Back to Journals » Drug Design, Development and Therapy » Volume 20
Novel Pyrazole-3-Cyano-2-Pyridinone Hybrids as Multitarget Anti-Inflammatory Agents: Synthesis, Computational Modeling, and Biological Evaluation
Authors Hussein BRM, Salem IM, Aziz HA ![]() , Alshazly O, Hofny HA, Mohassab AM
, Alshazly O, Hofny HA, Mohassab AM ![]() , Awan ZA
, Awan ZA ![]() , Ibrahim TS, Mahmoud EM, Bräse S
, Ibrahim TS, Mahmoud EM, Bräse S ![]() , Mohamed MFA
, Mohamed MFA ![]()
Received 28 September 2025
Accepted for publication 18 February 2026
Published 1 April 2026 Volume 2026:20 570813
DOI https://doi.org/10.2147/DDDT.S570813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Yan Zhu
Bahgat RM Hussein,1 Ibrahim M Salem,2 Hossameldin A Aziz,3,4 Omar Alshazly,5,6 Heba A Hofny,5,6 Aliaa M Mohassab,7,8 Zuhier A Awan,9 Tarek S Ibrahim,10 Elsayed M Mahmoud,11,12 Stefan Bräse,13 Mamdouh FA Mohamed3– 5
1Department of Chemistry, Faculty of Science, Sohag University, Sohag, 82524, Egypt; 2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Sphinx University, New Assuit City, Assiut, 71515, Egypt; 3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, New Valley University, New Valley, 72511, Egypt; 4Department of Pharmaceutical Chemistry, Faculty of Pharmacy, New Valley National University, New Valley, 72511, Egypt; 5Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Sohag University, 82524, Sohag, Egypt; 6Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Sohag National University, Sohag, 82524, Egypt; 7Department of Medicinal Chemistry, Faculty of Pharmacy, Minia University, Minia, 61519, Egypt; 8Medicinal Chemistry Department, Faculty of Pharmacy, Minia National University, New Minia, Egypt; 9Department of Clinical Biochemistry, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia; 10Department of Pharmaceutical Chemistry, Faculty of Pharmacy, King Abdulaziz University, Jeddah, 21589, Saudi Arabia; 11Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Zagazig University, Zagazig, 44519, Egypt; 12Course of Applied Life Science, Faculty of Applied Biological Sciences, Gifu University, Gifu, 501-1193, Japan; 13Institute for Biological and Chemical System, Karlsruhe Institute of Technology, Karlsruhe, 76131, Germany
Correspondence: Mamdouh FA Mohamed, Email [email protected] Stefan Bräse, Email [email protected]
Introduction: The development of multi-target anti-inflammatory agents represents a promising strategy to improve therapeutic efficacy while minimizing adverse effects associated with single-target drugs. In this study, a rational hybridization approach was employed to design pyrazole/3-cyano-2-pyridinone hybrids aimed at modulating key inflammatory mediators.
Methods: A novel series of pyrazole/3-cyano-2-pyridinone hybrids was synthesized and evaluated for anti-inflammatory activity. Nitric oxide (NO) production and iNOS activity were assessed in LPS-stimulated RAW 264.7 macrophages. COX-1/COX-2, LOX (5-LOX and 15-LOX), PGE2, and TNF-α inhibition assays were performed. Cytotoxicity was determined using MTT assays. Molecular docking and 100-ns molecular dynamics (MD) simulations were conducted to investigate binding modes and stability, while in silico ADME profiling was used to predict pharmacokinetic properties.
Results: Compounds 5f, 5g, 5k, and 5m significantly inhibited NO production and iNOS activity, with compound 5m showing the strongest effect (IC50 = 203.9 μM). COX inhibition assays revealed selective COX-2 activity, with compound 5k exhibiting the highest potency (IC50 = 0.92 μM) and a selectivity index of 19.6. Compound 5g most effectively suppressed PGE2 production (IC50 = 152.7 pg/mL), while 5m markedly reduced TNF-α levels, comparable to ibuprofen. In LOX assays, compound 5f showed potent inhibition of both 5-LOX (IC50 = 0.34 μM) and 15-LOX (IC50 = 0.21 μM), outperforming zileuton. All tested compounds exhibited low cytotoxicity (IC50 > 85 μM). Docking and MD simulations confirmed stable and favorable binding interactions of 5k, 5f, and 5m with COX-2, 5-LOX, and iNOS, respectively. In silico pharmacokinetic analysis predicted good oral bioavailability and drug-like properties.
Conclusion: The synthesized pyrazole/3-cyano-2-pyridinone hybrids demonstrated promising multi-target anti-inflammatory activity with favorable safety and pharmacokinetic profiles, highlighting their potential as lead candidates for further development.
Keywords: Pyrazole, 3-Cyano-2-pyridinone, Anti-inflammatory, Healthcare, COX2, iNOS, TNF-α, 5-LOX
Introduction
Inflammation constitutes a vital physiological defense mechanism that is activated in response to infection, tissue injury, or other harmful stimuli.1 While acute inflammation is essential for restoring tissue integrity and initiating repair processes, persistent or dysregulated inflammatory responses are implicated in the pathogenesis of numerous chronic diseases,2,3 including rheumatoid arthritis,4,5 inflammatory bowel disease,6 and various malignancies.7,8 At the cellular level, macrophages and other immune cells orchestrate the inflammatory cascade through the production of a wide array of mediators, such as prostaglandins (PGs),9 leukotrienes (LTs),10 nitric oxide (NO),11 and proinflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6).12 These inflammatory mediators are biosynthesized through distinct yet interconnected enzymatic pathways: PGs are generated via cyclooxygenases (COX-1 and COX-2), LTs through the 5-lipoxygenase (5-LOX) pathway, and NO via inducible nitric oxide synthase (iNOS).13,14 Conventional non-steroidal anti-inflammatory drugs (NSAIDs) exert their effects by non-selectively inhibiting COX enzymes, which effectively attenuates inflammation but also results in adverse gastrointestinal (GI) and renal effects due to the suppression of COX-1, a constitutive isoform involved in cytoprotection.15 To mitigate these limitations, selective COX-2 inhibitors were introduced, offering improved GI safety profiles while retaining anti-inflammatory efficacy.16 Nevertheless, prolonged use of COX-2 inhibitors has been associated with an increased risk of cardiovascular (CV) events, thereby restricting their clinical utility.17 Furthermore, both COX and 5-LOX pathways utilize arachidonic acid (AA) as a common substrate. Selective inhibition of COX enzymes can inadvertently redirect AA metabolism toward the 5-LOX pathway, resulting in elevated LT production and potentially exacerbated inflammatory responses.18 Similarly, aberrant NO production via iNOS contributes to tissue injury and chronic inflammation, making it an attractive target for therapeutic intervention.19 In light of these complexities, there is growing interest in the development of novel multi-target anti-inflammatory agents capable of concurrently inhibiting COX-2, 5-LOX, and iNOS.20,21 Such a poly pharmacological approach holds the potential to deliver enhanced therapeutic efficacy with a reduced incidence of GI and CV adverse effects. Among the chemical scaffolds investigated, pyrazole derivatives have demonstrated considerable promise in modulating these key inflammatory targets. Celecoxib (compound I) remains one of the most clinically successful examples of a selective COX-2 inhibitor within this class, incorporating benzene sulfonamide for protruding inside the COX-2 side pocket.22,23 Building upon this scaffold, several analogues, such as compound II, that exhibited selective COX-2 inhibition potential (IC50 = 0.32 µM),24 as well as compound III, that elucidated dual inhibition of COX-2 and 5-LOX, alongside potent in vivo anti-inflammatory and analgesic activities.25 In contrast, although the pyrazole-chalcone compound IV lacks a sulfonamide pharmacophore, it demonstrated significant anti-inflammatory activity through selective inhibition of COX-2 (IC50 = 5.13 µM), 5-LOX (IC50 = 5.88 µM), and iNOS (IC50 = 4.93 µM). Additionally, it markedly suppressed NO and PGE2 production in LPS-stimulated RAW 264.7 macrophages. These findings suggest a unique COX-2 binding interaction for compound IV, despite the absence of the conventional sulfonamide moiety26 (Figure 1).
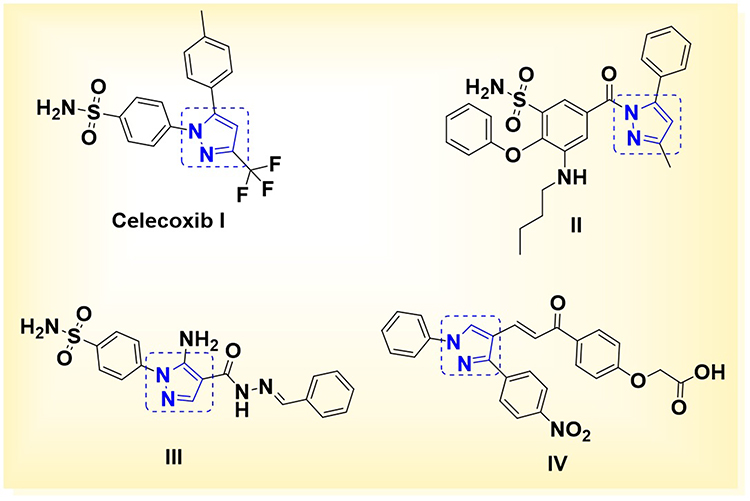 |
Figure 1 Chemical structures of pyrazole-benzene sulfonamide-based anti-inflammatory compounds (celecoxib derivatives I, II, and III), along with pyrazole-based anti-inflammatory compound IV, which lacks the benzene sulfonamide moiety. |
On the other hand, the U.S. Food and Drug Administration (FDA) approved etoricoxib (compound V) as a selective COX-2 inhibitor, a pyridine-based heterocyclic compound featuring a methyl sulfonyl-substituted phenyl moiety designed to engage the COX-2 side pocket.27 This structural configuration confers potent COX-2 inhibitory activity while exhibiting a favorable safety profile, with minimal gastrointestinal (GI) and cardiovascular (CV) adverse effects.28 Notably, the replacement of the pyridine ring by an electron-deficient 3-cyano-2-pyridinone scaffold, as in compound VI, resulted in an enhanced COX-2 selectivity index, superior anti-edematous efficacy, and a markedly reduced ulcerogenic liability in comparison to celecoxib.29 The 3-cyanopyridine-based compounds VII and VIII exhibited notable in vivo anti-inflammatory activity, achieving 64% and 56.73% inhibition, respectively, in the carrageenan-induced paw edema model. These results are comparable to the 77% inhibition observed with the reference drug indomethacin, and 53.95% inhibition with ibuprofen, indicating the potential of these compounds despite the absence of the methyl sulfonyl moiety typically required for COX-2 side pocket binding30,31 (Figure 2).
