Back to Journals » International Journal of Nanomedicine » Volume 14
Novel SN38 derivative-based liposome as anticancer prodrug: an in vitro and in vivo study
Authors Wu C, Zhang Y, Yang D, Zhang J ![]() , Ma J, Cheng D, Chen J, Deng L
, Ma J, Cheng D, Chen J, Deng L
Received 18 September 2018
Accepted for publication 29 November 2018
Published 20 December 2018 Volume 2019:14 Pages 75—85
DOI https://doi.org/10.2147/IJN.S187906
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Chan Wu,1,* Yang Zhang,1,2,* Daoqiu Yang,3,* Jinfeng Zhang,4 Juanjuan Ma,1 Dan Cheng,1 Jianming Chen,1,5 Li Deng1
1Department of Pharmaceutics, School of Pharmacy, Second Military Medical University, Shanghai 200433, People’s Republic of China; 2Department of Pharmacy, Shanghai Tenth People’s Hospital, School of Medicine, Tongji University, Shanghai 200072, People’s Republic of China; 3Department of Dermatology, 107th Hospital of PLA, Yantai 264000, People’s Republic of China; 4Department of Traditional Chinese Medicine, Shanghai Hospital of Chinese Integrative Medicine, Shanghai, People’s Republic of China; 5Tasly Diyi Pharmaceutical Group Co., Ltd, Jiangsu 223002, People’s Republic of China
*These authors contributed equally to this work
Background: Many novel drug delivery systems have been extensively studied to exploit the full therapeutic potential of SN38, which is one of the most potent antitumor analogs of camptothecins (CPTs), whose clinical application is seriously hindered by poor water solubility, low plasmatic stability, and severe toxicity, but results are always unsatisfactory.
Methods: In this study, combining the advantages of prodrug and nanotechnology, a lipophilic prodrug of SN38, SN38-PA, was developed by conjugating palmitic acid to SN38 via ester bond at C10 position, and then the lipophilic prodrug was encapsulated into a long-circulating liposomal carrier by film dispersion method.
Results: The SN38-PA liposomes were characterized as follows: an average particle size of 80.13 nm, an average zeta potential of -33.53 mv, and the entrapment efficiency of 99%. Compared with CPT-11, SN38-PA liposome was more stable in close lactone form, more efficient in conversion rate to SN38, and more potent in cytotoxicity against tumor cells. Pharmacokinetic study showed that SN38-PA liposome had significantly enhanced plasma half-life (t1/2) value of SN38 and increased area under the curve (AUC) of SN38, which was 7.5-fold higher than that of CPT-11. Biodistribution study showed that SN38-PA liposome had more active metabolite SN38 in each tissue. Finally, the pharmacodynamic study showed that SN38-PA liposome had higher antitumor effect with the antitumor inhibition rate of 1.61 times than that of CPT-11.
Conclusion: These encouraging data merit further investigation on this novel SN38-PA liposome.
Keywords: SN38, lipophilic prodrug, long-circulating liposome, pharmacokinetics, biodistribution, pharmacodynamics, CPT-11
Introduction
Camptothecin (CPT) is a naturally occurring alkaloid with potent antineoplastic activity against a broad spectrum of solid tumors (eg, primary and metastatic colon carcinoma, small cell lung carcinoma, and ovarian, breast, pancreatic, and stomach cancers), which inhibits DNA topoisomerase I.1 SN38 (7-ethyl-10-hydroxyl camptothecin) is a semisynthetic analog of CPT in which the hydroxyl group at the C10 position and the ethyl group at the C7 position help to stabilize SN38 in physiological environments. SN38 shows enhanced anticancer potency, making it one of the most potent CPT analogs and attracting more and more scientific attention.2
However, the clinical application of SN38 is hindered by chemical and pharmacological challenges.3 SN38 is insoluble in water (11–38 μg/mL) as well as in most of the pharmaceutically acceptable solvents and oils, thus limiting its formulation as a solution.2 Moreover, as with CPTs, the closed lactone ring of SN38, which is essential for anticancer activity, shows reversible pH-dependent hydrolysis. In physiological environment at pH 7.4, the lactone ring of SN38 shows instability and favors the open lactone form, with the carboxylate form of SN38 showing no therapeutic effect.4 The poor solubility of SN38 and the instability of the active drug molecule at physiological pH (pH 7.4) are major hurdles in achieving effective therapy with SN38.5
The Food and Drug Administration (FDA)-approved water-soluble prodrug of SN38 is irinotecan, or CPT-11, which is converted to the more active metabolite SN38 after administration in vivo and is 100–1,000 times less potent than SN38.6 However, the in vivo conversion rate of CPT-11 to SN38 is very low (<8% of injected dose of CPT-11) and highly variable between patients, and the administration of higher doses of CPT-11 is infeasible because of severe dose-limiting toxicity.7 There is therefore an urgent need to develop efficient ways of using SN38 in tumor therapy. To exploit the full therapeutic potential of SN38, a number of drug delivery systems, eg, PEGylation (PEG, polyethylene glycol), polymer micelle, liposome formulation, and dendrimers, have been investigated extensively to improve the water solubility and bioavailability of SN38.6,8–12 Unfortunately, some inherent drawbacks are associated with these drug delivery systems, such as low drug loading, high toxicity, and difficult preparation.13
As recommended by experts, the challenges in SN38 drug delivery may be overcome by two approaches: ensuring multiple layers of protection against degradation and slow but sustained release of the therapeutically effective drug.14,15 In our design, we combined the advantages of prodrug with nanotechnology and obtained a long-circulating liposomal drug delivery system, in which a lipophilic prodrug of SN38, 7-ethyl-10-hydroxycamptothecin-10-palmitic acid ester (named SN38-PA), was incorporated. Liposomes are well-studied biocompatible carriers that play a key role in nanotechnology-based drug delivery. Several liposomal drug formulations have been approved in the past 20 years.16,17 Liposomes protect the encapsulated drugs from structural transformation or chemical degradation by isolating them from the surrounding environment and can provide targeted therapy to tumor cells.18 It is difficult to formulate the SN38 liposome with high encapsulation efficiency (EE) because of the poor solubility of SN38 and its low affinity to the lipid membrane. The lipophilic prodrug has the benefits of improving drug loading in lipid-based formulations, stabilizing the lactone form and protecting it from hydrolysis, and improving the permeability of the parent drug through cell membranes.4 In addition, loading the lipophilic prodrug in a lipid-based carrier system can lead to slow and sustained release of the drug, thus reducing its high toxicity. Through preliminary experiments, palmitic acid was selected as the appropriate fatty acid to conjugate SN38 at the C10 position via an ester bond, as the generated prodrug SN38-PA has an appropriate partition coefficient (log P) of 7.92, making it easy to incorporate into liposomes with high EE of approximately 99.3% and drug loading of approximately 3 mg/mL. The lipophilic prodrug SN38-PA was then entrapped in PEGylated long-circulating liposomes, which not only protect the lactone form of SN-38 but also prolong its circulating time to achieve passive targeting of tumors through enhanced permeability and retention (EPR) effects. To compare the hydrophilic prodrug CPT-11, a series of in vitro and in vivo studies were conducted to evaluate SN38-PA liposomes, including conversion rate to parent drug, antitumor cytotoxicity, stability of lactone form, pharmacokinetics, biodistribution, and antitumor efficacy in xenograft animals. Our data demonstrated that the SN38-PA liposome showed improved stability with high drug loading, high conversion rate, excellent antitumor activity, and tumor targeting, thus showing promise as a potent anticancer drug.
