfactant, was heated to the same temperature and then mixed with the melted phase, using probe sonication (Bandelin Sonopuls HD 220, Bandelin Electronics, Berlin, Germany) for 3 minutes at 40% voltage efficiency. The emulsion formed was then dispersed in chilled distilled water (containing 1% glucose as a cryoprotector) by stirring for 3 minutes using a magnetic stirrer. The SLN obtained were stabilized using a freeze dryer (Alpha 1–4 LD Plus, Martin Christ Gefriertrocknugsanlagen GmbH, Osterode am Harz, Germany) at −60°C for 3 days.4,10,15 The composition of each SLN formulation is shown in Table 1.
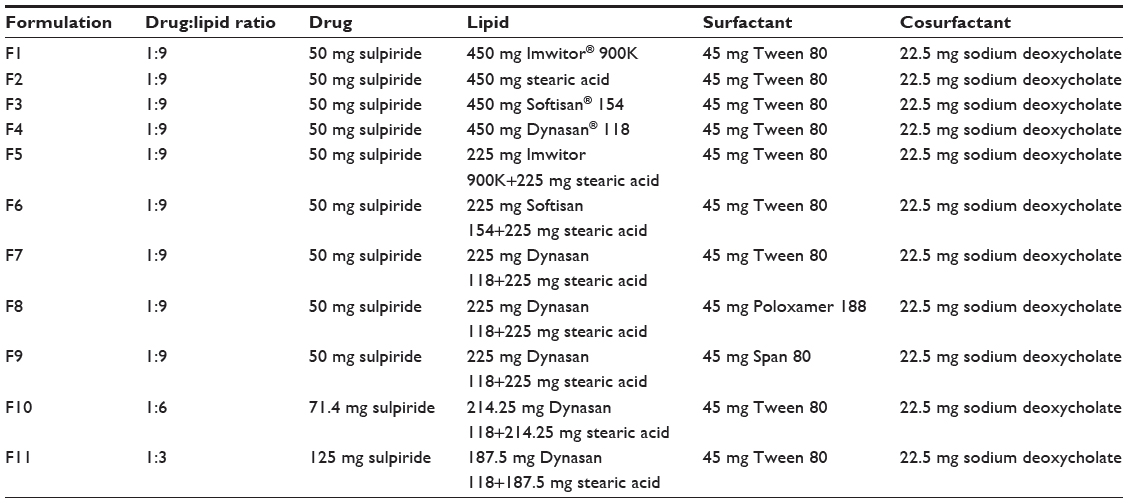 | Table 1 Composition for each of the prepared solid lipid nanoparticle formulations |
The composition of the formulation was intentionally varied to enable investigation of the effect of the type of lipid used (F1, F2, F3, and F4), combination with stearic acid (F5, F6, and F7), type of surfactant used (F7, F8, and F9), and the drug-to-lipid ratio (F7, F10, and F11) on the properties of the prepared SLN.
Analytical method
A simple validated high-pressure liquid chromatography (HPLC) method for analyzing sulpiride was employed, applying minor modifications and utilizing a complete system from Jasco (Tokyo, Japan).16 Metoclopramide was used as the internal standard. The HPLC system consisted of a 5 μm (4.6 × 100 mm) cyanopropyl column and a freshly prepared mobile phase consisting of 50% (5 mmol) phosphate buffer, 25% methanol, and 25% acetonitrile. The flow rate was adjusted to 1 mL per minute and the ultraviolet detector wave length was set at 232 nm. This method enabled determination of concentrations in the range of 0.1–20 μg/mL.
Evaluation of prepared SLN
Measurement of particle size
The mean particle size and the polydispersity index of the size distribution for each SLN formulation were measured by photon correlation spectroscopy, using a 90 Plus particle size analyzer (Brookhaven Instruments Corporation, Holtsville, NY, USA). The SLN dispersions were subjected to a dilution of 1:1,000 using distilled water. The measurements were performed at 25°C at an angle of detection of 90°.
Measurement of zeta potential
The zeta potential for samples from each formulation, diluted to the concentration of 1:1,000 lipid dispersion in distilled water, were determined using a 90 Plus particle size analyzer (Brookhaven Instruments Corporation).
Measurement of drug entrapment efficiency and drug loading
Samples from each SLN formulation were centrifuged at 50,000 rpm and 4°C for 30 minutes using an Optima MAX-E ultracentrifuge (Beckman Coulter, Inc., Nyon, Switzerland). The amount of nonentrapped sulpiride in the supernatant obtained after centrifugation was determined using HPLC. The percent entrapment efficiency (%EE) and percent drug loading (%DL) were calculated according to Equations [1] and [2], respectively, where Wtotal drug is the total amount used, Wfree drug is the free amount present in the aqueous phase of supernatant, and Wtotal lipid is the total amount of lipid used.
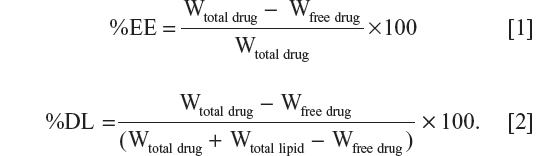
Differential scanning calorimetry
The thermal behavior of the freeze-dried SLN formulations and sulpiride powder was investigated by differential scanning calorimetry using a Shimadzu DSC-60 (Shimadzu Corporation, Tokyo, Japan). Samples of 4–7 mg were weighed and a heating rate of 10°C per minute in the range of 25°C–200°C was employed.
Fourier transform infrared spectroscopy
The Fourier transform infrared spectrum for each formulation was compared with that of pure sulpiride and each type of lipid material used in the formulation. The method reported by Yassin et al17 was used, whereby Fourier transform infrared spectra were recorded on a Nicolet 380 FT-IR (Thermo Electron Corporation, Madison, WI, USA) and a potassium bromide disk technique was used. Small amounts of each sample were mixed with potassium bromide in a clean glass pestle and mortar. Compression was applied in order to obtain a disk. The baseline was corrected and the samples were scanned at a wave number ranging from 400 to 4,000 cm−1.
Particle morphology
The morphologic characteristics of the selected SLN were examined under a scanning electron microscope (JSM-6360 LV, JEOL, Tokyo, Japan). The SLN samples were mounted on carbon tape and sputter-coated with a thin gold palladium layer under an argon atmosphere using a gold sputter module in a high-vacuum evaporator (JFC-1100 fine coat ion sputter; JEOL). The coated samples were then scanned and photomicrographs were taken at an acceleration voltage of 20 kV.
The morphologic characteristics of the selected SLN formulations were also investigated using an atomic force microscope (Nanosurf AG Inc, Liestal, Switzerland). The SLN were deposited on mica, and their shape, diameter, and size distribution were examined.
In vitro release profile study
Certain weights from each formulation equivalent to 1 mg of sulpiride were dispersed in 1 mL of medium and placed in dialysis tubing made of seamless regenerated cellulose with a typical molecular weight cutoff of 12,000–14,000 Da. The sealed bags were immersed in a beaker with 40 mL of phosphate buffer at pH 6.8. The beakers were covered with paraffin films and maintained in a shaking water bath (SW22, Julabo, Allentown, PA, USA) at 37°C and a speed of 80 rpm. Next, 1 mL aliquots were withdrawn from the medium at certain time intervals, replaced with an equal volume of fresh buffer, and preheated to 37°C in order to maintain a sink condition. The amount of drug released was determined using HPLC.