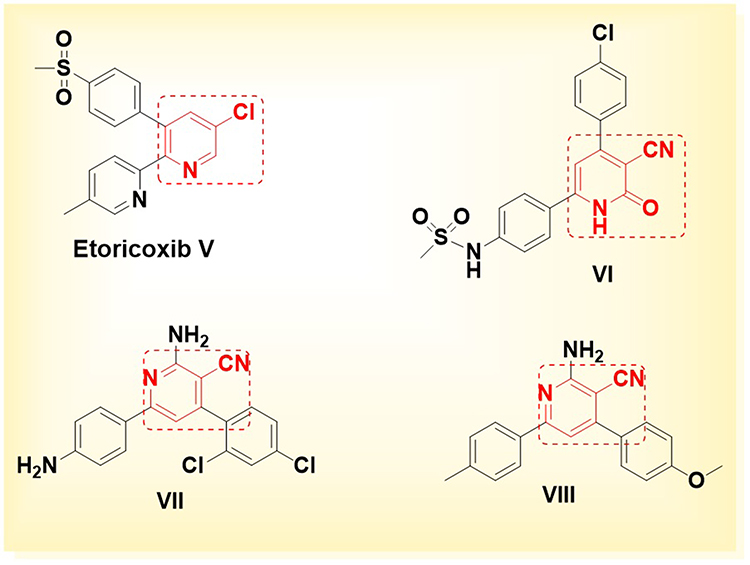 |
Figure 2 Chemical structures of anti-inflammatory compounds: pyridine-based etoricoxib (compound V) and 3-cyano-2-pyridinone (compound VI), featuring a methyl sulfonyl functional group, and 3-cyanopyridine derivatives (compounds VII and VIII), which lack the methyl sulfonyl key moiety. |
Building upon our previous work in the synthesis and development of pyridine-based heterocyclic compounds,32–36 this study continues our efforts through the rational design of hybrid molecules that incorporate both pyrazole and 3-cyano-2-pyridinone pharmacophores into a single, compact structure. This strategy aims to integrate and synergize the individual therapeutic benefits of each moiety, potentially yielding more effective anti-inflammatory agents. These hybrid compounds are anticipated to act as dual or multi-target inhibitors of COX-2, 5-LOX, nitric oxide (NO) production, prostaglandin E (PGE), and tumor necrosis factor-alpha (TNF-α), while also exhibiting improved safety profiles (Figure 3).
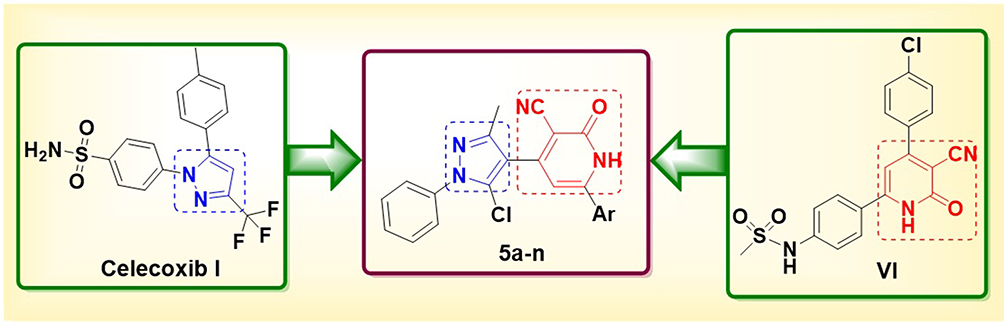 |
Figure 3 Rationale design of the investigated pyrazole-cyanopyridine derivatives 5a-n as anti-inflammatory agents as analogues for Celecoxib I and compound IV. |
Materials and Methods
Chemistry
General Information
All information about reagents and spectral analyses is shown in the Supporting Information (supplementary Figures 1–48).
General procedure for synthesis of 6-(4-aryl)-4-(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile derivatives 5a-n
A mixture of pyrazole aldehyde 1 (2.5 mmol, 0.55 g), ethyl cyanoacetate 2 (2.5 mmol, 0.28 g), ammonium acetate 3 (10 mmol, 0.76 g), and 2.5 mmol of the respective aryl/heteroaryl/methyl ketones 4a-n in 30 mL of ethanol was refluxed for 3–5 hrs. (Monitored by TLC). The reaction mixture was concentrated, then poured into 40 mL of ice-cold water. The formed solid product was filtered off, washed with water several times, dried, and crystallized from ethanol.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-2-oxo-6-phenyl-1,2-dihydropyridine-3-carbonitrile (5a)
Yield (87%); white solid; m.p.: 232–234 °C. IR (ATR) νmax 3139 (NH), 3069 (C–H aromatic), 2926, 2899 (C–H aliphatic), 2220 (C≡N), 1699 (C=O), 1651 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.86 (s, 1H, NH, disappeared by D2O), 7.89, 7.88 (d, J = 6.2 Hz, 2H, CHarom)., 7.62–7.55 (m, 8H, CHarom)., 6.88 (s, 1H, CHarom)., 2.35 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 162.23 (C=O), 150.88, 148.26, 137.77, 132.67, 131.76, 129.88, 129.46, 129.36, 128.20, 126.46, 125.74, 125.45, 116.51 (CN), 115.67, 107.56, 13.53 (CH3); Dept-135 NMR (100 MHz, DMSO-d6) δ 131.76, 129.88, 129.45, 129.36, 128.20, 125.44, 13.53; Anal. Calcd. for C22H15ClN4O (386.83): C, 68.31; H, 3.91; N, 14.48. Found: C, 68.18; H, 3.69; N, 14.59.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(3-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5b)
Yield (82%); yellow solid; m.p.: 198–200 °C. IR (ATR) νmax 3172 (NH), 3024 (C–H aromatic), 2999, 2968 (C–H aliphatic), 2218 (C≡N), 1669 (C=O), 1621 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H, NH, disappeared by D2O), 7.62–7.46 (m, 8H, CHarom)., 7.14 (s, 1H, CHarom)., 6.94 (s, 1H, CHarom)., 3.85 (s, 3H, OCH3), 2.35 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 162.13 (C=O), 160.02, 152.09, 150.87, 148.27, 137.79, 133.90, 130.60, 129.87, 129.33, 126.46, 125.42, 120.46, 118.06, 116.49 (CN), 115.69, 113.33, 113.04, 107.57, 55.90 (OCH3), 13.54 (CH3); Anal. Calcd. for C23H17ClN4O2 (416.85): C, 66.27; H, 4.11; N, 13.44. Found: C, 66.08; H, 4.31; N, 13.67.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(4-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5c)
Yield (89%); yellow solid; m.p.: 278–280 °C. IR (ATR) νmax 3180 (NH), 3043 (C–H aromatic), 2933, 2842 (C–H aliphatic), 2217 (C≡N), 1674 (C=O), 1612 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.82 (s, 1H, NH, disappeared by D2O), 7.89 (s, 2H, CHarom)., 7.62–7.55 (m, 5H, CHarom)., 7.10 (s, 2H, CHarom)., 6.85 (s, 1H, CHarom)., 3.85 (s, 3H, OCH3), 2.35 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 162.35 (C=O), 151.95, 150.92, 148.23, 137.79, 129.87, 129.72, 129.32, 127.56, 126.37, 125.43, 125.16, 116.69 (CN), 115.81, 114.96, 114.49, 114.37, 56.02 (OCH3), 13.52 (CH3); Anal. Calcd. for C23H17ClN4O2 (416.85): C, 66.27; H, 4.11; N, 13.44. Found: C, 66.42; H, 4.28; N, 13.71.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(4-methylphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5d)
Yield (81%); yellow solid; m.p.: > 300 °C. IR (ATR) νmax 3187 (NH), 3034 (C–H aromatic), 2916, 2876 (C–H aliphatic), 2221 (C≡N), 1685 (C=O), 1637 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.89 (s, 1H, NH, disappeared by D2O), 7.62–7.22 (m, 9H, CHarom)., 6.86 (s, 1H, CHarom)., 2.36, 2.34 (ss, 6H, 2CH3); 13C NMR (100 MHz, DMSO-d6) δ 148.24, 142.03, 138.21, 137.79, 130.05, 129.87, 129.73, 129.52, 129.34, 128.77, 128.10, 126.42, 126.07, 125.44, 125.16, 116.58 (CN), 115.73, 21.39 (CH3), 13.53 (CH3); Anal. Calcd. for C23H17ClN4O (400.86): C, 68.91; H, 4.27; N, 13.98. Found: C, 68.69; H, 4.08; N, 14.16.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(4-chlorophenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5e)
Yield (93%); white solid; m.p.: 296–298 °C. IR (ATR) νmax 3141 (NH), 3013 (C–H aromatic), 2920, 2846 (C–H aliphatic), 2222 (C≡N), 1671 (C=O), 1614 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.87 (s, 1H, NH, disappeared by D2O), 7.94, 7.93 (d, J = 4.5 Hz, 2H, CHarom)., 7.62–7.55 (m, 7H, CHarom)., 6.95 (s, 1H, CHarom)., 2.35 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 162.34 (C=O), 151.51, 150.68, 148.28, 137.77, 136.63, 131.74, 130.89, 130.06, 129.87, 129.45, 129.35, 126.47, 125.43, 116.37 (CN), 115.61, 108.08, 13.52 (CH3); Anal. Calcd. for C22H14Cl2N4O (421.27): C, 62.72; H, 3.35; N, 13.30. Found: C, 62.95; H, 3.51; N, 13.49.