Materials and methods
Materials
SN38 and 10-hydroxy CPT were purchased from Chenguang Pharma Ltd. (Chengdu, China). CPT-11 was purchased from Hengrui Pharma Ltd. (Jiangsu, China). Egg yolk phospholipids (PC-98T), synthetic phospholipids (DSPE-PEG2000), and cholesterol were purchased from Advanced Vehicle Technology Pharma Ltd. (Shanghai, China). Stearoyl chloride, dichloromethane, and N,N-diisopropylethylamine were supplied by Sinopharm Chemical Reagent Ltd. (Shanghai, China). DMEM and 10% FBS were obtained from Gibco Laboratories (New York, NY, USA). Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Laboratories (Kumamoto, Japan). Chromatography grade water, methanol, and formic acid were supplied by EMD Millipore Corporation (Billerica, MA, USA). All other reagents were of analytical grade.
Mouse ascites tumor S180 cells, mouse Lewis lung cancer (LLC) cells, human breast cancer MCF-7 cells, and human colon cancer HCT-116 cells were obtained from the Chinese Academy of Sciences Shanghai Institute of Cell Bank (Shanghai, China). Institute of Cancer Research mice and Sprague Dawley rats were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and maintained under standard conditions. All animal experiments were performed in accordance with guidelines approved by the ethics committee of the Second Military Medical University and were conducted in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Methods
Synthesis of SN38-PA
SN-38 (392.4 mg, 1 mmol) was mixed with 200 mL dichloromethane, before the addition of N,N-diisopropylethylamine (1.2 mmol). The mixture was stirred at 0°C for 10 minutes. Palmitoyl chloride (1.2 mmol) was added to the mixture dropwise and stirred for a further 15 minutes at 0°C, before raising the reaction temperature to room temperature and stirring for a few hours until the solution became a transparent yellow liquid. The solution was evaporated under reduced pressure, and the light yellow solid SN38-PA (yield 98%), as characterized by nuclear magnetic resonance (NMR) spectroscopy and liquid chromatography–mass spectrometry (LC/MS), was obtained. MS (electrospray ionization−) m/z: 629 (M–H+). 1H NMR (CDCl3, 600 MHz, ppm) δ: 8.22 (1H, d, J=9.12 Hz), 7.81 (1H, d, J=2.46 Hz), 7.64 (1H, s), 7.54 (1H, dd, J=9.12 and J=2.46 Hz), 5.74 (1H, d), 5.30 (1H, d), 5.25 (2H, s), 3.86 (1H, s), 3.15 (2H, q), 2.65 (2H, t), 1.95–1.85 (2H, m), 1.84–1.79 (2H, m), 1.50–1.26 (27H, m), and 1.03 (3H, t), 0.87 (3H, t).
Determination of partition coefficients (log P)
The partition coefficients (log P) of SN38-PA and SN38 were determined using a shake flask method. Before use, 1-octanol and buffer solutions (0.1 M citric acid buffer, pH 3.0) were mutually saturated for 24 hours by shaking at 600 rpm on mechanical shaker at 25°C. Samples with known concentration of test compounds in 1-octanol (5 mL) and buffer solution (5 mL) were shaken on mechanical shaker for 24 hours at 600 rpm and 25°C, and the phases were allowed to separate through centrifugation at 5,000 rpm for 10 minutes. The concentration of the test compounds in both phases was determined precisely by HPLC. Each experiment was repeated thrice, and the mean value was used to calculate log P. Log P was calculated using the logarithm of the ratio of concentrations of compound (solute) in 1-octanol to buffer solution: log P=lg ([solute]1–octanol/[solute]buffer solution).
Liposome preparation
SN38-PA liposomes were prepared using a thin film hydration method. Briefly, hydrophobic excipients, including phospholipid (3% PC-98T and 0.4% DSPE-PEG2000), cholesterol (0.3%), and SN38-PA (0.3%), were dissolved in dichloromethane and transferred to a suitable round bottom flask. The flask was then connected to a Buchi R205 rotary evaporator (Flawil, Switzerland) and water bath (Buchi B-409) with temperature maintained at 40°C–45°C. Vacuum was applied to the flask to evaporate the dichloromethane and form a homogeneous lipid film on the flask wall. The dry lipid film was maintained overnight under vacuum to remove traces of dichloromethane. The lipid film was then hydrated using a solution containing 7%–10% sucrose by ultrasound until the lipid film was completely detached. Liposomes were then formed, ultrasonicated using a cell disruptor (Ultrasonics 450; Branson Ultrasonics Corp., Danbury, CT, USA) for 3 minutes, and passed through a microfluidizer (M-110EH; Microfluidics Inc., Newton, MA, USA) at 15,000 psi for three cycles at an operating temperature of 25°C. The resulting liposome-based drug formulation was then sterile filtered through a 0.2 μm filter and transferred to 10 mL vials.
Characterization of liposomes
The size distribution and zeta potential of SN38-PA liposomes were measured using a Malvern Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK). The liposomes were diluted to appropriate concentrations and maintained at 25°C. The liposomal EE was determined by passing through a Sephadex G-50 column as described in the following sections. Briefly, 0.5 mL of SN38-PA liposomes was loaded on the column and eluted with water containing 3% (w/v) Tween-80. The free drug fraction was then collected for further investigation, and EE was calculated according to the following equation: %EE = (1−amount of free SN38-PA/total SN38-PA added) ×100%.
In vitro lactone stability
Because the lactone (ring E) is a moiety critical for CPT activity and is reported to be unstable under physiological conditions (pH 7.4), the stability of the lactone in SN38-PA liposomes and CPT-11 was determined.
To evaluate lactone stability, a saline-buffered solution (10 mM, pH 7.4) containing 4% human serum albumin was incubated at 37°C in the presence of SN38-PA liposomes and CPT-11.19 HPLC was used to analyze the decrease in the concentration of lactone over time.