Intestinal permeability assessment
The optimum SLN formulation was compared with the sulpiride dispersion using the everted sac technique. Studies were carried out using the intestines of healthy rats weighing 200–250 g. All of the rats were being used for the first time as experimental animals. Each rat was anesthetized with ether, and its abdomen was opened by a midline incision. The jejunum was rapidly removed and divided into segments, each about 5–7 cm in length. All rats were euthanized while still under anesthesia. The isolated segments of jejunum were immersed immediately in oxygenated Krebs solution (pH 6.5). The composition of the Krebs solution was 7 g/L sodium chloride, 0.34 g/L potassium chloride, 1.8 g/L glucose, 0.251 g/L disodium hydrogen phosphate, 0.207 g/L sodium dihydrogen phosphate, and 46.8 mg/L magnesium chloride.18 Tween 80 1% was added to help enhance the wettability of the SLN, simulating the action of bile salts present in the biological intestinal medium.19 Each segment was tied on one side with a braided silk suture, and 0.5 mL of the same medium containing a dispersed amount of sulpiride equivalent to 200 μg of the tested formulation was inserted using a 1 mL syringe.20 Each segment was then tied on the other side. All sealing ligatures were firm enough to prevent leaking but not so tight as to cause damage to the tissue. Each segment was immersed in a 10 mL incubation flask (or tube) containing Krebs solution at 37°C. Aliquots of 1 mL were withdrawn at certain time intervals and replaced with an equal fresh preheated aliquot at 37°C.18,21 Samples were collected from the surrounding medium at 0.5, 1, 1.5, and 2 hours. The amount of drug transported through the intestinal segments was determined using HPLC. The apparent permeability was then calculated based on the following equation:21
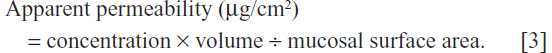
To calculate the mucosal surface area, the intestine was considered a cylinder and the following equation was used:
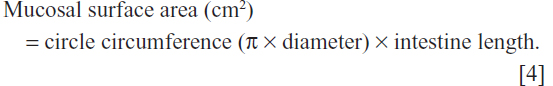
Preparation of the everted sac
Each segment was washed with cold Krebs solution to remove any debris. A narrow glass rod was inserted into one end of the intestine. The sac was then everted by gently pushing the rod through the whole length of the intestine. The glass rod was then removed, and the intestine was placed in Krebs solution at room temperature. A few centimeters of length of the intestine was tied off with a thread, and an open sac was cut from the main length. The test preparation was introduced as described earlier, and the other end was firmly tied with a thread.18,21
Statistical analysis
The difference between obtained values (mean ± standard error of the mean) for the prepared formulations was determined using one-way analysis of variance with a Tukey-Kramer post hoc test, with the paired t-test used to determine a difference between any two formulations. A statistical software pack (GraphPad InStat version 3.05, GraphPad Software, San Diego, CA, USA) was used. A P-value less than 0.05 was deemed to indicate a statistically significant difference.
Results
Particle size and zeta potential
The mean particle size, zeta potential, %EE, and %DL for all the prepared formulations are shown in Table 2. The polydispersity index measures the width of the particle dispersion, providing information on the homogeneity of the particle size distribution of the system. The literature reports that polydispersity indices less than 0.3 are considered ideal and are referred to as indicating a narrow size distribution,22,23 whereas polydispersity values less than 0.1 are referred to as monodispersed.24 Therefore, according to Table 2, all the formulations had a narrow size dispersion. Formulations F2–F8 had a polydispersity index of only 0.005, indicating a very narrow distribution and clear monodispersion. Figures 1–3 show a comparison of the mean particle size, polydispersity index, and zeta potential for all the prepared formulations. The particle size of the prepared SLN ranged from 147.8 nm to 298.8 nm, depending on the lipid type, surfactant type, and drug-to-lipid ratio. F4 containing Dynasan 118 had the lowest mean particle size (147±17.7 nm), whereas the largest mean particle size was seen with F1 (241.3±4.1 nm). The type of lipid used also had a significant (P<0.05) effect on the zeta potential (−33.86 mV in F1, −25.45 mV in F2, −2.21 mV in F3, and −2.84 mV in F4), using one-way analysis of variance. However, no statistically significant difference (P>0.05) was found between F3 and F4 using the paired t-test. A combination of lipids and stearic acid, ie, F5, F6, and F7, increased the particle size of all three formulations. In addition, the zeta potential was increased to the range of the upper 30s (−35.91 mV to −38.35 mV) for these formulations.
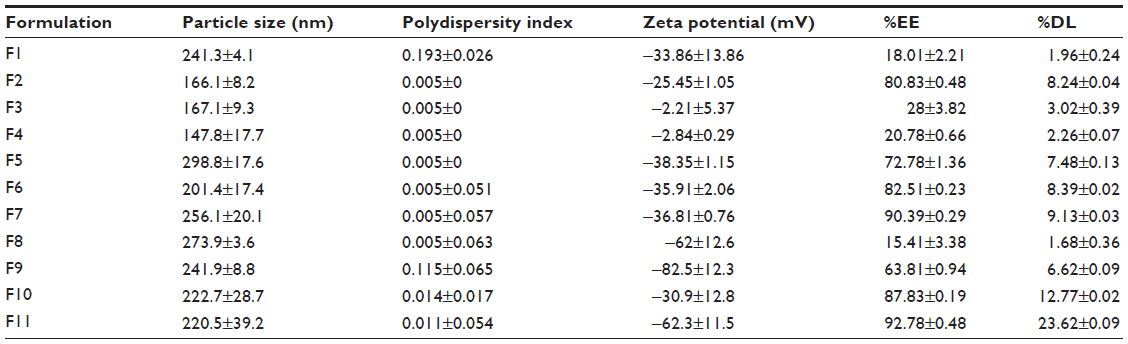 | Table 2 Mean particle size, zeta potential, percent entrapment efficiency, and percent drug loading measurements for all formulations |
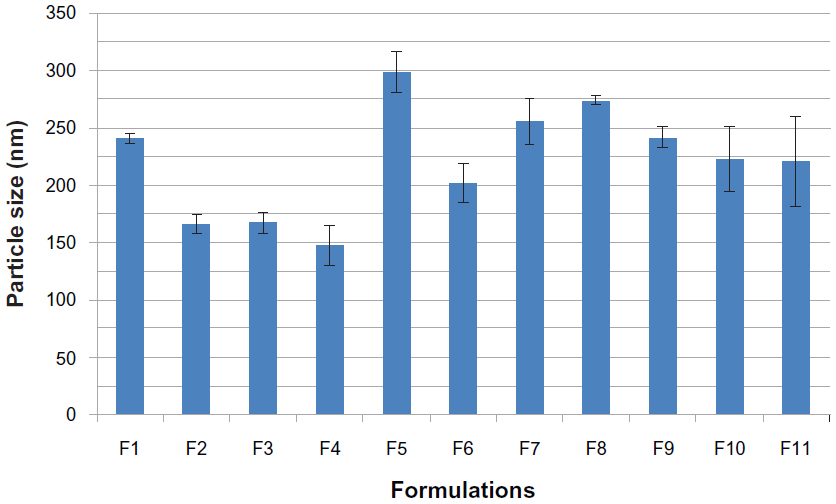 | Figure 1 Histogram of mean particle sizes for all solid lipid nanoparticle formulations. |
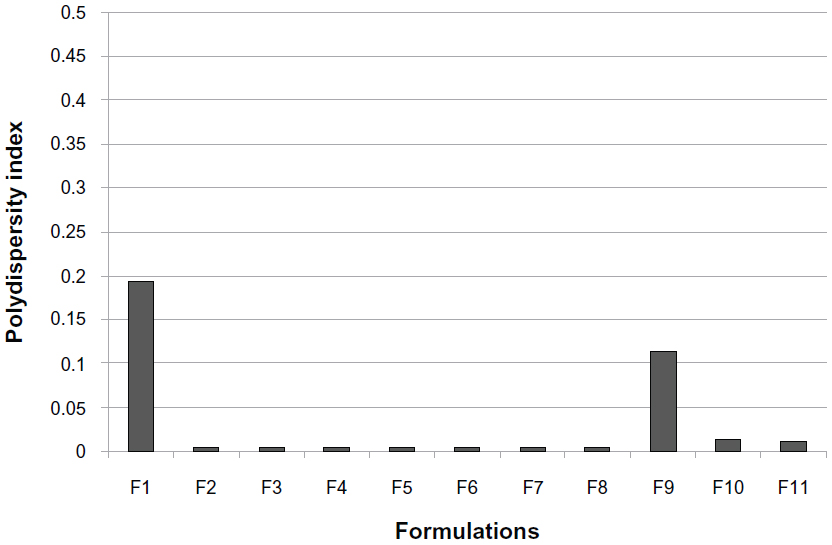 | Figure 2 Histogram of polydispersity index for all solid lipid nanoparticle formulations. |