6-(4-Bromophenyl)-4-(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5f)
Yield (85%); white solid; m.p.: 287–289 °C. IR (ATR) νmax 3132 (NH), 3009 (C–H aromatic), 2915, 2846 (C–H aliphatic), 2217 (C≡N), 1681 (C=O), 1636 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.88 (s, 1H, NH, disappeared by D2O), 7.86, 7.85 (d, J = 6.2 Hz, 2H, CHarom)., 7.76, 7.74 (d, J = 7.3 Hz, 2H, CHarom)., 7.62–7.54 (m, 5H, CHarom)., 6.95 (s, 1H, CHarom)., 2.35 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 162.32 (C=O), 150.69, 148.30, 137.71, 132.40, 131.89, 130.25, 129.89, 129.75, 129.37, 126.49, 125.53, 125.44, 125.14, 116.46 (CN), 115.56, 108.10, 13.55 (CH3); Anal. Calcd. for C22H14BrClN4O (465.73): C, 56.74; H, 3.03; N, 12.03. Found: C, 56.88; H, 3.29; N, 12.31.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(4-hydroxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5g)
Yield (78%); brownish red solid; m.p.: > 300 °C. IR (ATR) νmax 3356 (OH), 3129 (NH), 3011 (C–H aromatic), 2927 (C–H aliphatic), 2219 (C≡N), 1675 (C=O), 1621 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H, NH, disappeared by D2O), 8.01, 7.99 (d, J = 7.8 Hz, 2H, CHarom)., 7.59–7.53 (m, 7H, HO+6CHarom)., 6.95, 6.93 (d, J = 7.4 Hz, 2H, CHarom)., 2.50 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 187.26 (C=O), 162.66, 149.88, 137.74, 131.53, 131.37, 129.78, 129.53, 129.28, 128.53, 125.45, 121.28, 115.98, 114.32 (CN), 14.45 (CH3); Anal. Calcd. for C22H15ClN4O2 (402.83): C, 65.59; H, 3.75; N, 13.91. Found: C, 65.37; H, 3.92; N, 13.73.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(4-nitrophenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5h)
Yield (75%); yellow solid; m.p.: 281–283 °C. IR (ATR) νmax 3139 (NH), 3083 (C–H aromatic), 2967, 2926 (C–H aliphatic), 2217 (C≡N), 1681 (C=O), 1637 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H, NH), 8.57–7.13 (m, 5H, CHarom)., 7.64–7.57 (m, 4H, CHarom)., 7.12 (s, 1H, CHarom)., 2.38 (s, 3H, CH3); Anal. Calcd. for C22H14ClN5O3 (431.83): C, 61.19; H, 3.27; N, 16.22. Found: C, 61.42; H, 3.33; N, 16.39.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(2-furyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5i)
Yield (81%); brown solid; m.p.: 266–268 °C. IR (ATR) νmax 3110 (NH), 3039 (C–H aromatic), 2897, 2840 (C–H aliphatic), 2218 (C≡N), 1669 (C=O), 1644 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.84 (s, 1H, NH, disappeared by D2O), 8.01 (s, 1H, CHarom)., 7.63–7.55 (m, 6H, CHarom)., 6.83 (s, 1H, CHarom)., 6.79 (s, 1H, CHarom)., 2.34 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 148.13, 147.70, 137.78, 129.85, 129.36, 126.36, 125.50, 116.51 (CN), 115.53, 115.10, 113.75, 13.49 (CH3); Anal. Calcd. for C20H13ClN4O2 (367.79): C, 63.75; H, 3.48; N, 14.87. Found: C, 63.47; H, 3.26; N, 14.59.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-2-oxo-6-(2-thienyl)-1,2-dihydropyridine-3-carbonitrile (5j)
Yield (80%); yellow solid; m.p.: 271–273 °C. IR (ATR) νmax 3117 (NH), 3031 (C–H aromatic), 2975, 2927 (C–H aliphatic), 2214 (C≡N), 1667 (C=O), 1639 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.97 (s, 1H, NH), 8.08 (s, 1H, CHarom)., 7.90 (s, 1H, CHarom)., 7.62–7.54 (m, 6H, CHarom)., 7.27 (s, 1H, CHarom)., 2.33 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 148.25, 137.80, 132.12, 130.40, 130.09, 129.86, 129.50, 129.32, 126.40, 125.44, 116.31 (CN), 115.48, 13.42 (CH3); Anal. Calcd. for C20H13ClN4OS (392.86): C, 61.14; H, 3.34; N, 14.26. Found: C, 61.32; H, 3.53; N, 14.47.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-oxo-1,6-dihydro-2,3’-bipyridine-5-carbonitrile (5k)
Yield (85%); yellow solid; m.p.: 272–274 °C. IR (ATR) νmax 3154 (NH), 3040 (C–H aromatic), 2924, 2849 (C–H aliphatic), 2221 (C≡N), 1674 (C=O), 1638 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.99 (s, 1H, NH, disappeared by D2O), 9.08 (s, 1H, CHarom)., 8.74 (s, 1H, CHarom)., 8.29, 8.28 (d, J = 4.2 Hz 1H, CHarom)., 7.62–7.56 (m, 6H, CHarom)., 7.04 (s, 1H, CHarom)., 2.36 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 162.38 (C=O), 152.08, 150.70, 150.37, 148.87, 148.34, 137.73, 135.86, 129.89, 129.38, 129.05, 126.55, 125.42, 124.19, 116.30 (CN), 115.51, 108.58, 101.36, 13.49 (CH3); Anal. Calcd. for C21H14ClN5O (387.82): C, 65.04; H, 3.64; N, 18.06. Found: C, 65.28; H, 3.40; N, 18.35.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-oxo-1,6-dihydro-2,4’-bipyridine-5-carbonitrile (5l)
Yield (91%); pale yellow solid; m.p.: 276–278 °C. IR (ATR) νmax 3165 (NH), 3021 (C–H aromatic), 2837, 2810 (C–H aliphatic), 2206 (C≡N), 1663 (C=O), 1609 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H, NH, disappeared by D2O), 8.66 (s, 2H, CHarom)., 7.94 (s, 2H, CHarom)., 7.62–7.52 (m, 5H, CHarom)., 6.89 (s, 1H, CHarom)., 2.30 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 151.01 (C=O), 150.83, 150.11, 148.33, 140.97, 140.78, 137.77, 129.90, 129.37, 126.49, 125.44, 122.00, 116.45 (CN), 115.60, 109.07, 106.61, 13.49 (CH3); Anal. Calcd. for C21H14ClN5O (387.82): C, 65.04; H, 3.64; N, 18.06. Found: C, 65.16; H, 3.49; N, 18.23.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(1-naphthyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5m)
Yield (89%); pale yellow solid; m.p.: > 300 °C. IR (ATR) νmax 3161 (NH), 3008 (C–H aromatic), 2922 (C–H aliphatic), 2223 (C≡N), 1678 (C=O), 1636 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.10 (s, 1H, NH, disappeared by D2O), 8.14–8.01 (m, 4H, CHarom)., 7.72–7.57 (m, 9H, CHarom)., 2.38 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 148.32, 137.76, 133.60, 132.90, 131.14, 130.56, 129.85, 129.37, 129.02, 128.67, 128.31, 128.12, 127.95, 127.57, 127.08, 126.56, 125.70, 125.48, 125.06, 124.72, 116.62 (CN), 115.72, 115.52, 13.62 (CH3); Anal. Calcd. for C26H17ClN4O (436.89): C, 71.48; H, 3.92; N, 12.82. Found: C, 71.64; H, 3.79; N, 12.60.
4-(5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)-6-(2-naphthyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (5n)
Yield (90%); yellow solid; m.p.: > 300 °C. IR (ATR) νmax 3172 (NH), 3053 (C–H aromatic), 2920 (C–H aliphatic), 2221 (C≡N), 1680 (C=O), 1636 (C=N) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 13.06 (s, 1H, NH, disappeared by D2O), 8.57 (s, 1H, CHarom)., 8.15–7.95 (m, 4H, CHarom)., 7.64–7.56 (m, 7H, CHarom)., 7.08 (s, 1H, CHarom)., 2.38 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 162.29 (C=O), 150.77, 148.29, 137.80, 134.39, 132.91, 129.88, 129.73, 129.36, 129.08, 128.67, 128.50, 128.12, 127.96, 127.55, 127.10, 126.47, 125.45, 125.16, 124.72, 124.09, 116.53 (CN), 115.74, 13.56 (CH3); Anal. Calcd. for C26H17ClN4O (436.89): C, 71.48; H, 3.92; N, 12.82. Found: C, 71.72; H, 4.11; N, 12.98.
Biological Evaluation
Inhibitory Activity of NO Over-Production
NO colorimetric assay kit (ab65328, Abcam Co., Waltham, MA, USA) was used to analyze the content of NO in brain tissue homogenate, following the manufacturer’s instructions. The kit measures the total nitrate/nitrite content in a simple two-step process. The first step converts nitrate to nitrite utilizing nitrate reductase. The second step uses Griess Reagents to convert nitrite to a deep purple azo compound. The amount of the azochromophore accurately reflects the nitric oxide amount in samples.
In vitro Cyclooxygenase (COX) Inhibition Assay
The tested compounds and celecoxib were evaluated for their ability to inhibit COX-1/COX-2 isozymes using a fluorometric enzyme kit as recommended by the manufacturer (catalog no. K548-100 for COX-1, and K547-100 for COX-2, Bio Vision, Milpitas, CA, U.S.A).37
Inhibitory Activity on PGE2 Production
ELISA kit (catalog No. KGE004B, R&D Systems, Minneapolis, MN) was used to measure the concentration of prostaglandin E2 (PGE2) in the culture media as recommended by the manufacturer.38 This assay is based on the forward sequential competitive binding technique in which PGE2 present in a sample competes with horseradish peroxidase (HRP)-labeled PGE2 for a limited number of binding sites on a mouse monoclonal antibody. PGE2 in the sample is allowed to bind to the antibody in the first incubation. During the second incubation, HRP-labeled PGE2 binds to the remaining antibody sites. Following a wash to remove unbound materials, a substrate solution is added to the wells to determine the bound enzyme activity. The color development is stopped, and the absorbance is read at 450 nm. The intensity of the color is inversely proportional to the concentration of PGE2 in the sample.
Assessment of Tissue TNF-α Levels
To detect the levels of TNF-α (cat. no. ab181421; Abcam) in the cell supernatants, related ELISA human TNF alpha Simple Step assay kits were used according to the manufacturer’s recommendations.39 Moreover, the murine macrophage cell line RAW 264.7 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA; catalog no. TIB-71) and cultured according to the supplier’s instructions.