Conversion study of SN38-PA in plasma
To evaluate the conversion rate of SN38-PA to SN38, 0.1 mL of the sample solution (3 mg/mL CPT-11, 3 mg/mL SN38-PA liposome) was added to 1 mL rat plasma and incubated for 0, 0.5, 1.5, 3, 5, 12, and 24 hours.9 A 100 μL aliquot of plasma was immediately added into a 1.5 mL centrifuge tube with 20 μL 10-hydroxy CPT (internal standard, IS) working solution (50 μg/mL in methanol–0.2% formic acid water, 50:50, v/v), 50 μL formic acid, and 200 μL methanol. After centrifugation, the supernatant was analyzed via HPLC.
In vitro cytotoxicity assay
The cytotoxicity of SN38-PA liposomes was evaluated using a CCK-8.20 Briefly, MCF-7, HCT-116, LLC, and S180 cells were seeded (5×103 cells/well) in 96-well plates (BD, Franklin Lakes, NJ, USA), cultured for 24 hours, and then incubated with 10 μL various concentrations of SN38-PA liposome, CPT-11, or SN38 (dissolved in dimethyl sulfoxide, DMSO) in DMEM supplemented with 10% (v/v) FBS, 100 μg/mL streptomycin, and 100 IU penicillin in a humidified atmosphere with 5% CO2 at 37°C. After treatment for 24 hours, 10 μL of CCK-8 solution was added to each well, and the 96-well plates were further incubated at 37°C for 2 hours. The OD of each well was measured on a microplate reader (Multiskan; Thermo Fisher Scientific) at a wavelength of 450 nm to determine the cell viability. The assay was repeated three times. The cell viability was calculated as follows: ([As−Ab]/[Ac−Ab]) ×100%, where As, Ac, and Ab are defined as the absorbance of experimental samples, untreated samples, and blank controls, respectively.
In vivo pharmacokinetic study
Twelve Sprague Dawley rats (male, 180±20 g) were randomly assigned to two groups (six mice per group). CPT-11 (15.88 mg/kg, equivalent to 10 mg/kg SN38) and SN38-PA liposomes (16.06 mg/kg, equivalent to 10 mg/kg SN38) were administered intravenously (i.v.) as a single dose via injection in the tail vein.21 At different time points, 300 μL blood was collected from the retro-orbital sinus into heparinized tubes and immediately centrifuged to collect the plasma fraction. All plasma samples were pretreated as follows: 100 μL plasma was spiked with 20 μL IS (10-hydroxy CPT), 50 μL formic acid, and 200 μL methanol and vortexed for 5 minutes. After centrifugation, the supernatant was analyzed via HPLC.
Biodistribution study
S180 cells were collected from the intraperitoneal cavity of S180-bearing ICR mice. The mice (6 weeks old) were injected subcutaneously in the right armpit with S180 cell suspensions (2×107 cells/mL). After 4 days, 42 S180-bearing mice were randomly divided into two groups and i.v. injected with CPT-11 and SN38-PA liposomes at a dose of 15.88 mg/kg and 16.06 mg/kg (equivalent to 10 mg/kg SN38), respectively. The mice were sacrificed at different time points post injection. The tumors and organs of interest (heart, liver, spleen, lung, kidney, and rectum) were excised, and CPT-11, SN38-PA, and SN38 content were measured. An amount of 0.1 g organ tissue was homogenized with saline (1:4, mg/μL),22 and 20 μL 10-hydroxy CPT (IS) was added to the samples (100 μL). CPT-11, SN38-PA, and SN38 were extracted with 50 μL formic acid and 200 μL methanol by vigorous mixing for 5 minutes. After centrifugation at 12,500 rpm for 10 minutes, the organic layer was collected, transferred to an autosampler vial, and 20 μL supernatant was injected to quantify CPT-11, SN38-PA, and SN38 content via HPLC.
In vivo antitumor activity
In vivo anticancer activity against S180 solid tumors in ICR mice was evaluated.23 Briefly, ICR mice were inoculated with S180 cells in the right armpit, and mice were randomly divided into five groups for 24 hours later. The negative control group was treated with saline. CPT-11 injection (15.88 mg/kg, equivalent to 10 mg/kg SN38) and SN38-PA liposome (8.03 mg/kg, 16.06 mg/kg, or 32.12 mg/kg, equivalent to 5 mg/kg, 10 mg/kg, or 20 mg/kg SN38, respectively) were administered to the mice via tail vein injection. Administration was performed on days 0, 3, and 5. The body weights were measured every 2 days. Twenty-four hours after the last injection, mice were sacrificed after weighing, and the tumor was carefully excised and weighed. The tumor growth inhibition was calculated using the following formula: tumor growth inhibition (%) = (W1−W2)/W1×100%, where W1 and W2 refer to the mean weights of the negative control group and the drug-treated group, respectively.
HPLC analysis and quantification
Sample quantification was performed using an HPLC system consisting of an Agilent G1311C pump, a G1329B UV–visible detector, and an Agilent ZORBAX Eclipse Plus C18 column (250×4.6 mm, 5 μm). 10-Hydroxy CPT (50 μg/mL) was used as the IS. The mobile phase consisted of methanol (A) and water with 0.1% formic acid (B) at a flow rate of 1 mL/min. A gradient was used for SN38-PA liposome detection as follows: 0 minute, 70% A and 30% B; 6 minutes, 70% A and 30% B; 7 minutes, 100% A; 17 minutes, 100% A; and 17.5 minutes, 70% A and 30% B. The mobile phases for CPT-11 injection consisted of 55% A and 45% B, and the injection volume was 20 μL. This quantification method was fully validated, and the inter-day and intraday accuracy of samples ranged narrowly between 98% and 102%, while both intraday and inter-day relative SDs were below 5%.
Statistical analyses
All quantitative data are presented as mean ± SD, and Student’s t-test was used for the evaluation of statistical significance. A P-value of <0.05 was considered significant, while a P-value of <0.01 was considered highly significant.
Results and discussion
Log P of SN38-PA between 1-octanol and water
The lipophilic prodrug SN38-PA was obtained from the reaction of SN38 and palmitoyl chloride with a yield of 98%. The synthesis route is illustrated in Figure 1. The chemical structure was confirmed via NMR spectroscopy and LC/MS. The drug SN38, with a log P of 2.65, exhibited both poor hydrophilicity and lipophilicity and could not be integrated into liposomes. The log P of SN38-PA was 7.92, and its lipophilicity was significantly enhanced compared with that of SN38. Increasing the lipid solubility of SN38 through prodrug formulation has the benefit of improving drug loading in lipid-based formulations, possible stabilization of the lactone form from hydrolysis, and improving the permeability of the parent drug through cell membranes. A liposome is a lipid-based formulation, and the EE of liposomes correlated well with the partition coefficient of the drug. A study previously demonstrated that liposomes favorably encapsulate drugs with either a low or high log P, but do not hold drugs having an intermediate log P of 2–3.24 Through conjugation of SN-38 with palmitic acid, the lipophilic prodrug SN38-PA, showing an appropriate log P for encapsulation into liposomes, was obtained.