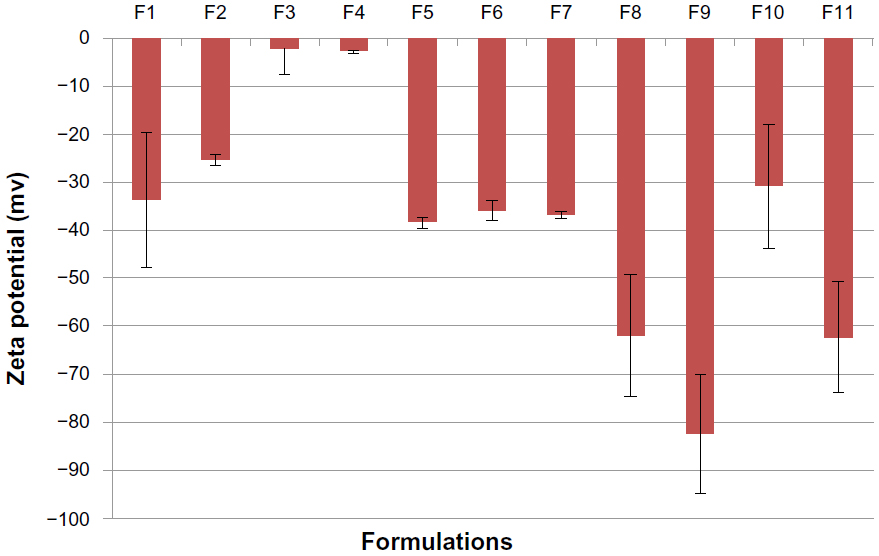 | Figure 3 Histogram of mean zeta potential for all solid lipid nanoparticle formulations. |
Changing the type of surfactant in formulations F7, F8, and F9 did not have a significant effect (P>0.05) on particle size, but a significant change (P<0.05) was seen for the zeta potential using one-way analysis of variance. The particle size changed slightly from 256.1 nm in F7 to 273.9 nm in F8 and to 241.9 nm in F9. The zeta potential increased in F8 (−62.0 mV) and F9 (−82.5 mV) when compared with F7 (−36.81 mV). The effect of the drug-to-lipid ratio is seen when comparing F11 (1:3), F10 (1:6), and F7 (1:9). No significant increase (P>0.05) in particle size was seen (222.7 nm in F10 and 220.5 nm in F11) compared with F7 using one-way analysis of variance. However, the zeta potential increased for F11 (−62.3 mV), which had the lowest drug-to-lipid ratio (1:3).
Drug entrapment efficiency and drug loading
The type of lipid used had a significant effect (P<0.05) on the %EE and %DL using one-way analysis of variance. F2 containing stearic acid alone had the highest %EE (80.83%) and %DL (8.24%). All of the other three formulations containing a single lipid component showed a low %EE and %DL. Combination of lipids and stearic acid (formulations F5, F6, and F7) resulted in a significant increase (P<0.05) in %EE and %DL. The %DL increased in parallel with the %EE when lipids were combined with stearic acid. The %DL values were 1.96%, 3.02%, and 2.26% for formulations containing single lipids, ie, F1, F3, and F4, respectively, and increased to 7.48%, 8.39%, and 9.13%, respectively, for formulations containing a combination of lipids and stearic acid (F5, F6, and F7). The %EE and %DL were also significantly affected (P<0.05) by the type of surfactant used, because the %EE and %DL were reduced to 15.41% (%EE) and 1.68% (%DL) for F8 (containing Poloxamer 188), whereas it was slightly reduced in the case of Span 80 (F9), with values of 63.81% (%EE) and 6.62% (%DL). The effect of the drug-to-lipid ratio can be demonstrated by comparing F11 (1:3), F10 (1:6), and F7 (1:9). No changes were seen in %EE (90.39% in F7, 87.83% in F10, and 92.78% in F11). However, the %DL was markedly increased (P<0.05), with a reduction in lipid content (9.13% in F7, 12.77% in F10, and 23.62% in F11), which was expected.
Differential scanning calorimetry
A comparison of the thermal behavior of the pure drug, Imwitor 900K, Softisan 154, and Dynasan 118 with the thermograms for the different lyophilized SLN formulations was performed using a heat range of 25°C–200°C. The thermograms are shown in Figure 4. A sharp melting endotherm was seen with pure sulpiride at approximately 180.62°C, which is in agreement with the British Pharmacopeia.25 It is reported in the literature that differential scanning calorimetric analysis showing an absence of the melting endotherm of a drug after incorporation into SLN would indicate that the drug was completely solubilized inside the lipid matrix of the SLN and that it was converted to the amorphous form.26 The peak disappeared in all formulations, indicating possible transformation of the drug to the amorphous form. Thermograms for the pure lipids, ie, Imwitor 900K, stearic acid, Softisan 154, and Dynasan 118, showed a characteristic sharp peak at 64.69°C±0.42°C, 71.59°C±0.65°C, 55.03°C±0.33°C, and 73.19°C±0.50°C, respectively. Such peaks appeared in all thermograms for the SLN formulations containing lipids, thereby confirming the solid crystalline state of these lipids inside the prepared formulations.
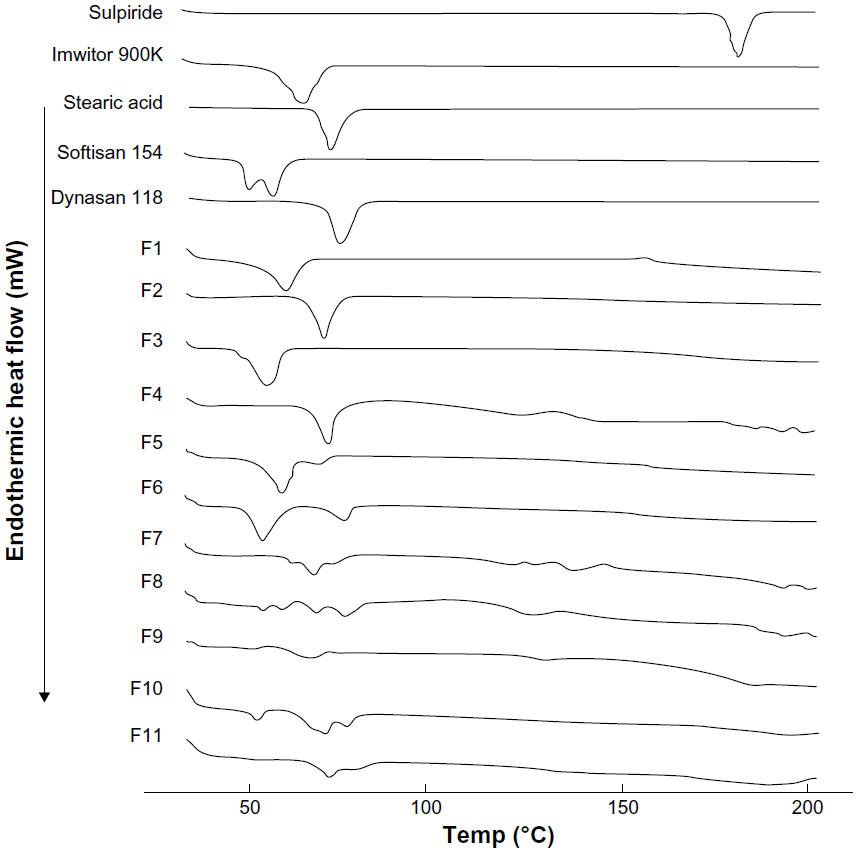 | Figure 4 Differential scanning calorimetric thermograms for sulpiride, Imwitor® 900K, stearic acid, Softisan® 154, Dynasan® 118, and solid lipid nanoparticle formulations (F1–F11). |
Fourier transform infrared spectroscopy
The Fourier transform infrared spectra for sulpiride showed absorption bands with characteristic peaks in the regions of 3,385, 3,213, and 3,082 cm−1, corresponding to the N-H of sulfonamide, amide, and aromatic groups, respectively. The bands at 2,968–2,821 cm−1 represented the C-H of the methylene and methyl groups. The band at 1,643 cm−1 was for the C=O of the amide group. Skeletal stretching of the benzene ring was seen at 1,592 cm−1. The absorption bands at 1,547, 1,334, and 827 cm−1 were assigned to N-H, SO2 and C-H, respectively. The C-O of the methoxy group was seen at 1,172 and 1,150 cm−1. Similar results were reported by Peitre et al and Mohammed et al.27,28
It has been reported that disappearance of the characteristic absorption bands for a drug after incorporation into lipids indicates that the drug has transferred to the amorphous state.16 As shown in Figure 5, the spectra for all the study formulations showed significant changes in the absorption bands for sulpiride, which diminished significantly in the fingerprint region of the drug. This shows that the drug changed to an amorphous state after it had been encapsulated in the lipid.29 These changes could be related to those observed by differential scanning calorimetry. The disappearance of the absorption bands for the lipids in F10 and F11 could be attributed to the smaller amounts of lipids used in these formulations.