In vitro 5- and 15-LOX Inhibition Assay
The ability of the tested compounds to inhibit the 5- and 15-LOX enzymes was evaluated using the 5-LOX inhibitory fluorometric and 15-LOX-2 human recombinant screening assay kits (kit catalog number K980 for 5-LOX, and kit catalog number 10011263 for 15-LOX, Cayman Chemical, Ann Arbor, MI), following the manufacturer’s instructions.40
Inhibitory Activity of iNOS Enzymatic Activity
iNOS in vitro simple step ELISA kit (cat. No. ab253219, abcam, USA) was used for the quantitative measurement of iNOS protein in mouse cell extract samples, following the manufacturer’s instructions.41
MTT Cytotoxicity Assay
An in vitro toxicology assay kit was used to determine the cell number spectrophotometrically as a function of mitochondrial dehydrogenase activity in living cells (Sigma TOX-1), following the manufacturer’s instructions.42 See
Docking Methodology
Molecular docking analyses were conducted using the Maestro suite (Schrödinger Release 2024.1). The most stable conformations of the lead compounds 5k, 5f, and 5m were docked into the respective target enzymes as follows:1 compound 5k was docked into the active site of the COX-2 enzyme (PDB ID: 5F19),43 alongside the reference inhibitors celecoxib and etoricoxib;2 compound 5f was docked into the active site of the 5-LOX enzyme (PDB ID: 3V99),44 in comparison with the reference drug zileuton and the native ligand arachidonic acid; and3 compound 5m was docked into the active site of the iNOS enzyme (PDB ID: 1R35),45 along with its native ligand I58. The three-dimensional structures of COX-2, 5-LOX, and iNOS were retrieved from the RCSB Protein Data Bank. All essential steps of the docking protocol, including ligand preparation,46,47 protein preparation,48 receptor grid generation, and molecular docking,49 were carried out following standard methodologies.3,50
Molecular Dynamics Simulations
The stability of the most active 3-cyano-2-pyridinone compounds 5k, 5f, and 5m, in complex with the active sites of COX-2, 5-LOX, and iNOS, respectively, was assessed through molecular dynamics (MD) simulations. Selected reference compounds were also included for comparative analysis. These simulations were performed using the OPLS3e force field within the Desmond software package (Desmond 2024–1, Schrödinger LLC).51,52 The MD workflow encompassed essential preparatory steps, including system setup, solvation, charge neutralization, and molecular dynamics optimization under standard temperature and pressure conditions (NPT ensemble).53,54 A comprehensive account of the simulation protocol is consistent with previously established methodologies.
Results and Discussion
Chemistry
Herein, we report the synthesis of a novel series of 3-cyanopyridinone derivatives 5a-n hybridized with a pyrazole moiety via a one-pot, four-component reaction involving pyrazole aldehyde derivative 1, ethyl cyanoacetate 2, ammonium acetate 3, and the respective aryl/heteroaryl/methyl ketones 4a-n in ethanol (Scheme 1).
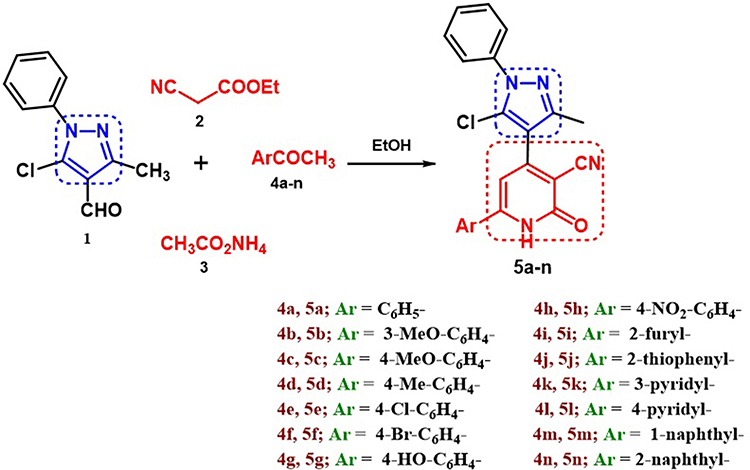 |
Scheme 1 The synthesis of cyanopyridines 5a-n via one-pot, four-component reaction. |
The chemical structures of the newly synthesized pyrazole/3-cyano-2-pyridinone hybrids 5a-n were confirmed by the spectral analyses IR, 1H NMR, 13C NMR, Dept-135. Detailed results in Supporting Information Introduction (supplementary Figures 1–48)., and elemental analyses. The IR spectrum of pyrazole/3-cyano-2-pyridinone hybrids 5a (as a representative example) showed characteristic absorption bands corresponding to NH at 3139 cm−1, CH aromatic at 3069 cm−1, CH aliphatic at 2926 and 2899 cm-1, cyano group at 2220 cm-1, C=O group at 1699 cm-1, and C=N group at 1651 (C=N) cm−1. The 1H NMR spectrum of 5a displayed three singlet signals at 12.86, 6.88, and 2.35 ppm, corresponding to NH, CHpyridyl, and CH3, respectively; Additionally, a doublet at 7.89 and 7.88 ppm was observed for two aromatic protons with a coupling constant of 6.2 Hz, along with a multiplet at 7.62–7.55 ppm for eight aromatic protons. The 13C NMR of compound 5a revealed characteristic three signals of C=O, cyano, and CH3 at 162.23, 116.51, and 13.53 ppm, respectively; The remaining aromatic and sp2-hybridized carbons were observed at 150.88, 148.26, 137.77, 132.67, 131.76, 129.88, 129.46 (2CH), 129.36, 128.20, 126.46, 125.74, 125.45, 115.67, and 107.56 ppm. The Dept-135 NMR spectrum confirmed the absence of all quaternary carbon signals and showed positive signals corresponding to aromatic CH groups and the methyl group at 131.76, 129.88, 129.45, 129.36, 128.20, 125.44 (2CH), and 13.53 ppm, respectively.
Biological Evaluation
Inhibitory Activity of NO Over-Production
Nitric oxide (NO) is an ancient messenger. It is one of the most prominent inflammatory and regulatory mediators. It is a multifaceted molecule crucial for diverse pathological and physiological body processes, including neurotransmission, bronchodilation, vasodilatation, and most significantly, modulation of the host immune response.55,56 Nitric oxide (NO) is a signaling molecule generated endogenously in mammalian cells by nitric oxide synthase (NOS), which catalyzes NADPH-dependent oxidation of L-arginine into NO and L-citrulline.57 NOS exists in three isoforms; two of them, endothelial NOS (eNOS) and neuronal NOS (nNOS), are constitutively expressed, while the third one is inducible (iNOS).19 An inappropriately high concentration of NO resulting from overexpression or dysregulation of iNOS, on the other hand, can engender toxic effects and is associated with a wide variety of human diseases, including inflammation, carcinogenicity, autoimmune disorders, cardiac dysfunction, and diabetes.58,59
The dual activity of iNOS-related NO (beneficial vs detrimental) is eminently concentration-dependent. Accordingly, regulation of NO production is crucial for both maintaining its proper physiological functions and controlling its detrimental effects.19,60
Therefore, all the synthesized cyanopyridine derivatives 5a-n were evaluated for their potential abilities to decrease the release of NO, which indicates their abilities to inhibit iNOS activity and expression. The results in Table 1 displayed that compound 5g (Ar = 4-HO-C6H4-) is the most potent inhibitor of NO release among all the tested compounds. It decreased the level of NO from 8.60 nmol/mL to 3.42 nmol/mL (60.23% inhibition) with a 0.39-fold decrease in NO production. Compound 5m (Ar = 1-naphthyl-) comes next with a 0.42-fold decrease in NO production (3.67 nmol/mL, 57.35%), followed by compound 5f (Ar = 4-Br-C6H4-) with a 0.43-fold decrease in NO production (3.71 nmol/mL, 56.90%). On the other hand, compound 5k (Ar = 3-pyridyl-) exhibited moderate inhibition of NO with a 0.48-fold decrease (4.16 nmol/mL, 51.60%). All other derivatives possessed weak inhibitory activity on NO production with a range of 22.14%-30.49% percent inhibition.
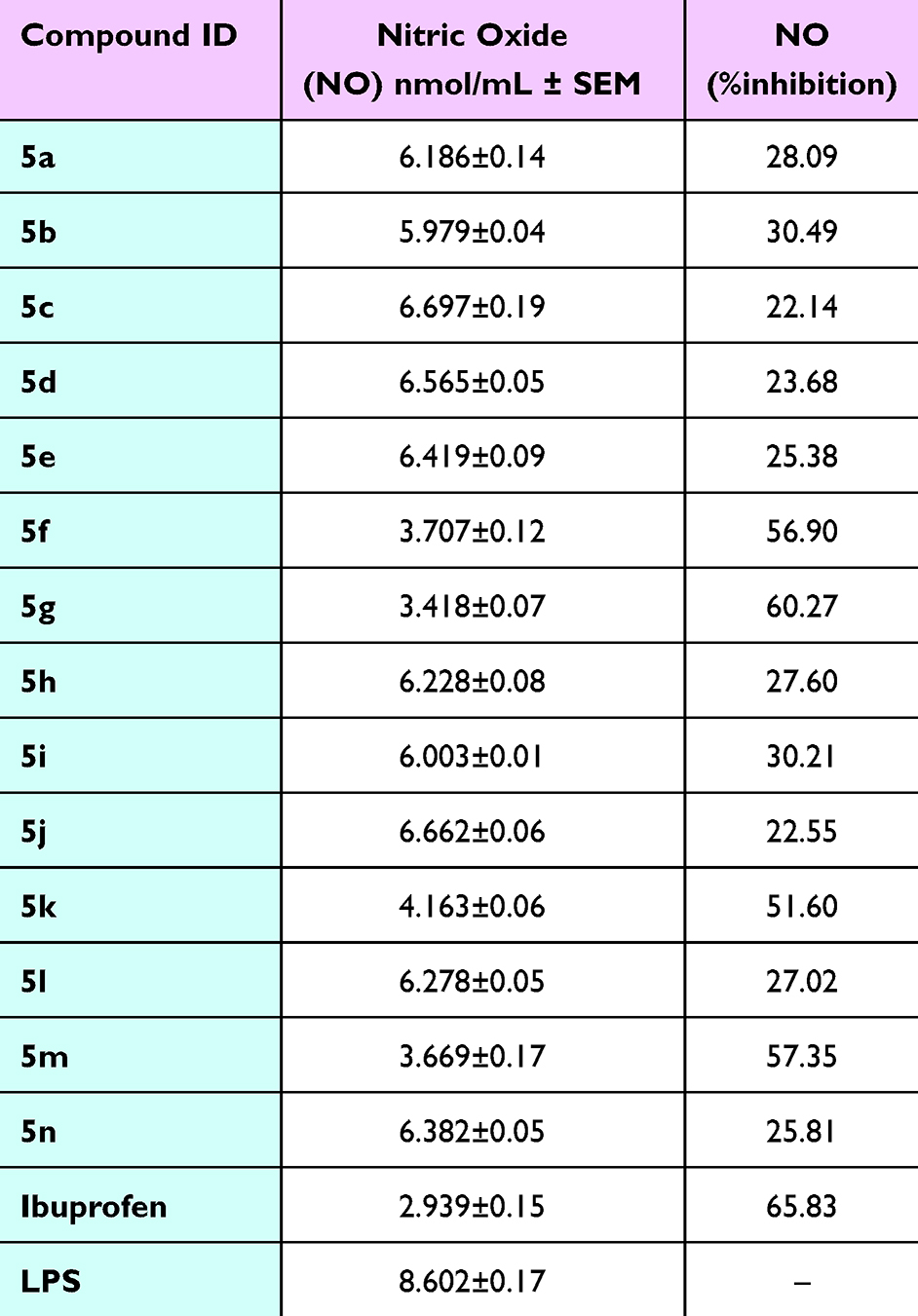 |
Table 1 The Inhibitory Activity of Cyanopyridine Derivatives 5a-n (10 μg/mL) in LPS-Induced Raw 264.7 Cells on NO Production |
Therefore, it could be implied that the ability of these compounds to decrease the NO overproduction may be due to their ability to inhibit overactivity and/or overexpression of iNOS. Consequently, the iNOS inhibitory activity of these compounds was assessed. Detailed results in Supporting Information Materials and Methods.