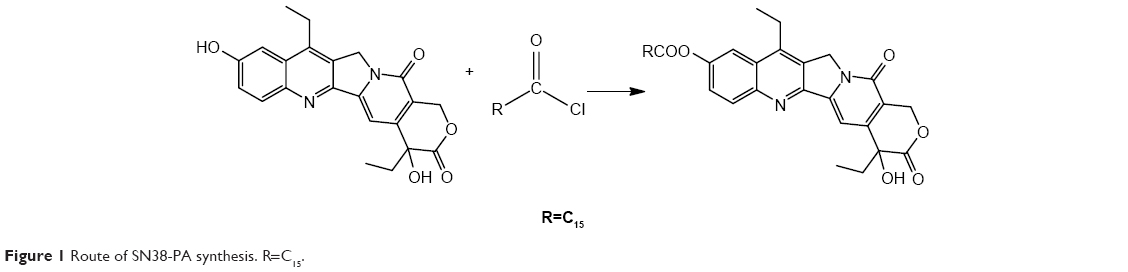 | Figure 1 Route of SN38-PA synthesis. R=C15. |
Characterization of SN38-PA liposomes
SN38-PA liposomes were prepared using a thin film hydration method in which 0.4% DSPE-PEG2000 was added to improve in vivo longevity. The hydrodynamic diameter of the prepared liposome was approximately 80.13±0.94 nm with polydispersity index of 0.094 (Figure 2), and the zeta potential was approximately −33.53±2.57 mV (Figure 3). The EE of SN38-PA liposomes was approximately 99.34%, and drug loading was approximately 3 mg/mL, while SN38 liposomes prepared using the conventional thin film hydration method showed only 5.6% EE and 0.5 μg/mL drug loading. These results demonstrated that using the lipophilic prodrug SN38-PA, we obtained stable liposomes with satisfactory drug loading and high entrapment efficiency.
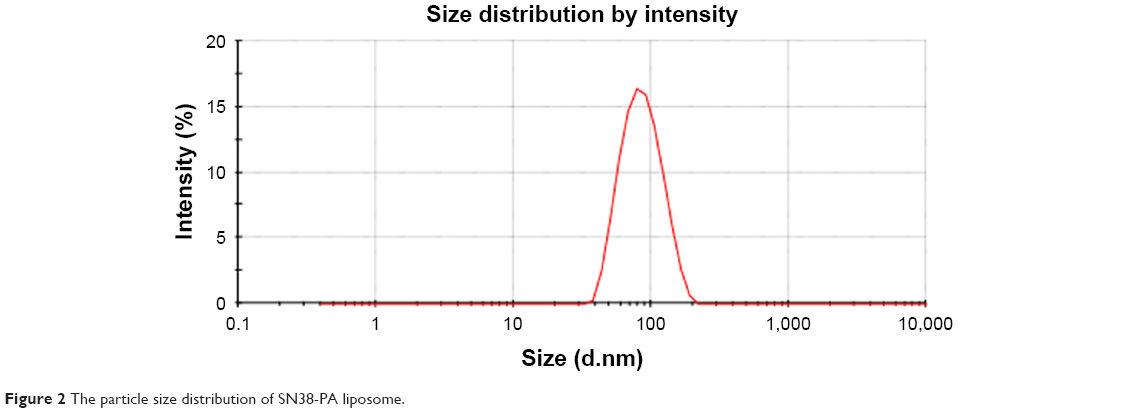 | Figure 2 The particle size distribution of SN38-PA liposome. |
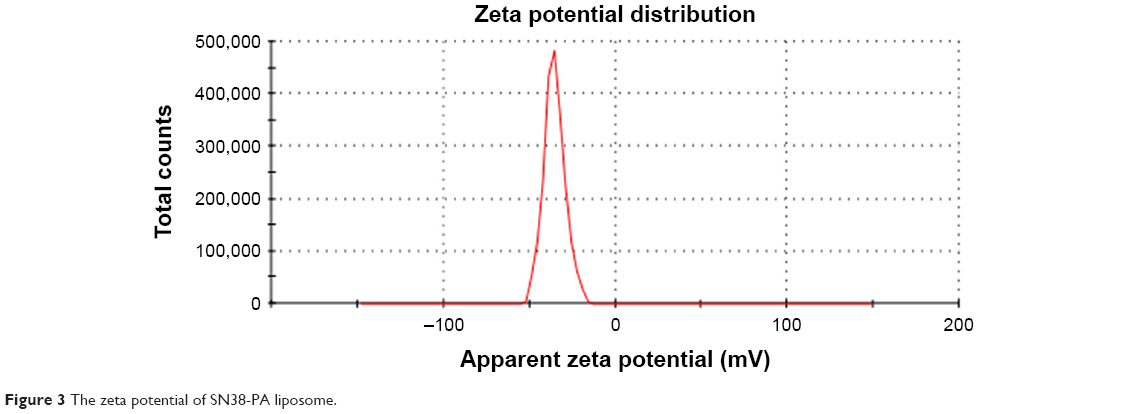 | Figure 3 The zeta potential of SN38-PA liposome. |
In vitro lactone stability
As with SN38, the lactone ring essential for bioactivity shows reversible pH-dependent hydrolysis. The lactone ring of SN38 is stable at pH ≤4.5 and hydrolyzes completely to form the carboxylate form at pH >9.0.25 The instability of the active drug molecule at physiological pH (pH 7.4) is a major hurdle in attaining effective therapy with SN38, as the open carboxylate form of SN38 has no therapeutic effect.5
The percent change in the concentration of the lactone form over time for CPT-11 injection and SN38-PA liposome, in the simulated physiological environment in vitro, is shown in Figure 4. The results showed that the SN38-PA liposome holds a more stable closed lactone form than CPT-11. After 12 hours, the prevalence of the lactone ring of the SN38-PA liposome was more than 95%, while that of the lactone ring of CPT-11 was approximately 20% only. The stability of the lactone form of the SN38-PA liposome is greater than that of CPT-11. This dramatic increase in the stability of the lactone may result from the protective effect of the liposome by isolating the encapsulated drug from the surrounding environment.