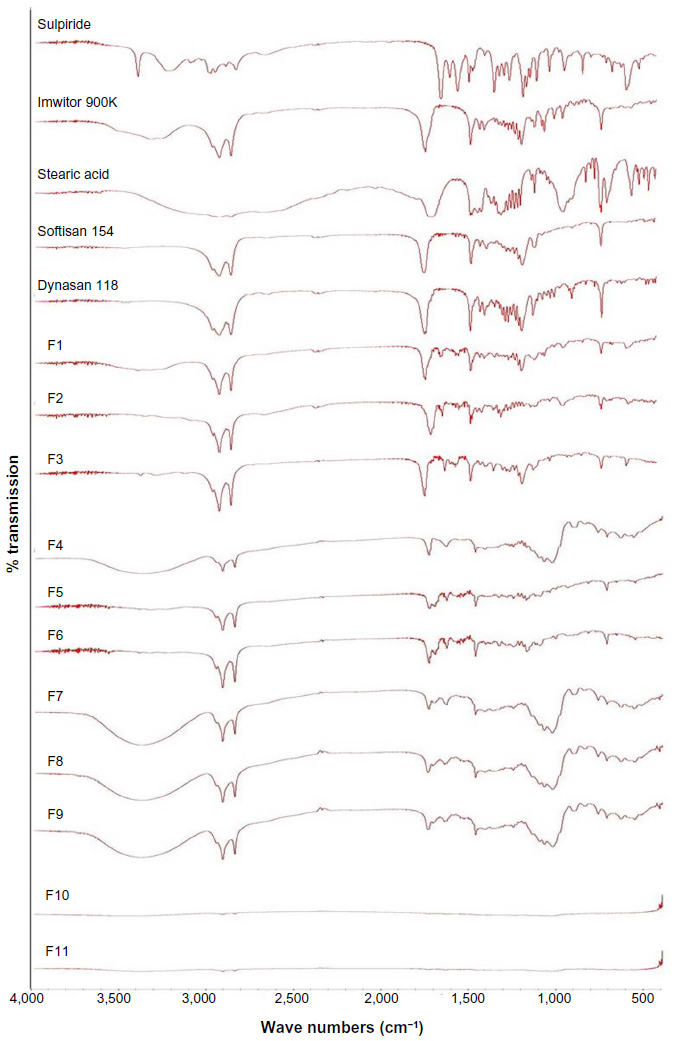 | Figure 5 Fourier transform infrared spectra for sulpiride, Imwitor® 900K, stearic acid, Softisan® 154, Dynasan® 118, and solid lipid nanoparticle formulations (F1–F11). |
Particle morphology
The scanning electron microscopic images of SLN from the F2 and F7 formulations are shown in Figure 6. It is obvious from all the images that the sulpiride-loaded SLN had a spherical and oval shape with the presence of some aggregates. The images for F7 in Figure 6C and D show a larger fraction of particles with smaller sizes. Generally, the particle sizes appeared in the same range as that determined by the light scattering technique.
 | Figure 6 Scanning electron micrographs for F2 and F7 solid lipid nanoparticle formulations. (A and B) fields containing different particle sizes from F2 using 10,000× magnification power, (C) a field containing different particle sizes from F7 using 16,000× magnification power, and (D) a field containing different particle sizes from F7 using higher magnification power (27,000×). |
Figure 7 shows atomic force microscopic images for the SLN particles in F2 deposited on mica. The quality of the images is better than for the scanning electron microscopic images, and enables better investigation of the particle morphology. Some of the particles appeared to be spherical in shape and some had irregular surfaces. The size distribution appeared to be variable, with the majority of particles in the size range of 150–160 nm and a second particle population in the range of 50–60 nm. The line profile passing through three particles with variable size showed that the particle height was in the range of 7–16 nm and proportional to size, thus confirming the spherical nature of the particles.
 | Figure 7 Atomic force microscopic images for the F7 solid lipid nanoparticle formulation on mica. (A) Lower magnification, (B) intermediate magnification, and (C) highest magnification. The blue line represents the topography profile of crossed particles having different sizes. |
In vitro release
A comparison of the release profiles for formulations F1, F2, F3, and F4, containing four different lipids, ie, Imwitor 900K, stearic acid, Softisan 154, and Dynasan 118, respectively, is shown in Figure 8. Except for F4, all the formulations show a high burst release, within the first half hour, with cumulative percent release values of 33.35%±1.52%, 60.02%±1.55%, and 45.79%±3.12% for F1, F2, and F3, respectively. F2, which contained stearic acid, showed the strongest burst effect and the most rapid release rate, with almost 60% of its sulpiride released within the first half hour. On the other hand, F4, which contained Dynasan 118, showed a slow release profile, with a low burst of about 19.61%±4.18% within the first half hour.
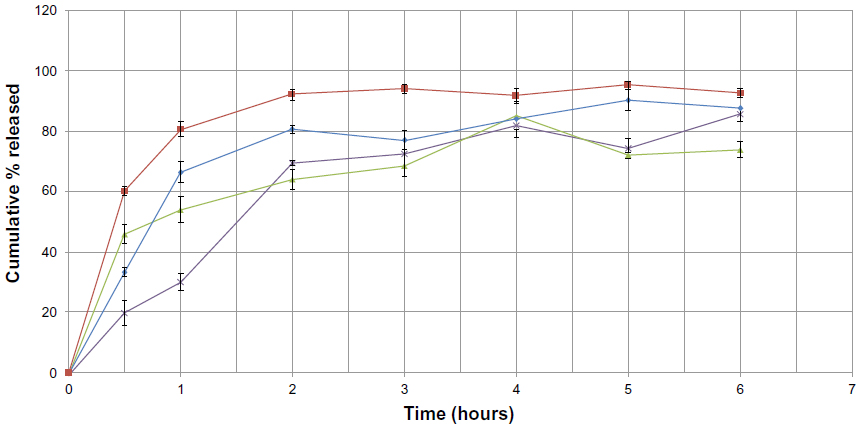 | Figure 8 Cumulative percent release of sulpiride from solid lipid nanoparticle formulations against time, depending on type of lipid used (♦=F1, ■=F2, ▴=F3, X=F4). |
This may be attributed to the rapid dissolution of the sulpiride molecules present in the surface layer of the particles, which was greater in F2 and lower in F4. This supports the idea that lipids containing triglycerides with long chains (such as Dynasan 118 in formula F4) transform more slowly than do triglycerides with short chains (such as Imwitor 900K in F1, stearic acid in F2, and Softisan 154 in F3), from the less stable form (α-form) to the more stable form (β-form), resulting in drug expulsion as it becomes more stable.30 This resulted in a prolonged release profile for F4 and a faster release profile in formulations having shorter chains (F1, F2, and F3). The biphasic drug-release pattern, with a high burst effect followed by a slow release profile, is very common in SLN and has been reported elsewhere.31,32
Another comparison of the release profile for formulations F5 (containing a mixture of stearic acid with Imwitor 900K), F6 (containing a mixture of stearic acid with Softisan 154), and F7 (containing a mixture of stearic acid with Dynasan 118) is shown in Figure 9. A burst effect with values around 71.75%±1.18%, 51.78%±3.23%, and 13.11%±0.62% is seen within the first half hour in formulations F5, F6, and F7, respectively. Formula F2, containing stearic acid, had a %EE of 80.83% and a burst effect of about 60.02%±1.55%, whereas formula F4 containing Dynasan 118 had a %EE of 20.78% and a burst effect of about 19.61%±4.18%. A 1:1 combination of these lipids (F7), having the benefits of both lipids, resulted in a higher %EE of about 90.39% and a decrease in burst effect of about 13.11%±0.62%. An increased %EE was also observed in other formulations containing mixtures with stearic acid, but with higher burst effects (71.75% in F5 and 51.78% in F6 compared with 33.35% in F1 and 45.79% in F3, respectively). These results show that F7 (containing Dynasan 118) enabled better embedding of the drug inside the lipid layers, resulting in a lower burst effect compared with F5 and F6.