In vitro Cyclooxygenase (COX) Inhibition Assay
The in vitro biological activity assay was used to investigate the ability of compounds to inhibit both bovine COX-1 and human recombinant COX-2 isozymes. The potency of the tested compounds was calculated to be the concentration causing 50% inhibition of the enzyme (IC50). A fluorometric enzyme kit was used to monitor the isozyme-specific inhibition.61 Moreover, selectivity indices (SI values) against COX-2 were estimated as IC50 (COX-1)/IC50 (COX-2) and compared with celecoxib as the reference drug.62
The four most potent derivatives, 5f, 5g, 5k, and 5m, which exhibited considerable inhibition of NO production, were assessed for their ability to inhibit both COX1 and COX-2 isozymes in vitro. As illustrated in Table 2, all the tested compounds displayed a notable selectivity toward COX-2 (IC50 = 0.92–4.94 μM range) more than COX-1 (IC50 = 6.83–18.07 μM range) with selectivity indices (SI) of 2.95–19.6 using celecoxib as reference drug (COX-1 IC50 = 21.48 μM; COX-2 IC50 = 0.391 μM; COX-2 selectivity index = 53.74).63
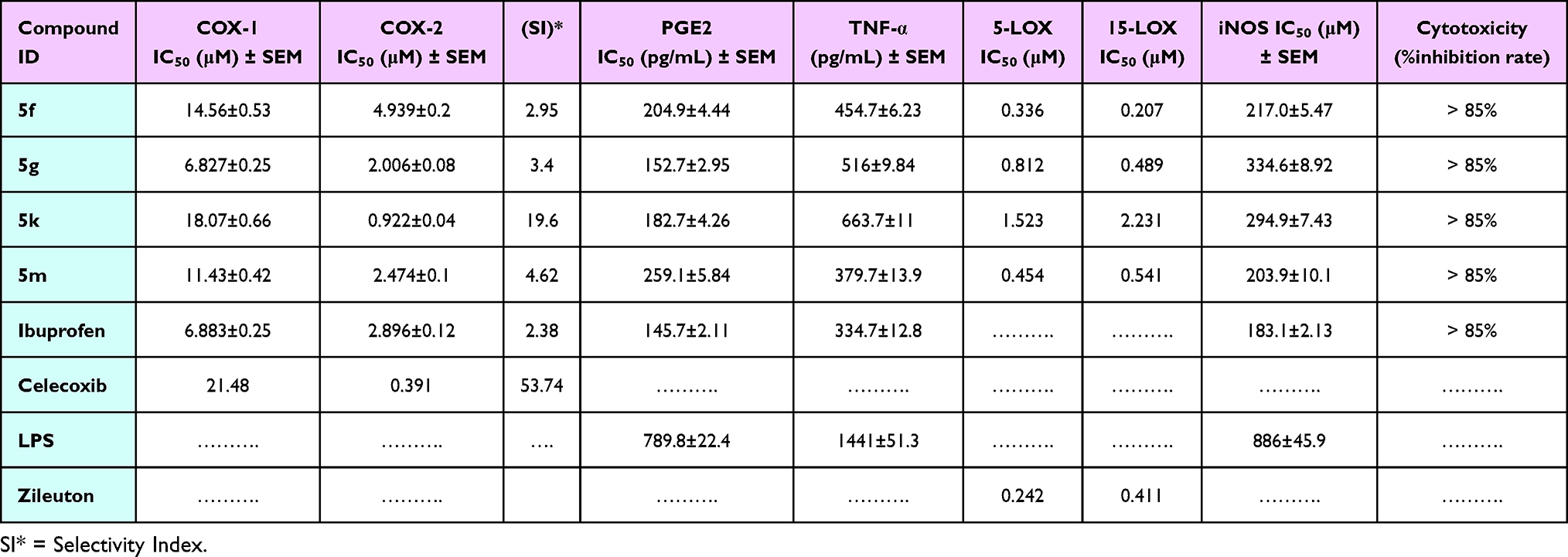 |
Table 2 In vitro Inhibitory Activity of the Investigated Compounds 5f, 5g, 5k, and 5m on COX-1, COX-2, PGE2, TNF-α, 5-LOX, 15-LOX, and iNOS Isozymes, Along with Their Evaluated Cytotoxicity Percentages |
Among the four tested compounds, compound 5k (Ar = 3-pyridyl-) was the most potent. It displayed significant inhibitory activity against COX-2 (IC50 = 0.92 μM) compared to the reference drug, celecoxib (COX-2 IC50 = 0.391 μM) and COX-1 (IC50 = 18.07 μM) with a significantly elevated selectivity index (SI) of 19.6. On the other hand, compounds 5m (Ar = 1-naphthyl-) and 5g (Ar = 4-HO-C6H4-) possessed IC50 values of 2.47 and 2.00 μM, respectively, against COX-2, which are comparable to that of celecoxib (COX-2 IC50 = 0.391 μM), and displayed moderate selectivity indices (4.62 and 3.4, respectively).
Moreover, compound 5f (Ar = 4-Br-C6H4-) was the least potent derivative with an IC50 value of 4.94 μM against COX-2 isozyme and a SI of 2.38. Based on the abovementioned results, the beneficial selectivity of all the tested compounds may predict promising anti-inflammatory activity with minimal undesirable side effects associated with COX-1 inhibition, Supporting Information Results and Discussion.
Inhibitory Activity on PGE2 Production
Prostaglandin E2 (PGE2) is a valuable physiologically active lipid, which is biosynthesized from arachidonic acid (AA) by COX-1, COX-2, and PGE synthases.26 PGE2 is abundantly produced in the region of inflammation, causing vasodilation that contributes to the vascular manifestations of inflammation and induces edema formation.64 Therefore, the most active four derivatives 5f, 5g, 5k, and 5m were further assessed for their effect on the level of PGE2 in LPS-induced RAW 264.7 cells. Results revealed that the four tested compounds, 5f, 5g, 5k, and 5m, exhibited inhibitory activity on the PGE2 production with IC50 values of 204.9, 152.7, 182.7, and 259.1 pg/mL, respectively, compared to ibuprofen (IC50 = 145.7 pg/mL), as shown in Table 2. The maximum PGE2 inhibition was observed for compound 5g (Ar = 4-HO-C6H4) with an IC50 value of 152.7 pg/mL (80.6% inhibition), which is 0.19-fold less than the PGE2 produced in LPS-induced cells. Compound 5k (Ar = 3-pyridyl) comes next in PGE2 inhibition with an IC50 value of 182.7 pg/mL and 76.87% percent inhibition. However, compounds 5f (Ar = 4-Br-C6H4) and 5m (Ar = 1-naphthyl) had lower PGE2 inhibitory potency with percent inhibition of 74.06% and 67.19%, respectively. Detailed results in Supporting Information Results and Discussion.
Assessment of Tissue TNF-α Levels
TNF-α is a key proinflammatory mediator, which has a significant role in the initiation of the inflammatory cascade and prolongation of the duration of inflammation.65 The promising in vitro anti-inflammatory potency of the four tested compounds motivated us to investigate their possible mechanism of action. Therefore, TNF-α levels were assessed using an ELISA kit in LPS-induced RAW 264.7 cells to determine the ability of these compounds to block the action of TNF-α.66 As shown in Table 2, it was revealed that all the tested cyanopyridine derivatives 5f, 5g, 5k, and 5m displayed a significant reduction in serum content of TNF-α. Compound 5m (Ar = 1-naphthyl) exhibited remarkable TNF-α inhibitory activity (379.7±13.9 pg/mL), which was nearly the same TNF-α inhibitory activity exerted by ibuprofen (334.7±12.8 pg/mL). At the same time, compound 5f (Ar = 4-Br-C6H4) comes next in reducing TNF-α serum content (454.7±6.23 pg/mL), which is 1.35-fold less than ibuprofen. Nonetheless, compounds 5g (Ar = 4-HO-C6H4) and 5k (Ar = 3-pyridyl) exerted the least inhibitory activity (516±9.84 and 663.7±11 pg/mL, respectively) compared to ibuprofen. These outcomes suggested that the anti-inflammatory activity of the tested compounds may be attributed to their ability to inhibit the serum content of TNF-α. Detailed results in Supporting Information Results and Discussion.
In vitro 5- and 15-LOX Inhibition Assay
5-LOX was reported to be associated with inflammatory reactions and diverse human diseases, such as atherosclerosis, colon cancer, pulmonary arterial hypertension, and asthma.67 Furthermore, 15-LOX-1 plays a dual role in inflammatory reactions as its activity produces mediators that function both in the progression and in the resolution of inflammation.68 Therefore, 5- and 15-LOX inhibitory activities of the four most potent compounds 5f, 5g, 5k, and 5m were assessed using zileuton as the reference drug. The IC50 values of the tested compounds are displayed in Table 2. The results revealed that compound 5f (Ar = 4-Br-C6H4) suppressed the 5-LOX enzymatic activity with an IC50 value of 0.34 μM, compared to zileuton (IC50 = 0.24 μM). Moreover, compound 5f significantly inhibited the 15-LOX enzyme with an IC50 value of 0.21 μM, which is 0.5-fold lower than the reference drug zileuton (IC50 = 0.41 μM). At the same time, compound 5g (Ar = 4-HO-C6H4) exhibited almost an equipotent ability to inhibit the 15-LOX enzyme compared to the reference zileuton, with IC50 values of 0.49 and 0.41 μM, respectively. However, it showed weaker 5-LOX inhibitory activity with an IC50 value of 0.81 μM in comparison to zileuton (IC50 = 0.24 μM). On the other hand, compounds 5k (Ar = 3-pyridyl) and 5m (Ar = 1-naphthyl) possessed the weakest 5-LOX inhibitory activity with IC50 values of 1.52 and 0.45 μM, respectively. Also, they exhibited the weakest inhibition of 15-LOX with IC50 values of 2.23 and 0.54 μM, respectively. Detailed results in Supporting Information Results and Discussion.