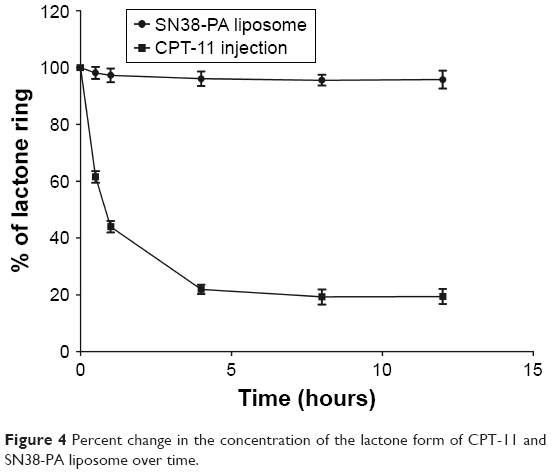 | Figure 4 Percent change in the concentration of the lactone form of CPT-11 and SN38-PA liposome over time. |
Conversion study of SN38-PA
CPT-11 is a water-soluble prodrug of SN38 that has been approved by the FDA for chemotherapy. CPT-11 is 100–1,000 times less potent than SN38, and only a small fraction of CPT-11 (approximately 2%–8%) converts to SN38 in vivo after i.v. infusion.7,26,27 Using CPT-11 injection as comparison, the conversion rate of SN38-PA in rat plasma was investigated. As shown in Figure 5, the percentage conversion of SN38-PA was significantly higher than that of CPT-11 injection after incubation with plasma. Twelve hours later, approximately 87% of SN38-PA liposomes were converted to the more active metabolite SN38, and only approximately 6% of CPT-11 was converted to SN38, demonstrating that the SN38-PA liposome resulted in more efficient conversion to SN38.
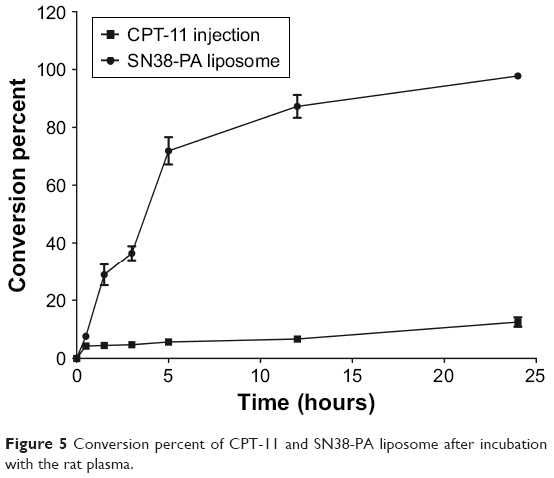 | Figure 5 Conversion percent of CPT-11 and SN38-PA liposome after incubation with the rat plasma. |
In vitro cytotoxicity assay
SN38-PA liposomes demonstrated significant in vitro antitumor activity comparable with that of SN38 dissolved in DMSO, but far better than that of CPT-11, against a large panel of tumor cell lines, including S180, MCF-7, LLC, and HCT-116 cells. The 50% inhibitory concentrations (IC50) of SN38-PA liposomes in the above mentioned cell lines were 0.08, 0.34, 0.18, and 0.12 μM, respectively, or approximately 1.26- to 8-fold greater than those of SN38 in the same cell lines. These data indicate that SN38-PA liposomes released SN38 effectively. In the four cell lines tested, SN38-PA liposomes exhibited significantly higher antitumor activity than CPT-11. The IC50 values of CPT-11 against the tested cell lines were 11.12, 5.50, 59.11, and 12.26 μM, respectively, or 16.18- to 328.39-fold higher than that of SN38-PA liposomes.
Pharmacokinetics
The pharmacokinetic profiles of CPT-11 injection and SN38-PA liposomes in Sprague Dawley rats are shown in Figure 6A and B. Compared with CPT-11, SN38-PA showed greater concentrations in plasma and was rapidly metabolized to SN38 in vivo. The animals treated with SN38-PA liposomes showed an obvious increase in plasma SN38 concentration compared with those treated with CPT-11. The main pharmacokinetic parameters were calculated using a non-compartmental model, as summarized in Table 1. The plasma half-life (t1/2) of SN38 was significantly enhanced (P<0.05), and the clearance of SN38 decreased