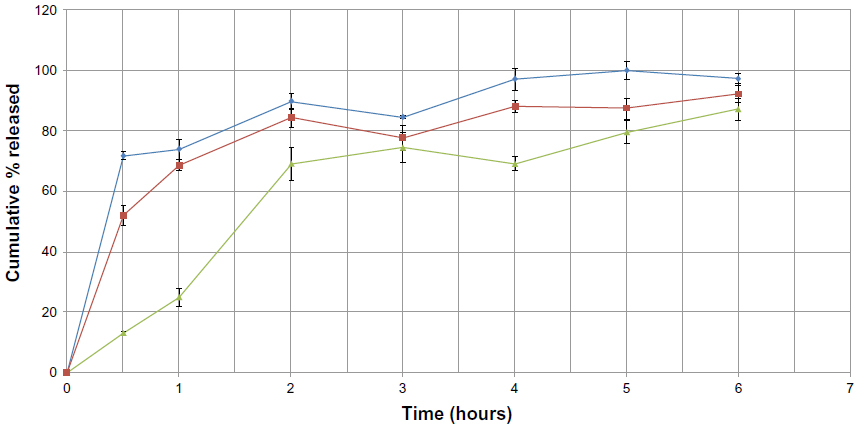 | Figure 9 Cumulative percent release of sulpiride from solid lipid nanoparticle formulations against time, using a combination of lipids (♦=F5, ■=F6, ▴=F7). |
In an attempt to optimize the F7 formulation, the type of surfactant was changed. Poloxamer 188 and Span 80 were used instead of Tween 80 in formulations F8 and F9, respectively. This is shown in Figure 10. A burst effect with values around 28.47%±0.68% and 13.92%±8.63% is seen in formulations F8 and F9, respectively. F8 showed a greater burst effect than F7, whereas similar bursts were seen in F7 and F9. However, the amount of release in F9 was higher than in F7 after one hour. In general, changing the surfactant from Tween 80 to Poloxamer 188 or Span 80 neither decreased the burst effect nor enhanced the %EE.
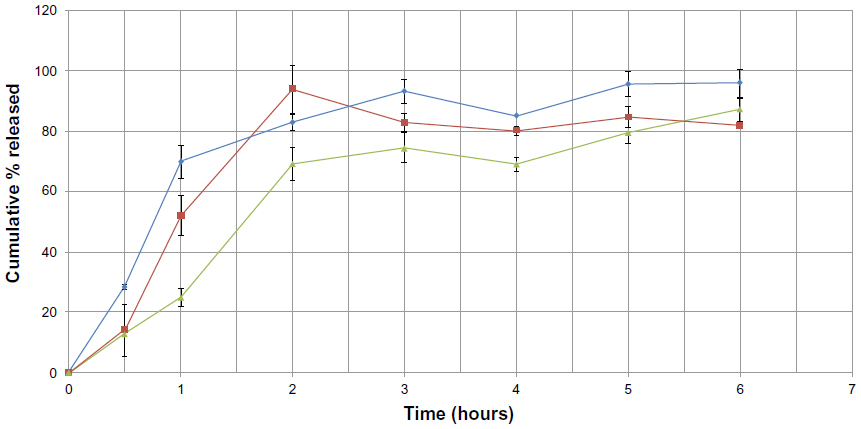 | Figure 10 Cumulative percent release of sulpiride from solid lipid nanoparticle formulations against time, depending on type of surfactant used (▴=F7, ♦=F8, ■=F9). |
Different drug-to-lipid ratios were used in formulations F10 and F11 and were compared with the F7 formulation. F7 had a drug-to-lipid ratio of 1:9, whereas the F10 and F11 formulations had ratios of 1:6 and 1:3, respectively, and are shown in Figure 11. Burst effects of 44.10%±2.61% and 29.45%±2.12% were seen in formulations F10 and F11, respectively. Statistical analysis by one-way analysis of variance showed that there was a significant difference (P<0.05) in the cumulative percentage released from F7, F10, and F11. This may be attributed to the large change in the drug-to-lipid ratio between F7 and F10 and between F7 and F11, which was smaller when comparing F10 with F11. The percentage sulpiride release after 48 hours was less than 60% in formulation F10 and less than 50% in formulation F11. F10 and F11 showed a greater burst effect compared with F7. It was observed that decreasing the lipid ratio in formulation F10 resulted in a decrease in the total amount of sulpiride released to about 56.86%±1.35%. A further decrease in the amount of lipid in formulation F11 resulted in a greater decrease in the total amount of sulpiride to about 44.54%±1.71%. In general, a decrease in the amount of lipid and an increase in the amount of drug in these formulations resulted in a decrease in the total percentage of sulpiride released over a 48-hour test period.
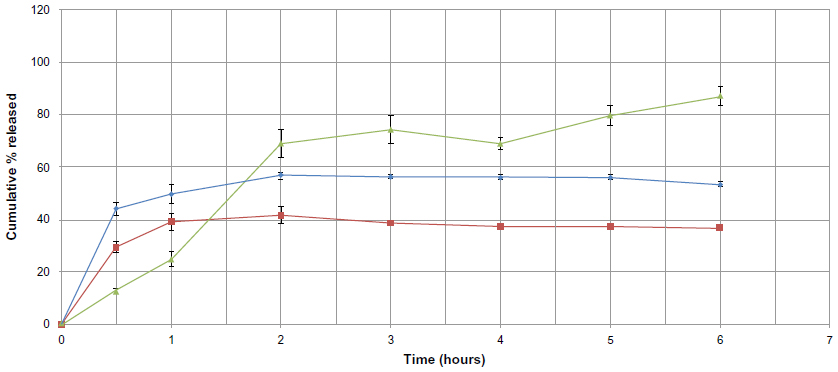 | Figure 11 Cumulative percent release of sulpiride from solid lipid nanoparticle formulations against time, depending on drug to lipid ratio used (▴=F7, ♦=F10, ■=F11). |
Intestinal permeability assessment
The apparent permeability of sulpiride transported from the sulpiride dispersion and formulation F7 using the everted sac technique is shown in Figure 12. In the noneverted control samples, the permeability was gradually increased with time from 1.610±0.076 μg/cm2 until it reached a value of 3.878±0.109 μg/cm2 after 2 hours. The same pattern was seen for the everted sacs, but the magnitude was higher, reaching a value of 6.609±0.947 μg/cm2 after 2 hours. The permeability of sulpiride from F7 was greatly enhanced compared with the control in both noneverted and everted states, reaching values of 7.691±0.159 μg/cm2 and 11.661±1.04 μg/cm2, respectively, after 2 hours. The reduction in permeability in the case of the noneverted sac compared with the everted sac is attributed to the expected efflux of sulpiride, which is a known P-glycoprotein substrate.1,33 Comparing F7 with the control (noneverted F7 with the noneverted control, and everted F7 with the everted control), the permeability was almost doubled and showed a significant difference (P<0.05) in both states.