Inhibitory Activity of iNOS Enzymatic Activity
The iNOS/NO system has garnered increasing attention over the last decade, as it is intricately involved in several biological processes. Many research studies reported that high activation of iNOS during inflammatory response stimulates a large and prolonged production of nitric oxide.69,70 Therefore, compounds 5f, 5g, 5k, and 5m that exhibited a remarkable suppression rate of NO production from RAW 264.7 cells were evaluated for their ability to inhibit the activity of iNOS using the in vitro SimpleStep ELISA Kit.41 As displayed in Table 2, treatment of LPS-induced RAW 264.7 cells with the selected compounds 5f, 5g, 5k, and 5m caused a good inhibition of iNOS activity with IC50 values of 217.0, 334.6, 294.9, and 203.9 μM, respectively, compared to ibuprofen (IC50 = 183.1 μM). Compound 5m (Ar = 1-naphthyl) was the most active derivative in suppressing iNOS enzymatic activity in LPS-induced RAW264.7 cells with an IC50 value of 203.9 μM. While compounds 5f (Ar = 4-Br-C6H4), 5g (Ar = 4-HO-C6H4), and 5k (Ar = 3-pyridyl) exhibited lower inhibitory activity against the iNOS enzyme with IC50 values of 217.0, 334.6, and 294.9 μM, respectively, in comparison to ibuprofen (IC50 = 183.1 μM). Detailed results in Supporting Information Results and Discussion.
MTT Cytotoxicity Assay
MTT assay was performed on compounds 5f, 5g, 5k, and 5m to assess the correlation between the cell viability and the inhibitory activity of these compounds on NO over-production. No noticeable cytotoxicity (IC50 > 85 μM) of these compounds was observed on RAW 264.7 cells with or without LPS when used at the same inhibitory concentration on LPS-induced inflammation. These findings revealed that the four tested compounds suppressed the inflammatory response induced by LPS without compromising cell viability (Table 2), Detailed results in Supporting Information Results and Discussion.
Molecular Docking Study
To achieve accurate validation of the molecular basis underlying the proposed biological activities, molecular docking analyses were conducted for the most active novel pyrazole/3-cyanopyridinone compounds. The evaluated docked complexes included: (1) compound 5k within the active site of COX-2, compared to the reference inhibitors celecoxib and etoricoxib; (2) compound 5f within the 5-LOX active site, aligned with the native ligand arachidonic acid and the reference drug zileuton; and (3) compound 5m bound to iNOS, in comparison to the native ligand I58.
Docking Analysis of the Pyrazole/3-Cyanopyridinone Hybrid 5k and the Celecoxib and Etoricoxib References Into the COX-2 Active Site
Although COX-2 possesses distinct cyclooxygenase and peroxidase active sites, which are functionally interconnected through a bridging heme group. During the biosynthesis of prostaglandin H2 (PGH2), arachidonic acid (AA) binds within the cyclooxygenase channel, positioning its carboxylate group near Arg-120 and Tyr-355 at the channel entrance. At the same time, its ω-end is accommodated within a hydrophobic groove.43,71 Following the redocking of the investigated compound 5k, alongside the reference drugs celecoxib and etoricoxib, into the cyclooxygenase (COX-2) active site, several key interactions were observed. Celecoxib adopted an inverted binding orientation that allowed it to engage the critical Arg120 residue via a π-cation interaction (5.62 Å), facilitated by its pyrazole core. Additionally, its sulfonamide moiety formed a hydrogen bond donor interaction with Ser353 (2.02 Å) through the primary amino group, as well as a hydrogen bond acceptor interaction with Phe218 (2.59 Å) via the sulfonyl oxygen. These interactions contributed to a notable binding affinity, with a docking score of −9.107 kcal/mol (Figure 4: A1, B1, and C1). In the case of etoricoxib (S= −5.333 kcal/mol), the molecule engaged residues located at the entrance of the cyclooxygenase channel through its sulfonyl oxygens. Specifically, it formed two hydrogen bond acceptor interactions with Arg120 (1.96 Å and 2.11 Å) and another with Tyr355 (1.84 Å). Furthermore, the pyridine ring of etoricoxib participated in π–π* stacking with Tyr115 (5.03 Å) (Figure 4: A2, B2, and C2). Similarly, compound 5k was accommodated within the COX-2 active site in a binding orientation comparable with that of etoricoxib, achieving a docking score of −5.776 kcal/mol. The amide functionality of its 2-pyridinone ring established a hydrogen bond acceptor interaction with Arg120 (2.18 Å), resembling the sulfonyl interactions observed in etoricoxib. Moreover, its pyrazole core facilitated interaction with Tyr115 through π-stacking (5.35 Å) (Figure 4: A3, B3, and C3). In summary, all three ligands engaged the critical Arg120 residue at the entrance of the COX-2 cyclooxygenase active site, a key interaction that may obstruct AA binding and subsequently inhibit PGH2 synthesis. These findings suggest that compound 5k may possess promising anti-inflammatory potential, warranting further biological validation.
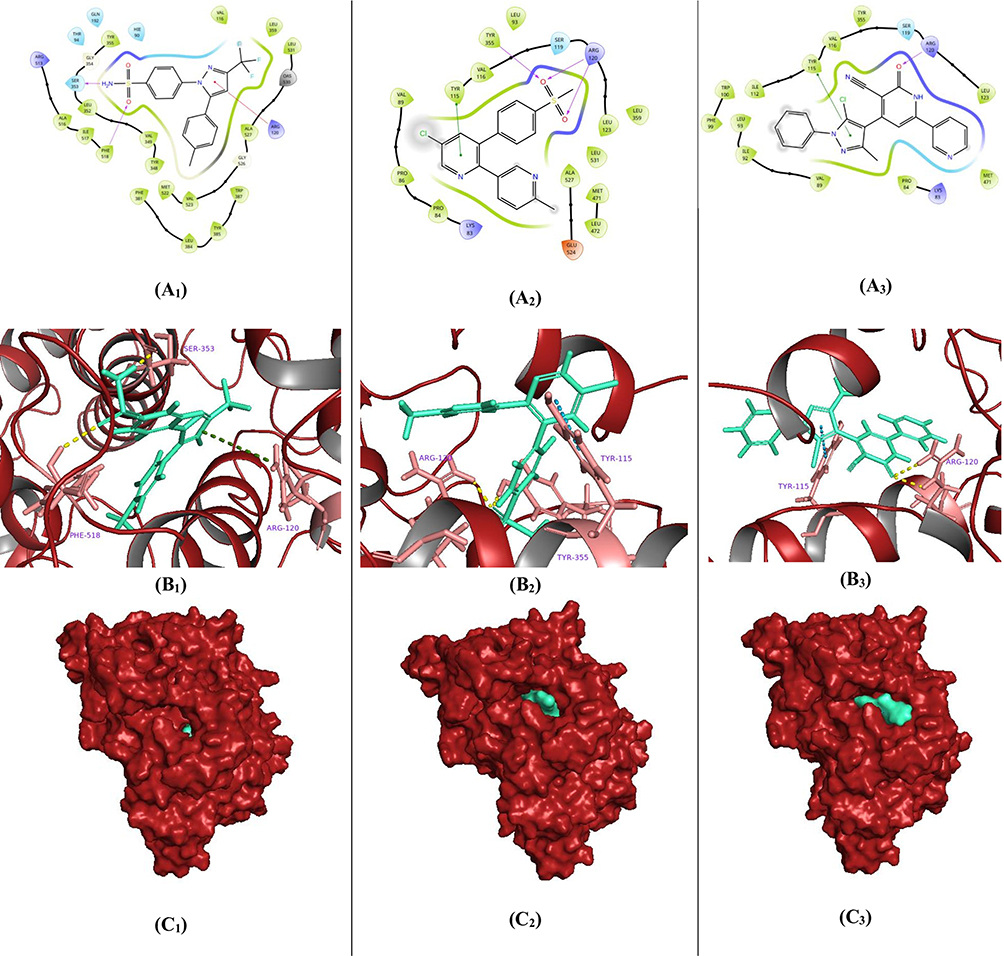 |
Figure 4 Binding conformations of the investigated most active pyrazole/3-cyanopyridinone candidate 5k (A3) related to celecoxib and etoricoxib references within the active site of the COX-2 enzyme. Two-dimensional schematic depicting key molecular interactions between celecoxib (A1), etoricoxib (A2), and 5k compound (A3) and the COX-2 active site. Three-dimensional representation illustrating the spatial orientations of compounds celecoxib (B1), etoricoxib (B2), and 5k (B3) (rendered in green cyan stick format) within the COX-2 binding pocket. Surface visualization of the COX-2 protein (depicted in brick red), demonstrating the surface-bound conformation of celecoxib (C1), etoricoxib (C2), and 5k (C3) compounds using a green cyan space-filling model. |
Docking Analysis of the Pyrazole/3-Cyanopyridinone Hybrid 5f Related to the Native Ligand Arachidonic Acid and the Reference Drug Zileuton Inside the 5-LOX Active Site
5-Lipoxygenase (5-LOX) plays a pivotal role in the biosynthesis of pro-inflammatory leukotrienes, utilizing arachidonic acid (AA) as its natural substrate and requiring the 5-LOX-activating protein (FLAP) as a cofactor for optimal enzymatic activity.46 In this study, molecular docking was performed to evaluate the binding interactions of AA, the reference inhibitor zileuton, and the investigated compound 5f within the active site of 5-LOX. Interestingly, both AA and compound 5f exhibited a similar binding orientation within the 5-LOX active site. Notably, compound 5f demonstrated a more favorable binding affinity with a docking score of −6.086 kcal/mol, surpassing that of AA (−5.699 kcal/mol). In the case of zileuton, the docking analysis revealed a distinct binding mode, characterized by a single prominent hydrogen bond acceptor interaction between its carboxamide oxygen and Val175 (2.56 Å) (Figure 5: A1, B1, and C1). Arachidonic acid formed two hydrogen bond acceptor interactions through its carboxylate moiety with Asp229 (1.76 Å) and Gly429 (1.84 Å) (Figure 5: A2, B2, and C2). In contrast, compound 5f engaged in a significant π–π* stacking with Phe359 with a bond length of 5.30 Å, contributing to its enhanced binding affinity (Figure 5: A3, B3, and C3). Zileuton showed a relatively lower binding score (−4.549 kcal/mol), suggesting a weaker affinity in the context of competitive binding at the active site. Overall, the findings suggest that compound 5f may act as a competitive inhibitor of AA at the 5-LOX active site due to its similar binding conformation and higher binding affinity. Conversely, zileuton may exert its inhibitory effect through an allosteric mechanism. Therefore, compound 5f holds promise as a potential anti-inflammatory agent by suppressing leukotriene biosynthesis via competitive inhibition of the AA substrate.