more than 9.7-fold in animals treated with SN38-PA liposomes, compared with that in animals treated with CPT-11 injection. The volume of distribution (V) was calculated to reflect the theoretical volume in which SN38 is evenly distributed after injection.28 The calculated V for SN38-PA liposomes was significantly smaller than that of CPT-11 injection, indicating that the concentration of active SN38 in plasma circulation was greater after the administration of SN38-PA liposomes. Moreover, the mean plasma area under the curve (AUC0–24 h) for SN38 after the administration of SN38-PA liposomes was 7.5-fold higher than that after CPT-11 administration. In conclusion, SN38-PA liposomes could slow down the elimination of SN38 and thereby extend the systemic circulation of SN38 in the blood.
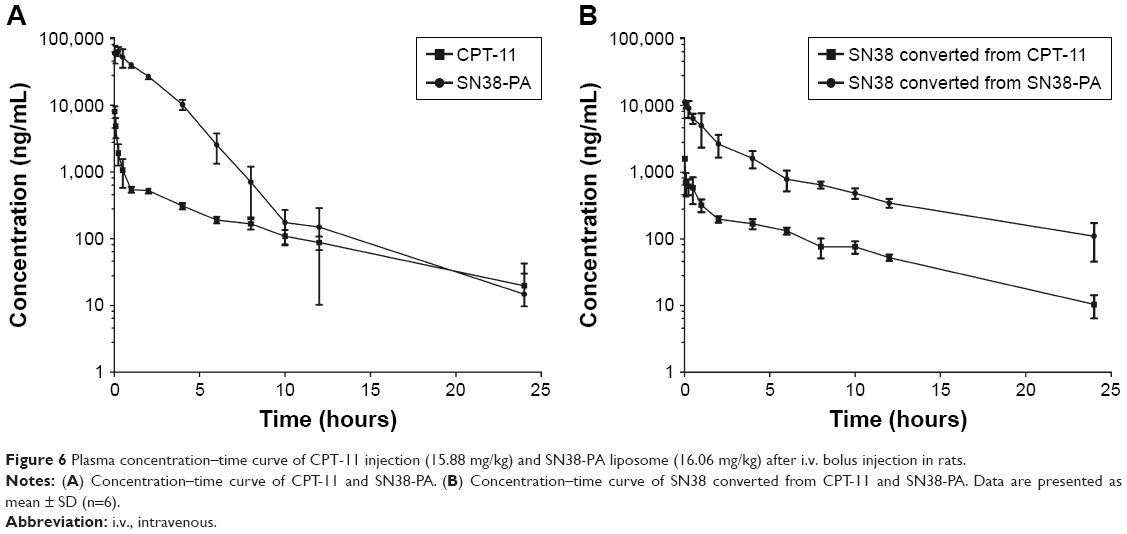 | Figure 6 Plasma concentration–time curve of CPT-11 injection (15.88 mg/kg) and SN38-PA liposome (16.06 mg/kg) after i.v. bolus injection in rats. |
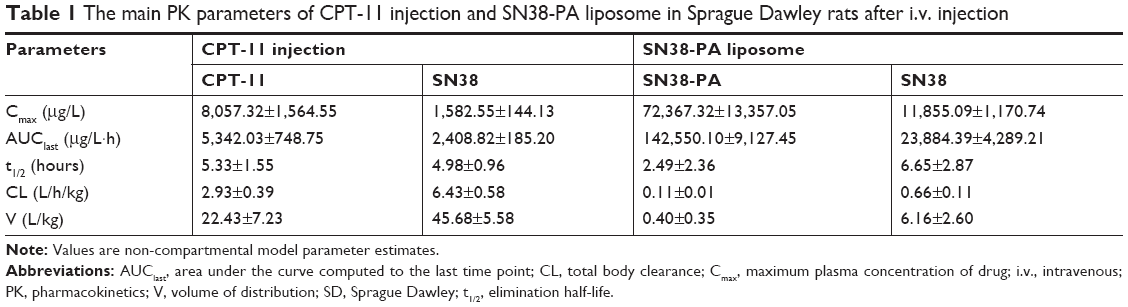 | Table 1 The main PK parameters of CPT-11 injection and SN38-PA liposome in Sprague Dawley rats after i.v. injection |
Biodistribution study
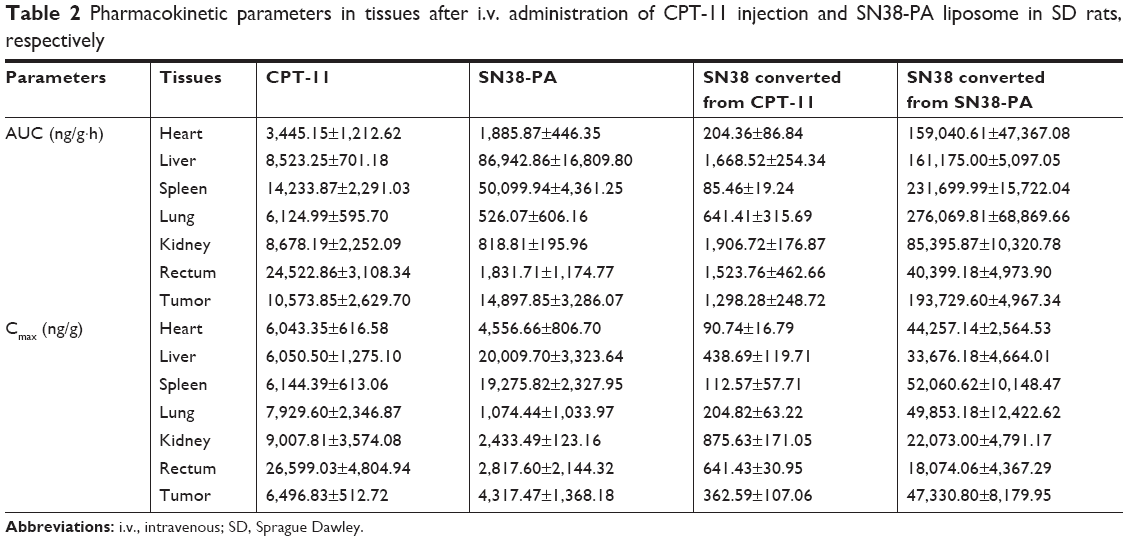
We subsequently monitored the time-dependent in vivo biodistribution of CPT-11 injection and SN38-PA liposomes after i.v. injection into ICR mice bearing S180 tumors.25 The mean concentration–time profiles of CPT-11 injection and SN38-PA liposomes in the main organs and tissues are shown in Figure 7, and the non-compartment pharmacokinetic parameters are listed in Table 2.
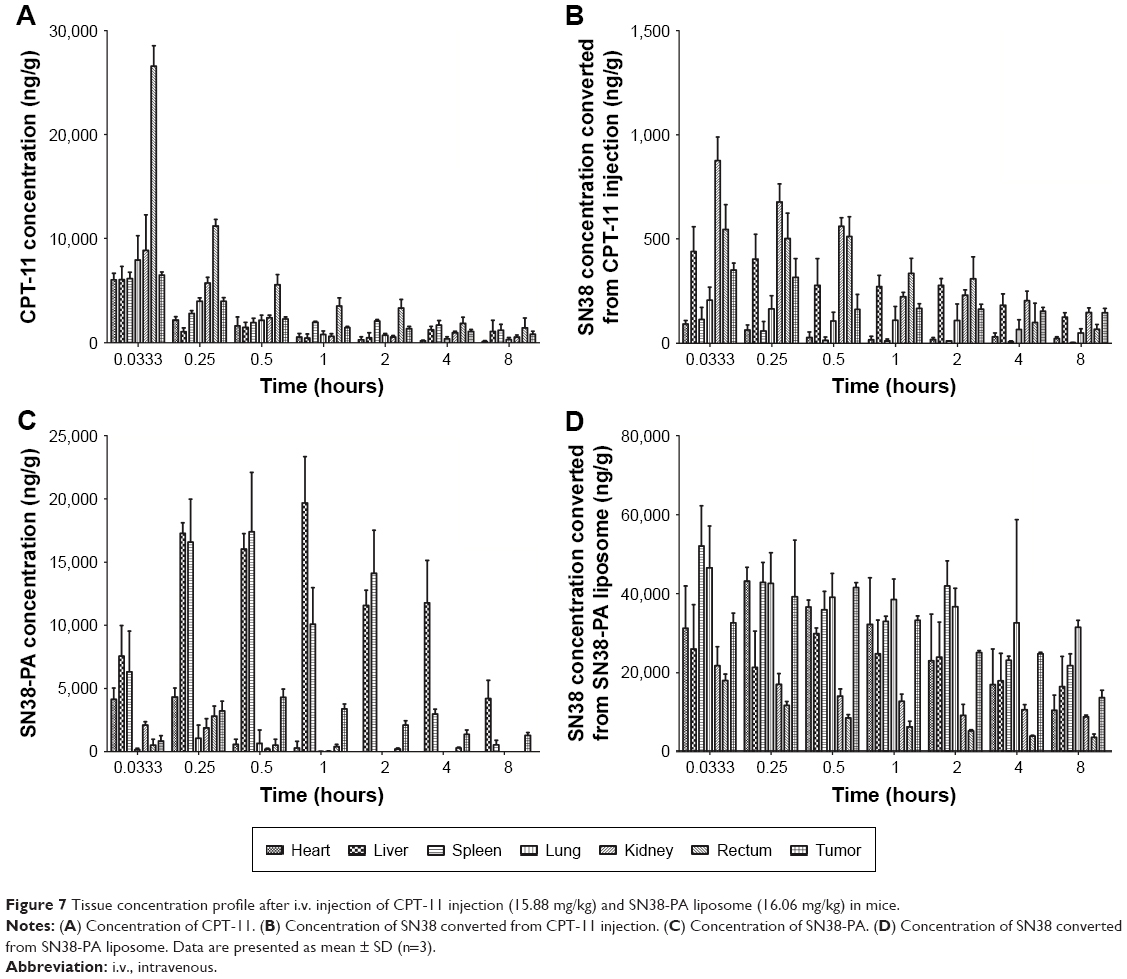 | Figure 7 Tissue concentration profile after i.v. injection of CPT-11 injection (15.88 mg/kg) and SN38-PA liposome (16.06 mg/kg) in mice. |
 | Table 2 Pharmacokinetic parameters in tissues after i.v. administration of CPT-11 injection and SN38-PA liposome in SD rats, respectively |
As shown in Figure 7A and C, CPT-11 distributed primarily to the rectum, kidney, and lung, while SN38-PA liposomes distributed primarily to the liver, spleen, and tumor. Administration of SN38-PA liposomes produced higher AUC values in the liver (approximately 10-fold, P<0.001), spleen (approximately 3.5-fold, P<0.01), and tumor (approximately 1.4-fold, P>0.05) when compared with CPT-11. SN38-PA formulated in PEGylated liposomal carrier reduced the disposition of no mononuclear phagocyte system (MPS) organs and achieved higher levels in tumor through prolonged systemic circulation and enhanced permeation and retention effect.