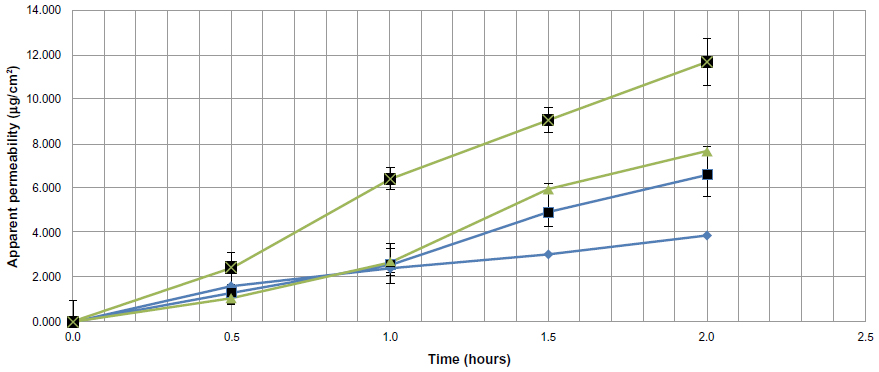 | Figure 12 Comparison between apparent permeability of sulpiride from a sulpiride dispersion and from formulation F7 using noneverted and everted sacs. (♦=Control [noneverted], ■=control [everted], ▴=F7 [noneverted], and ■=F7 [everted]). |
This means that incorporation of sulpiride into SLN resulted in enhanced permeability of sulpiride. This can be possibly attributed to the marked decrease in particle size and enhancement of surface area leading to a higher rate of drug dissolution and diffusion. In addition, the SLN can diffuse through the mucous layer and release the drug directly on the surface of the cell membrane. The higher permeability obtained with F7 in the everted sacs suggests that altering the P-glycoprotein efflux of sulpiride may not occur or may not be the key parameter.
Figures 13 and 14 show the correlation between the first 2 hours of in vitro release data and the cumulative percentage transported through the noneverted and everted rat intestinal sac. A very high correlation was noticed in the case of the noneverted sac, and is shown by a regression factor of R=0.999. In the case of the everted sac, the correlation was also good, with R=0.94.
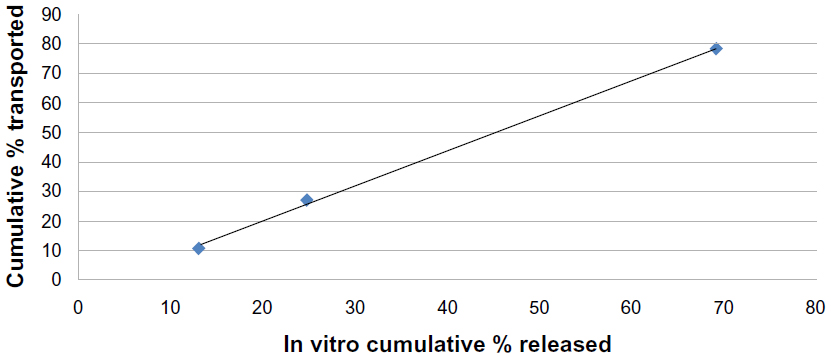 | Figure 13 Correlation between the in vitro cumulative percentage of solid lipid nanoparticles released and cumulative percentage transported across a noneverted rat intestinal sac for F7 (R=0.999). |
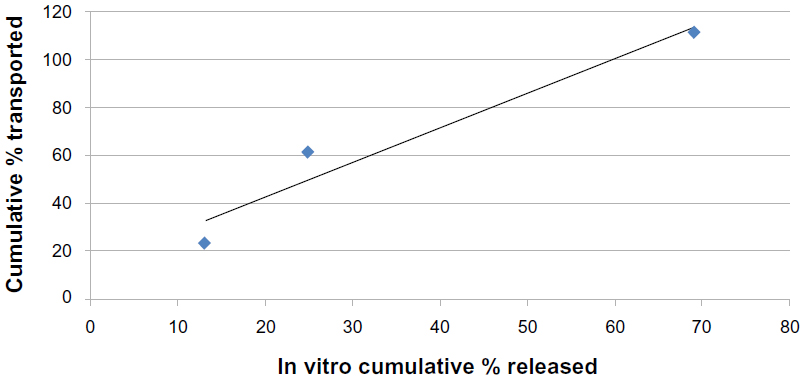 | Figure 14 Correlation between the in vitro cumulative percentage of solid lipid nanoparticles released and cumulative percentage transported across an everted rat intestinal sac for F7 (R=0.94). |
Discussion
The ultrasonic melt-emulsification method adopted in this work has a number of advantages, including simplicity, promptness, and minimum stress conditions. This method enabled the production of SLN with a low particle size ranging from 150 nm to 300 nm, depending mainly on the type of lipid used, and the most interesting features are the very narrow particle size distribution and low polydispersity values. This is in contrast with the work of other researchers claiming problems with poor polydispersity for both the ultrasonic and high shear methods.4,34,35 Bhaskar et al35 prepared flurbiprofen-loaded SLN by applying high shear (12,000 rpm) for 3 minutes followed by 15 minutes of probe sonication. However, the SLN that they prepared had polydispersity values of 0.3–0.5 and a particle size in the range of 250 nm–350 nm.
Another interesting issue regarding the SLN formulations in this work is the use of a low surfactant-to-lipid ratio, ie, around 0.15–1, without affecting the reproducibility of the method. This is considered a very low ratio compared with many reports in the literature.36 Triplett and Rathman36 optimized the ratio of total surfactant to lipids at almost 1.5–1.75 to 1, giving SLN with a particle size in the range of 180–500 nm.
Zeta potential measurements indicated that all the formulations were stable, except for F3 and F4. Usually, a value of −25 mV indicates that a sample is stable, whereas a value of −15 mV indicates the start of gelation phenomena. Muller et al reported similar values for the stability of parenteral fat emulsions.37 Anionic surfactants such as sodium deoxycholate were incorporated to help reduce the interfacial tension and to enhance particle stability by providing negative surface charges with high zeta potential values, consequently enhancing SLN stability and reducing particle aggregation. This was based on the recommendation made by Fillery-Travis et al which has been confirmed by a number of studies.38–40
Both differential scanning calorimetry and Fourier transform infrared spectroscopy indicated a possible transformation of the drug to the amorphous form. This assumption is based on several reports indicating that the absence of the melting endotherm of the drug in differential scanning calorimetry and disappearance of the absorption bands for the drug on Fourier transform infrared spectroscopy, after incorporation or encapsulation in lipid material, indicates that the drug was transformed to the amorphous state.17,26,29
Both scanning electron microscopy and atomic force microscopy were utilized for examining the shapes of the particles. The atomic force microscopic images showed better resolution even at a high magnification power; in addition, the topography of the particles, including particle height, could be determined. This is in agreement with what was reported by Mukherjee et al,41 who mentioned that ultrahigh resolution and the ability to map a sample according to its properties make atomic force microscopy a valuable tool.