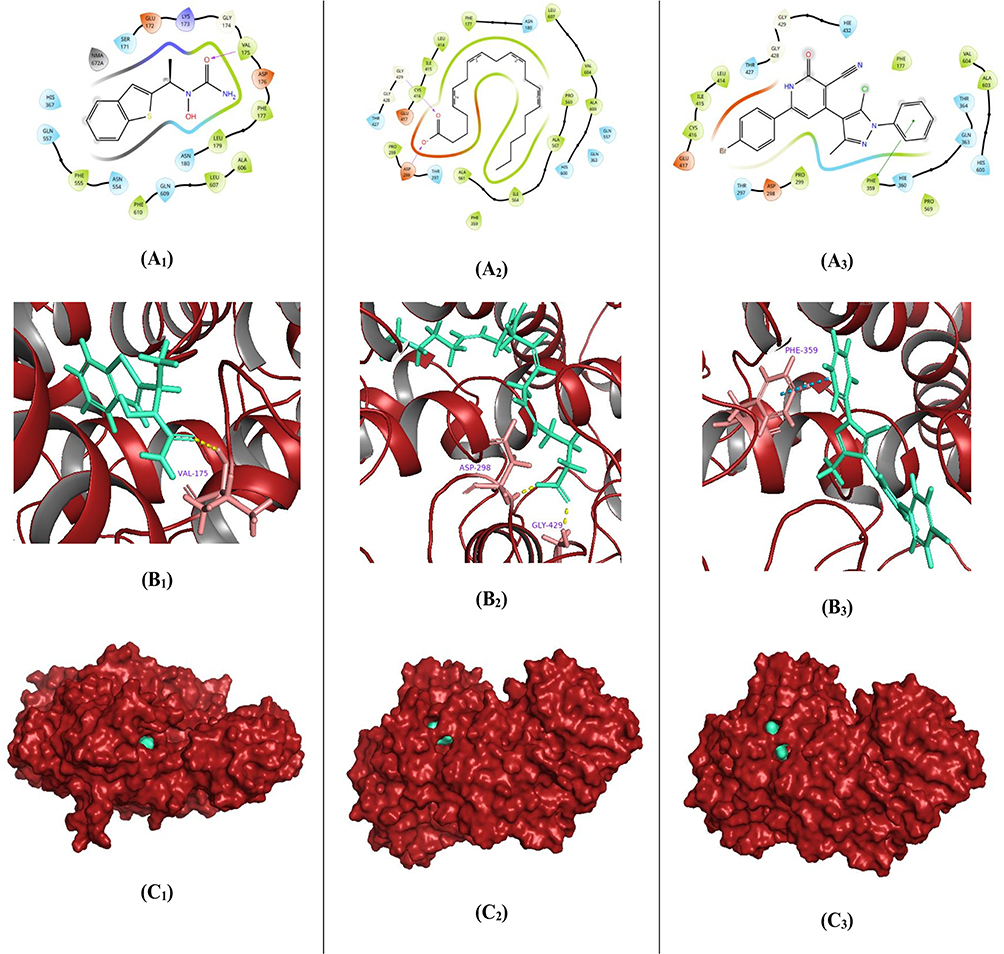 |
Figure 5 Binding conformations of the most active pyrazole/3-cyanopyridinone candidate 5f related to zileuton and arachidonic acid references within the active site of the 5-LOX enzyme. Two-dimensional schematic depicting key molecular interactions between zileuton (A1), arachidonic acid (A2), and 5f compounds (A3) and the 5-LOX active site. Three-dimensional representation illustrating the spatial orientations of compounds zileuton (B1), arachidonic acid (B2), and 5f (B3) (rendered in green cyan stick format) within the 5-LOX binding pocket. Surface visualization of the 5-LOX protein (depicted in brick red), demonstrating the surface-bound conformation of zileuton (C1), arachidonic acid (C2), and 5f (C3) compounds using a green cyan space-filling model. |
Docking Analysis of the Pyrazole/3-Cyanopyridinone Hybrid 5m and the Native Ligand I58 Inside the iNOS Active Site
Elevated levels of nitric oxide (NO), produced by the catalytic activity of inducible nitric oxide synthase (iNOS) on the L-arginine substrate, are known to contribute to cellular cytotoxicity and tissue damage through the formation of reactive NO-derived metabolites.72 The C4-fluorinated L-lysine derivative (I58), incorporating an amidine moiety as a functional isostere of the guanidine group in L-arginine, was previously introduced as a competitive substrate analog for iNOS. This compound was hypothesized to reduce NO production by preferential binding to the iNOS active site.45 Structurally, compound I58 is composed of three functional fragments: an amino acid-derived tail, a fluorinated aliphatic carbon chain as the central linker, and an amidine-functionalized head group. The molecule adopts a bent conformation, which facilitates multiple hydrogen bond donor (HBD) interactions. Specifically, the amidine NH groups form two HBD interactions with Glu371, with bond lengths of 1.77 Å and 1.83 Å, while the amino and carboxyl groups of the tail engage in dual HBD interactions with the key Asp376 residue, at 1.68 Å and 1.74 Å, respectively46 (Figure 6: A1, B1, and C1). Remarkably, the novel compound 5m exhibited a significantly improved binding affinity at the iNOS active site, achieving a docking score of −7.030 kcal/mol, compared to the redocked reference compound I58 (−2.869 kcal/mol). The enhanced affinity of compound 5m was attributed primarily to three strong π–π* stacking interactions mediated by its naphthyl ring: two interactions with Trp188 (of bond lengths 3.61 Å and 3.85 Å) and one with Phe363 (4.08 Å). Additionally, two halogen bonding interactions were observed between the chlorine substituent of 5m and Ile195 (2.78 Å) and Gly196 (3.45 Å) (Figure 6: A2, B2, and C2). Overall, these docking results indicate a favorable binding profile for compound 5m at the iNOS active site, suggesting its potential to effectively inhibit NO production by outcompeting the native substrate L-arginine, thereby contributing to its proposed anti-inflammatory activity.
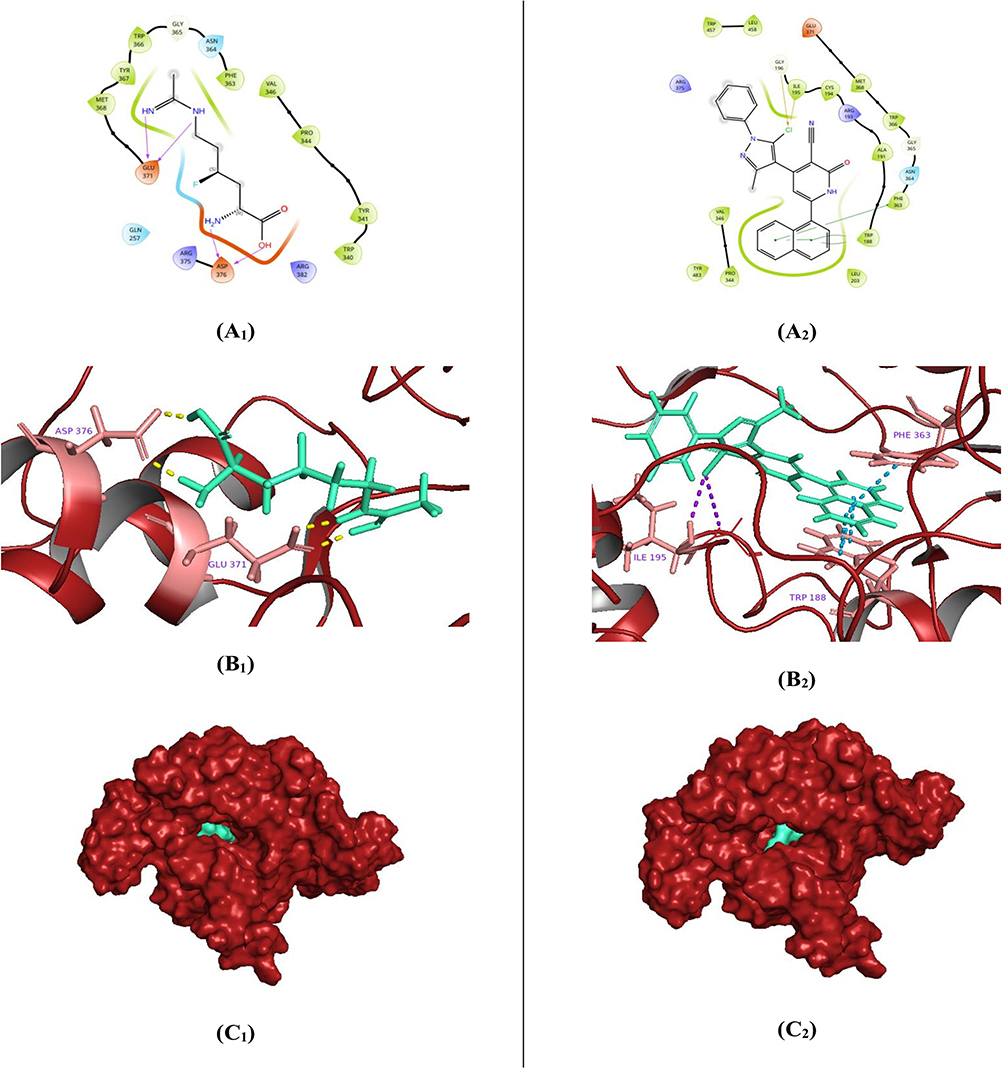 |
Figure 6 Binding conformations of the investigated most active pyrazole/3-cyanopyridinone candidate 5m related to I58 native ligand within the active site of the iNOS enzyme. Two-dimensional schematic depicting key molecular interactions between I58 (A1) and compound 5m (A2) and the iNOS active site. Three-dimensional representation illustrates the spatial orientations of I58 (B1) and compound 5m (B2) (rendered in green, cyan stick format) within the iNOS binding pocket. Surface visualization of the iNOS protein (depicted in brick red), demonstrating the surface-bound conformation of I58 (C1) and compound 5m (C2) using a green, cyan space-filling model. |
Molecular Dynamics Simulation
To elucidate the dynamic behavior and structural stability of the investigated biotargets, 100 ns molecular dynamics (MD) simulations were performed for COX-2, 5-LOX, and iNOS, each in complex with their respective most active candidate compounds and reference ligands. The simulated systems included: a) compound 5k in comparison with celecoxib and etoricoxib at the COX-2 active site, b) compound 5f in comparison with zileuton and arachidonic acid at the 5-LOX active site, and c) compound 5m in comparison with I58 at the iNOS active site. These simulations were conducted to provide insight into the conformational dynamics, binding stability, and interaction profiles of the selected complexes under physiological conditions.