Concentrations of SN-38, converted from CPT-11 injection and SN38-PA liposomes, measured in main organs and tissues are shown in Figure 7B and D, respectively. The concentration of SN38 converted from SN38-PA was much higher than that from CPT-11 in almost all tissues (Table 2). The AUC was approximately 778-fold in the heart (P<0.001), approximately 96-fold in the liver (P<0.001), approximately 2711-fold in the spleen (P<0.001), approximately 430-fold in the lungs (P<0.001), approximately 44-fold in the kidneys (P<0.001), approximately 26-fold in the rectum (P<0.001), and approximately 149-fold in the tumor (P<0.001), demonstrating that SN38-PA liposomes could convert to its active SN38 efficiently in all tissues, while CPT-11 could not convert to its active SN38 efficiently.
In vivo antitumor activity
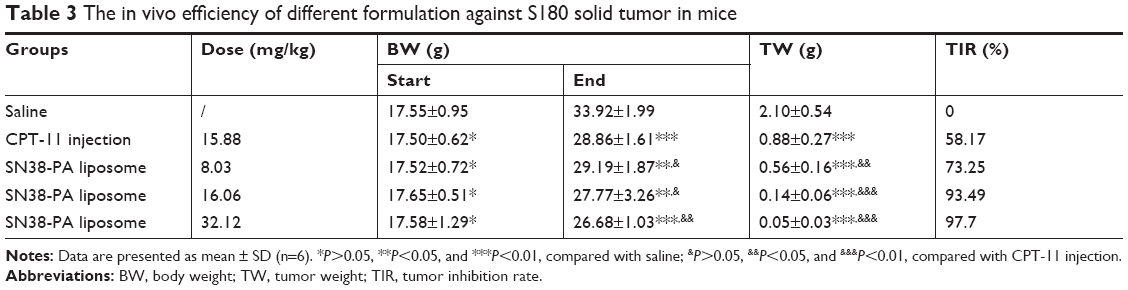
The in vivo antitumor activity of SN38-PA liposomes was investigated in ICR mice that were inoculated with S180 cells in the right armpit, with saline as the negative control, and with CPT-11 as the positive control. The results given in Table 3 summarizes that both CPT-11 and SN38-PA liposomes (in three dose formulations) showed significant tumor growth inhibition in comparison with the saline control (P<0.01), and dose-dependent tumor growth inhibition was observed with the administration of increasing doses of SN38-PA liposomes in the three treatment groups. Furthermore, tumor growth inhibition rate of SN38-PA liposome was 1.61 times that of CPT-11 injection at both doses equivalent to 10 mg/kg SN38 (P<0.01). Besides, SN38-PA liposomes were more efficient, resulting in higher tumor growth inhibition rates than CPT-11 injection at a lower dose (P<0.05).
 | Table 3 The in vivo efficiency of different formulation against S180 solid tumor in mice |
Images of tumor tissues stained with H&E were observed under microscope. As shown in Figure 8, in the negative control group, tumor cells grew well: they were tightly packed with adenoid hyperplasia, and the texture was clear with no obvious necrosis foci (Figure 8A). However, in tumors treated with CPT-11 injection (Figure 8B and C) and low dose of SN38-PA liposomes (8.03 mg/kg; Figure 8D), cell nuclei were partially ruptured and dissolved, the structures were unclear, and many necrotic foci appeared. Moreover, in tumors treated with medium (16.06 mg/kg; Figure 8E) and high-dose SN38-PA liposomes (32.12 mg/kg; Figure 8F), cell necrosis was obvious, tissue structure had disappeared, and more necrotic foci were visible. These results were consistent with the abovementioned therapeutic data indicating that both SN38-PA liposomes and CPT-11 injection possessed significant anticancer effects and that SN38-PA liposomes were more effective than CPT-11 injection at the same dose.
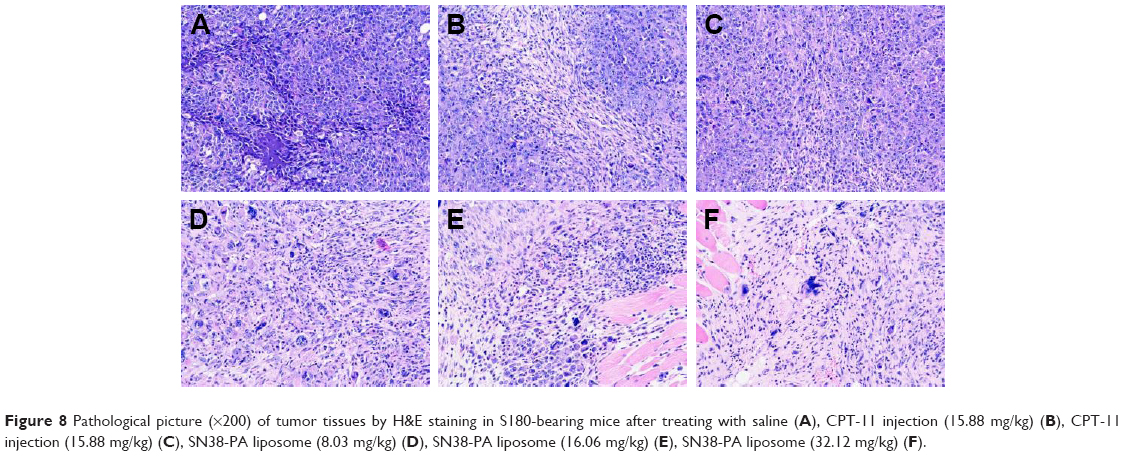 | Figure 8 Pathological picture (×200) of tumor tissues by H&E staining in S180-bearing mice after treating with saline (A), CPT-11 injection (15.88 mg/kg) (B), CPT-11 injection (15.88 mg/kg) (C), SN38-PA liposome (8.03 mg/kg) (D), SN38-PA liposome (16.06 mg/kg) (E), SN38-PA liposome (32.12 mg/kg) (F). |
According to the results summarized in Table 3, both the CPT-11 injection treatment group and the SN38-PA liposome treatment group exhibited body weight loss in comparison with the negative control group, indicating that both CPT-11 and SN38-PA liposome displayed certain toxicity. But we can see that the body weight of all animals increased steadily throughout the study. Besides, at the same dose, there was no significant difference in body weight between the SN38-PA liposome group and CPT-11 group. These results indicated that SN38-PA liposomes had no significant toxicity compared with CPT-11 injection. Further improvement will focus on the tumor targeting distribution of SN38-PA liposome, so as to reduce the toxicity of the formulation as far as possible.