The role of Tween 80 appeared to be vital in attaining a high %EE and %DL, given that replacement with Poloxamer 188 or Span 80 resulted in a reduction in %EE and %DL for the prepared SLN formulations. This is in agreement with reports by Abdelbary et al and Kumar et al.42,43 The higher %EE and %DL found in formulations containing stearic acid either alone or in combination with other lipid material is obvious, and can be explained by the higher solubility of sulpiride in acids.2,44 This was confirmed by the rapid dissolution of sulpiride in melted stearic acid during the preparation of SLN. Except for stearic acid, all the other lipids used do not have a free carboxylic group, so all formulations had relatively high %EE values except for F1, F3, and F4, which were free of stearic acid. This is in agreement with reports published by other researchers.45,46
The biphasic mode of release exhibited by all formulations is common to SLN and reported in many publications.17,47–49 Zur Mühlen et al50 discussed the possibility that during solidification of SLN, two layers are formed, ie, an inner core of lipid crystals containing a small amount of drug and an outer shell containing a high amount of drug in the form of a solid solution. Thus, a lack of homogeneity in drug concentration in the layers of SLN, with a drug-rich outer layer, might explain the biphasic mode of release and the relatively high burst release seen with most of the SLN formulations. The degree of the burst effect varies according to the ability of the lipid material to incorporate drug molecules within its crystal lattice. The release profile for sulpiride showed a minimum burst effect with F4 and F7 (both containing glyceryl tristearate, Dynasan 118), suggesting that triglycerides with a single fatty acid and longer chains enable better embedding of the drug inside the lipid layers. This is because triglycerides with long chains are transformed more slowly than are triglycerides with short chains from the less stable form (α-form) to the more stable form (β-form).30 The counterfactual mode of release seen with F10 and F11 compared with F7, containing a high sulpiride-to-lipid ratio, strongly supports the hypothesis of Zur Mühlen et al50 because the increase in drug-to-lipid ratio resulted in increased burst release and a smaller amount of the drug being released within the 48 hours of the test. This means that as the drug ratio increases, more drug goes to the outer shell layer, causing a greater burst effect, and more drug goes to the inner core, which may take longer to be released (more than the time course of our study), as shown by Zur Mühlen and Mehnert.51 These authors reported on 7 weeks of in vitro release of prednisolone from SLN made from different lipids, ie, glycerol behenat and cholesterol. In general, F10 and F11 could be useful when a longer duration is intended with delivery via other routes, such as parenteral, pulmonary, transdermal, and implantation. However, further studies are needed to investigate the release patterns for these formulations over extended periods.
Chitneni et al reported that sulpiride was reasonably stable in Krebs-Ringer buffer solutions throughout the duration of their experiment (2 hours).1 Meanwhile, Kohri et al20 reported that the aqueous solubility of sulpiride was 800 μg/mL, so a lower concentration of sulpiride (200 μg/0.5 mL) was used in their intestinal permeability studies. This low concentration ensured the maintenance of a sink condition throughout the duration of the study. This is similar to what was used in the study reported by Chitneni et al,1 in which 400 μg/mL of sulpiride was used to test the intestinal permeability of the drug when incorporated into a self-microemulsifying drug delivery system. The presence of surfactant, ie, Tween 80, was reported to induce membrane perturbation and P-glycoprotein inhibition that enhanced the permeability of sulpiride.52–54 It was also suggested that Tween 80 modulates P-glycoprotein efflux by inhibition of protein kinase C activity, causing a reduction in phosphorylation of P-glycoprotein and thereby reducing the transport function of P-glycoprotein.55 The results of our study cannot confirm this finding. The effects of this parameter have been neutralized, because the control contained the same ratio of Tween 80. However, sodium deoxycholate, added as a cosurfactant, has been reported to have a partial inhibitory effect on P-glycoprotein efflux,56 which could be related to the enhanced intestinal permeability of sulpiride-loaded SLN.
The high correlation between the in vitro release data and in situ absorption data can be considered a precision indicator for the experimental methods used. Enhancement of intestinal permeability by SLN has been demonstrated in a number of publications.57–59 Incorporation of γ-tocotrienol in SLN was reported to enhance intestinal permeability by three-fold compared with mixed micelles.57 Pandita et al59 showed that incorporation of paclitaxel in SLN increased drug exposure in plasma by ten-fold when compared with free paclitaxel solution. Another study showed that the apparent bioavailability of all-trans retinoic acid (a poorly soluble drug) was significantly increased when incorporated into SLN compared with a suitable aqueous drug dispersion as control.58
Conclusion
Keeping in mind the aim of this work, ie, to enhance the permeability of sulpiride by incorporation into a SLN formulation, it can be said that F7 is the optimum formulation that combines many positive attributes, including a relatively low particle size and polydispersity index, a high %EE, a high zeta potential, and optimal drug release profile, and is therefore potentially suitable for oral administration. Intestinal permeability assessment showed that incorporating sulpiride into an optimized formulation of SLN resulted in enhanced intestinal permeability of sulpiride. Therefore, SLN may be considered as potential candidates for enhancing the oral absorption of sulpiride. However, further studies are recommended to determine the mechanism of this enhancement and to investigate the bioavailability in the whole animal.
Acknowledgments
The authors gratefully acknowledge the generous financial support given by King Abdulaziz City for Science and Technology (grant A-S-12-0970). The authors also extend their thanks to the Department of Pharmaceutics, College of Pharmacy, King Saud University.
Disclosure
The authors report no conflicts of interest in this work.
References
Chitneni M, Peh KK, Darwis D, Abdulkarim M, Abdullah GZ, Qureshi MJ. Intestinal permeability studies of sulpiride incorporated into self microemulsifying drug delivery system (SMEDDS). Pak J Pharm Sci. 2001;24:113–121. | |
MedicinesComplete [homepage on the Internet]. London, UK: Pharmaceutical Press; 2013. Available from: http://www.medicinescomplete.com/. Accessed August 15, 2011. | |
Müller RH, Mehnert W, Lucks JS, et al. Solid lipid nanoparticles (SLN) - An alternative colloidal carrier system for controlled drug delivery. Eur J Pharm Biopharm. 1995;41:62–69. | |
Mehnert W, Mader K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196. | |
MacGregor KJ, Embleton JK, Lacy JE, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25:33–46. | |
Lim S, Kim C. Formulation parameters determining the physicochemical characteristics of solid lipid nanoparticles loaded with all-trans retinoic acid. Int J Pharm. 2002;243:135–146. | |
Jun H, Shi-wen Z. New research on development of solid lipid nanoparticles. Journal of Medical Colleges of PLA. 2007;22:385–390. | |
Kaur IP, Bhandari R, Bhandari S, Kakkar V. Potential of solid lipid nanoparticles in brain targeting. J Control Release. 2008;127:97–109. | |
Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42:11–18. | |
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. | |
Ponchel G, Montisci M, Dembri A, Durrer C, Duchene D. Mucoadhesion of colloidal particulate systems in the gastrointestinal tract. Eur J Pharm Biopharm. 1997;44:25–31. | |