Conclusion
In this study, by combining the advantages of lipophilic prodrugs and nanotechnology, we successfully designed and synthesized SN38-PA liposomes, thereby aiming to overcome the drawbacks of SN38 and the limitations of SN38 drug delivery systems.
Resultant SN38-PA liposomes exhibited significantly higher drug loading, more stable lactone form, more effective conversion rate, and improved in vivo pharmacokinetics, compared to CPT-11 injection.29 Further studies indicated that SN38-PA liposomes exhibited excellent antitumor activity against a large panel of cultured tumor cell lines, and favorable therapeutic effects in S180 tumor-bearing mice. Taking all these findings into consideration, the SN38-PA liposomes developed in this study display good druggability and show potential for clinical application.
Acknowledgment
This study was supported by the National Nature Science Foundation (81101141), Nanometer Science and Technology Foundation of Shanghai (13NM1401503), and Military Medical Scientific Research Project in 12th Five-Year (CWS11J122).
Disclosure
The authors report no conflicts of interest in this work.
References
Wall ME, Wani MC, Cook CE, Palmer KH, Mcphail AT, Sim GA. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata1,2. J Am Chem Soc. 1966;88(16):3888–3890. | ||
Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991;51(16):4187–4191. | ||
Monterrubio C, Pascual-Pasto G, Cano F, et al. SN-38-loaded nanofiber matrices for local control of pediatric solid tumors after subtotal resection surgery. Biomaterials. 2016;79:69–78. | ||
Lu J, Liu C, Wang P, et al. The self-assembling camptothecin-tocopherol prodrug: An effective approach for formulating camptothecin. Biomaterials. 2015;62:176–187. | ||
Bala V, Rao S, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172(1):48–61. | ||
Iyer R, Croucher JL, Chorny M, et al. Nanoparticle delivery of an SN38 conjugate is more effective than irinotecan in a mouse model of neuroblastoma. Cancer Lett. 2015;360(2):205–212. | ||
Slatter JG, Schaaf LJ, Sams JP, et al. Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab Dispos. 2000;28(4):423–433. | ||
Zhao H, Rubio B, Sapra P, et al. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem. 2008;19(4):849–859. | ||
Zhang H, Wang J, Mao W, et al. Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: in vitro and in vivo studies. J Control Release. 2013;166(2):147–158. | ||
Sadekar S, Thiagarajan G, Bartlett K, et al. Poly(amido amine) dendrimers as absorption enhancers for oral delivery of camptothecin. Int J Pharm. 2013;456(1):175–185. | ||
Omar R, Bardoogo YL, Corem-Salkmon E, Mizrahi B. Amphiphilic star PEG-Camptothecin conjugates for intracellular targeting. J Control Release. 2017;257:76–83. | ||
Xie J, Zhang X, Teng M, et al. Synthesis, characterization, and evaluation of mPEG-SN38 and mPEG-PLA-SN38 micelles for cancer therapy. Int J Nanomedicine. 2016;11:1677–1686. | ||
Sepehri N, Rouhani H, Tavassolian F, et al. SN38 polymeric nanoparticles: in vitro cytotoxicity and in vivo antitumor efficacy in xenograft balb/c model with breast cancer versus irinotecan. Int J Pharm. 2014;471(1–2):485–497. | ||
Palakurthi S. Challenges in SN38 drug delivery: current success and future directions. Expert Opin Drug Deliv. 2015;12(12):1911–1921. | ||
Botella P, Rivero-Buceta E. Safe approaches for camptothecin delivery: Structural analogues and nanomedicines. J Control Release. 2017;247:28–54. | ||
Olusanya TOB, Haj Ahmad RR, Ibegbu DM, Smith JR, Elkordy AA. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules. 2018;23(4):E907. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Sadzuka Y, Takabe H, Sonobe T. Liposomalization of SN-38 as active metabolite of CPT-11. J Control Release. 2005;108(2–3):453–459. | ||
Ci T, Li T, Chang G, Yu L, Ding J. Simply mixing with poly(ethylene glycol) enhances the fraction of the active chemical form of antitumor drugs of camptothecin family. J Control Release. 2013;169(3):329–335. | ||
Safra T, Muggia F, Jeffers S, et al. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11(8):1029–1033. | ||
Fang T, Dong Y, Zhang X, Xie K, Lin L, Wang H. Integrating a novel SN38 prodrug into the PEGylated liposomal system as a robust platform for efficient cancer therapy in solid tumors. Int J Pharm. 2016;512(1):39–48. | ||
Park JW. Liposome-based drug delivery in breast cancer treatment. Breast Cancer Res. 2002;4(3):95–99. | ||
Yang X, Zhao X, Phelps MA, et al. A novel liposomal formulation of flavopiridol. Int J Pharm. 2009;365(1–2):170–174. | ||
Nii T, Ishii F. Encapsulation efficiency of water-soluble and insoluble drugs in liposomes prepared by the microencapsulation vesicle method. Int J Pharm. 2005;298(1):198–205. | ||
Alferiev IS, Iyer R, Croucher JL, et al. Nanoparticle-mediated delivery of a rapidly activatable prodrug of SN-38 for neuroblastoma therapy. Biomaterials. 2015;51:22–29. | ||
Goff LW, Benson AB 3rd, LoRusso PM, et al. Phase I study of oral irinotecan as a single-agent and given sequentially with capecitabine. Invest New Drugs. 2012;30(1):290–298. | ||
Schoemaker NE, Kuppens IE, Huinink WW, et al. Phase I study of an oral formulation of irinotecan administered daily for 14 days every 3 weeks in patients with advanced solid tumours. Cancer Chemother Pharmacol. 2005;55(3):263–270. | ||
Signorell RD, Luciani P, Brambilla D, Leroux JC. Pharmacokinetics of lipid-drug conjugates loaded into liposomes. Eur J Pharm Biopharm. 2018;128:188–199. | ||
Lee PC, Chiou YC, Wong JM, Peng CL, Shieh MJ. Targeting colorectal cancer cells with single-walled carbon nanotubes conjugated to anticancer agent SN-38 and EGFR antibody. Biomaterials. 2013;34(34):8756–8765. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.