Freitas C, Mullera R. Spray drying of solid lipid nanoparticles (SLN™). Eur J Pharm Biopharm. 1998;46:145–151. | |
Almeida A, Souto E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv Drug Deliv Rev. 2007;59:478–490. | |
Schwarz C, Mehnert W, Lucks JS, Muller RH. Solid lipid nanoparticles (SLN) for controlled drug delivery: production, characterization and sterilization. J Control Release. 1994;30:83–96. | |
Sinha VR, Srivastava S, Goel H, Jindal V. Solid lipid nanoparticles (SLN’S) – trends and implications in drug targeting. Int J Adv Pharma Sci. 2010;1:212–238. | |
Hongjiu Z, Dong L, Xiaomin D, et al. Determination of sulpiride in human serum by HPLC. China Pharmaceuticals. 2005;12:067. | |
Yassin AE, Anwer MK, Mowafy HA, et al. Optimization of 5-fluorouracil solid-lipid nanoparticles: a preliminary study to treat colon cancer. Int J Med Sci. 2010;7:398–408. | |
Alam MA, Al-Jenoobi FI, Al-Mohizea AM. Everted gut sac model as a tool in pharmaceutical research: limitations and applications. J Pharm Pharmacol. 2012;64:326–336. | |
Simões SI, Tapadas JM, Marques CM, et al. Permeabilisation and solubilisation of soybean phosphatidylcholine bilayer vesicles, as membrane models, by polysorbate, Tween 80. Eur J Pharm Sci. 2005;26:307–317. | |
Kohri N, Naasani I, Iseki K, et al. Improving the oral bioavailability of sulpiride by a gastric-retained form in rabbits. J Pharm Pharmacol. 1996;48:371–374. | |
Mahmoud MR. Everted intestinal sacs as in vitro model for assessing absorptivity of L-histidine under the effect of aspirine and gum acacia in male rats. The Egyptian Journal of Hospital Medicine. 2004;16:14–28. | |
Pathak P, Nagarsenker M. Formulation and evaluation of lidocaine lipid nanosystems for dermal delivery. AAPS PharmSciTech. 2009;10:985–992. | |
Kashanian S, Azandaryani AH, Derakhshandeh K. New surface-modified solid lipid nanoparticles using N-glutaryl phosphatidylethanolamine as the outer shell. Int J Nanomedicine. 2011;6:2393–2401. | |
NanoComposix. nanoComposix’s guide to dynamic light scattering measurement and analysis. San Diego, CA, USA: Nanocomposix version 1.3; 2012. Available from: http://nanocomposix.com/sites/default/files/nanoComposix%20Guidelines%20for%20DLS%20Measurements%20and%20Analysis.pdf. Accessed November 7, 2013. | |
British Pharmacopeia Commission. British Pharmacopoeia. London, UK: Stationery Office; 2010. | |
Kotikalapudi LS, Adepu L, VijayaRatna J, et al. Formulation and in vitro characterization of domperidone loaded solid lipid nanoparticles. Int J Pharm Biomed Res. 2012;3:22–29. | |
Peitre D, Stradi R, Nathansohn G. Sulpiride. Analytical Profiles of Drug Substances. 1988;17:607–641. | |
Mohammed GG, Soliman MH. Synthesis, spectroscopic and thermal characterization of sulpiride complexes of iron, manganese, copper, cobalt, nickel, and zinc salts. Antibacterial and antifungal activity. Spectrochim Acta A Mol Biomol Spectrosc. 2010;76(3–4):341–347. | |
Illing A, Unruh T. Investigations on the flow behavior of dispersions of solid triglyceride nanoparticles. Int J Pharm. 2004;284:123–131. | |
Bunjes H, Westesen K, Koch MHJ. Crystallization tendency and polymorphic transitions in triglyceride nanoparticles. Int J Pharm. 1996;129:159–173. | |
Jain A, Jain SK. In vitro and cell uptake studies for targeting of ligand anchored nanoparticles for colon tumors. Eur J Pharm Sci. 2008;35:404–416. | |
Liu J, Gong T, Wang C, Zhong Z, Zhang Z. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: preparation and characterization. Int J Pharm. 2007;340:153–162. | |
Baluom M, Friedman M, Rubinstein R. Improved intestinal absorption of sulpiride in rats with synchronized oral delivery systems. J Control Release. 2001;70:139–147. | |
Bummer PM. Physical chemical considerations of lipid-based oral drug delivery-solid lipid nanoparticles. Crit Rev Ther Drug Carrier Syst. 2004;21:1–20. | |
Bhaskar K, Anbu J, Ravichandiran V, Venkateswarlu V, Rao YM. Lipid nanoparticles for transdermal delivery of flurbiprofen: formulation, in vitro, ex vivo and in vivo studies. Lipids Health Dis. 2009;8:6. | |
Triplett MD II, Rathman JF. Optimization of b-carotene loaded solid lipid nanoparticles preparation using a high shear homogenization technique. J Nanopart Res. 2009;11:601–614. | |
Muller RH, Heinemann S. Fat emulsions for parenteral nutrition. III. Lipofundin MCT/LCT regimens for parenteral nutrition (TPN) with low electrolyte load. Int J Pharm. 1994;101:175–189. | |
Fillery-Travis AJ, Foster LH, Robins MM. Stability of emulsions stabilized by two physiological surfactants: L-a-phosphatidylcholine and sodium taurocholate. Biophys Chem. 1995;54:253–260. | |
Baskin SI, Salem H. Oxidants, Antioxidants, and Free Radicals. Washington, DC: Taylor and Francis; 1997. | |
Bargoni A, Cavalli R, Zara GP, Fundarò A, Caputo O, Gasco MR. Transmucosal transport of tobramycin incorporated in solid lipid nanoparticles (SLN) after duodenal administration to rats. Part II – Tissue distribution. Pharmacol Res. 2001;43:497–502. | |
Mukherjee S, Ray S, Thakur RS. Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci. 2009;71:349–358. | |
Abdelbary G, Fahmy RH. Diazepam-loaded solid lipid nanoparticles: design and characterization. AAPS PharmSciTech. 2009;10:211–219. | |
Kumar V. Effect of surface modification of solid lipid nanoparticles in brain targeting. PhD thesis. Hyderabad, India: Indian Institute of Chemical Technology; 2010. | |
Ministry of Health, Labour and Welfare. The Japanese Pharmacopoeia, Fifteenth Edition. Tokyo, Japan: Ministry of Health, Labour and Welfare; 2006. Available from: http://jpdb.nihs.go.jp/jp15e/jp15e.pdf. Accessed December 3, 2013. | |
Radtke M, Muller RH. Comparison of structural properties of solid lipid nanoparticles (SLN) versus other lipid particles. Proc Int Symp Control Rel Bioact Mater. 2000;27:309–310. | |
Muller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drug. Int J Pharm. 2002;242:121–128. | |
Lukowski G, Hoell A, Dingler A, et al. Fractal surface of solid lipid nanoparticles. Proc Int Symp Control Rel Bioact Mater. 1997;24:631–632. | |
Liu J, Hu W, Chen H, et al. Isotretinoin-loaded solid lipid nanoparticles with skin targeting for topical delivery. Int J Pharm. 2007;328:191–195. | |
Rahman Z, Zidan S, Khan MA. Non-destructive methods of characterization of risperidone solid lipid nanoparticles. Eur J Pharm Biopharm. 2010;76:127–137. | |
Zur Mühlen A, Schwarz C, Mehnert W, Solid lipid nanoparticles (SLN) for controlled drug delivery – drug release and release mechanism. Eur J Pharm Biopharm. 1998;45:149–155. | |
Zur Muhlen A, Mehnert W. Drug release and release mechanism of prednisolone loaded solid lipid nanoparticles. Pharmazie. 1998;53:552–555. | |
Zhang H, Yao M, Morrison RA, Chong S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch Pharm Res. 2003;26:768–772. | |
Shono Y, Nishihara H, Matsuda Y, et al. Modulation of intestinal P-glycoprotein function by Cremophor EL and other surfactants by an in vitro diffusion chamber method using the isolated rat intestinal membranes. J Pharm Sci. 2004;93:877–885. | |
Wu W, Wang Y, Que L. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur J Pharm Biopharm. 2006;63:288–294. | |
Tang J, Sun J, He Z. Self-emulsifying drug delivery systems: strategy for improving oral delivery of poorly soluble drugs. Curr Drug Ther. 2007;2:85–93. | |
Lo YL, Huang JD. Effects of sodium deoxycholate and sodium caprate on the transport of epirubicin in human intestinal epithelial Caco-2 cell layers and everted gut sacs of rats. Biochem Pharmacol. 2000;59:665–672. | |
Abuasal BS, Lucas C, Peyton B, et al. Enhancement of intestinal permeability utilizing solid lipid nanoparticles increases γ-tocotrienol oral bioavailability. Lipids. 2012;47:461–469. | |
Hu L, Tang X, Cui F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J Pharm Pharmacol. 2004;56:1527–1535. | |
Pandita D, Ahuja A, Lather V, et al. Development of lipid-based nanoparticles for enhancing the oral bioavailability of paclitaxel. AAPS PharmSciTech. 2011;12:712–722. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.