Comparative MD Simulation of COX-2 Complexes with Compound 5k, Celecoxib, and Etoricoxib
To investigate the dynamic behavior of the COX-2 protein in complex with the most active 3-cyano-2-pyridinone derivative 5k and the reference drugs celecoxib and etoricoxib, 100 ns molecular dynamics (MD) simulations were performed. The simulations aimed to evaluate the stability of the ligand-protein complexes and confirm the robustness of their binding within the COX-2 active site. Root-mean-square deviation (RMSD) analysis of the Cα atoms in the COX-2-celecoxib complex revealed stable fluctuations around ~2.35 Å, following an initial deviation of 1.35 Å. Similarly, the COX-2-etoricoxib complex exhibited RMSD values stabilizing around 2.50 Å from an initial 1.10 Å, while the COX-2-5k complex showed consistent RMSD fluctuations within the 1.50–2.80 Å range. Providentially, all complexes remained well within the acceptable fluctuation threshold of 3.0 Å throughout the simulation period. Ligand RMSD analysis showed that celecoxib experienced early equilibration-related fluctuations, increasing from 0.25 Å to 1.55 Å within the first 45 ns, before stabilizing between 0.75 Å and 2.25 Å. At the same time, etoricoxib exhibited larger initial fluctuations (2.20–4.00 Å in the first 35 ns) before plateauing in the 12.00–14.50 Å range, albeit with minimal variability. Compound 5k maintained a relatively stable RMSD between 2.20 Å and 5.60 Å, indicating good accommodation within the COX-2 binding site (Figure 7: A1–A3). Minimal fluctuations observed in key active site residues suggest limited conformational rearrangements, supporting the ligands’ strong binding affinity. Root-mean-square fluctuation (RMSF) analysis showed that α-helices and β-strands (depicted in red and blue, respectively) exhibited less mobility compared to the loop regions (white background), in line with typical protein dynamics.47 Green vertical lines on the RMSF plots indicated residues directly involved in ligand interactions. Interestingly, all three ligands, celecoxib, etoricoxib, and compound 5k, shared interactions with only three common residues: Val116, Arg120, and Tyr355. However, compound 5k and etoricoxib both interacted with 13 residues, including Lys79, Leu80, Leu82, Lys83, Pro84, Thr85, Val89, Leu93, Thr115, Val116, Ser119, Arg120, and Tyr355, suggesting similar binding modes (Figure 7: B1–B3). The simulation interaction diagrams (Figures 7: C1–C3 and D1–D3) provided further insight into ligand-residue interactions. Celecoxib formed hydrogen bonds with Phe518 (59%), Ser353 (87%), and Leu352 (92%), as well as a water-bridged hydrogen bond with Arg513 (41%) and hydrophobic contacts with Val523 (55%) and Val349 (40%). Etoricoxib formed a hydrogen bond with Arg120 (40%) and a water-mediated hydrogen bond with Tyr355 (28%), along with hydrophobic interactions with Pro84 (32%), His122 (32%), and Tyr115 (30%). Compound 5d showed significant water-bridged interactions with Arg120 (91%) and Ser119 (31%), and hydrophobic contacts with Tyr115 (52%) and Ile112 (40%). Overall, the MD simulations confirmed that all ligands form stable and robust complexes within the COX-2 binding pocket and contact the key residue Arg120 at the channel entrance of the COX-2 active site. Notably, compound 5k and etoricoxib demonstrated similar binding behaviors, supporting the potential COX-2 inhibitory activity of compound 5k and warranting further exploration of its therapeutic application as an anti-inflammatory agent.
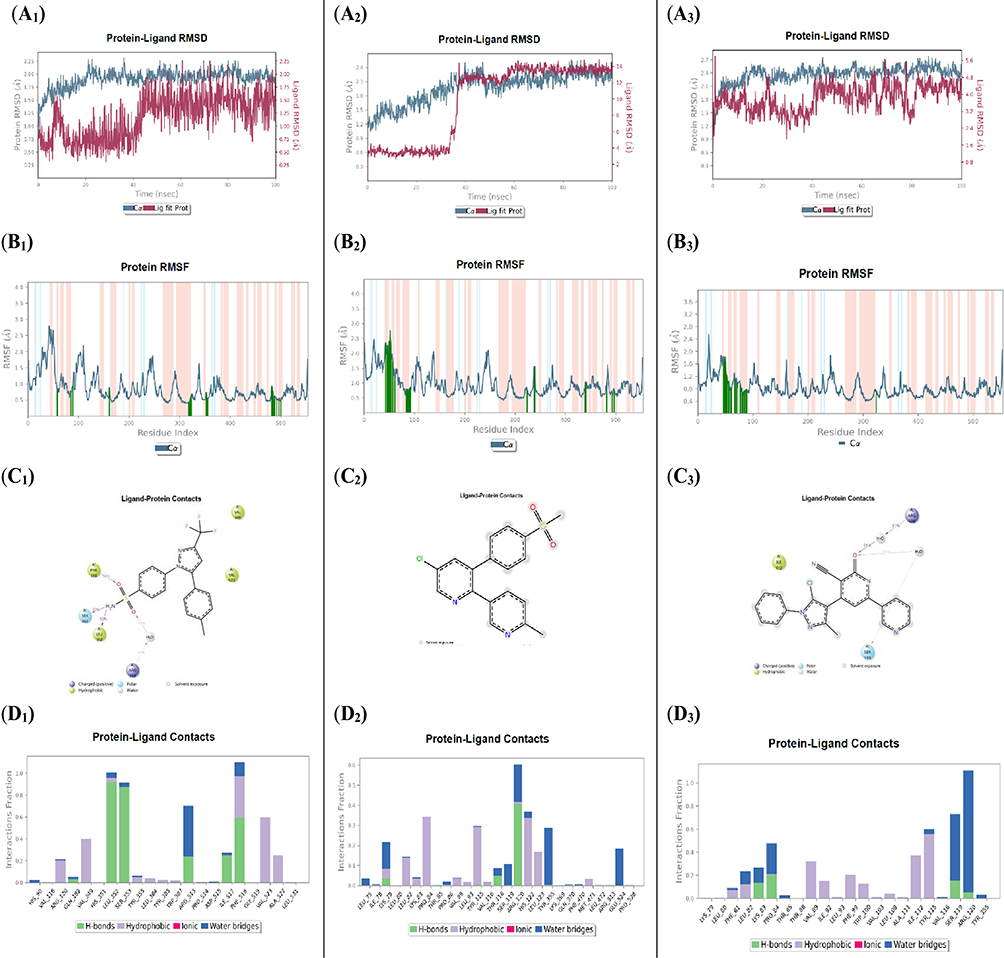 |
Figure 7 Molecular dynamics (MD) simulation analysis of compound 5k in comparison with the reference drugs celecoxib and, each bound to the COX-2 active site (celecoxib A1, etoricoxib A2, and compound 5k A3). Root Mean Square Deviation (RMSD) plots for the COX-2 protein (gray) and the respective ligands (red), illustrating the structural stability of the complexes over the simulation period. (celecoxib B1, etoricoxib B2, and compound 5k B3) Root Mean Square Fluctuation (RMSF) plots of COX-2 residues, highlighting regions of local flexibility influenced by ligand binding. (celecoxib C1, etoricoxib C2, and compound 5k C3) Two-dimensional interaction maps depicting key ligand–protein interactions were consistently maintained throughout the simulation. (celecoxib D1, etoricoxib D2, and compound 5k D3) Histograms showing the frequency and nature of ligand–protein interactions, offering insights into the stability and durability of binding over time. |
Comparative MD Simulation of 5-LOX Complexes with Compound 5k, Zileuton, and the Native Ligand Arachidonic Acid
The protein RMSD analysis of the 5-LOX active site complexed with the reference drug zileuton revealed consistent Cα atom deviations reaching up to 3.10 Å, beginning from an initial value of 1.60 Å. In comparison, the 5-LOX complex with the investigated 3-cyano-2-pyridinone compound 5f exhibited slightly lower deviations, peaking at 2.80 Å from the same initial value. Importantly, both remained within the acceptable fluctuation threshold of 3.0 Å throughout the simulation. Similarly, the native ligand arachidonic acid in complex with 5-LOX demonstrated an RMSD reaching 2.30 Å from an initial 1.52 Å. Regarding ligand stability, zileuton reached RMSD equilibrium at approximately 8.00 Å, having started at 5.00 Å. Arachidonic acid showed lower, steady fluctuations within the range of 5.20–8.70 Å, while compound 5f demonstrated stable fluctuations after the initial 17 ns, settling in the range of 4.30–5.80 Å with minimal deviation (Figure 8: A1–A3). These findings highlight the relative stability and lower fluctuation of the 5-LOX protein in complex with all tested ligands, particularly with compound 5f. The RMSF analysis of the protein revealed three shared interacting residues among all ligands: Phe177, Leu368, and Leu607, illustrating the different binding attitudes. Notably, compound 5f and arachidonic acid interacted with a broader set of 20 common residues, including Phe177, Lys296, Thr297, Asp298, Pro299, Phe359, Thr364, Leu368, Leu414, Ile415, Asn425, Ala426, Thr427, Gly428, Gly429, Gly430, Gly431, His432, Pro569, and Leu607. This extensive overlap underscores the similar binding patterns of compound 5f and arachidonic acid within the 5-LOX active site (Figure 8: B1–B3). Further insights from ligand-protein interaction histograms revealed that zileuton employed its benzothiophene moiety to engage in π–π* interaction with His372 (90%) and formed hydrogen bonds with Leu607 (69%) and Gly174 (67%). Arachidonic acid established hydrogen bonds with Thr297 (29%) and Asp298 (27%), an ionic interaction with Asp298 (20%), and water-bridged hydrogen bonds with Asp298 (45%) and His432 (38%). Compound 5f demonstrated π–π* interactions with Phe359 (32%) and His432 (49%), as well as a strong hydrogen bond with Thr297 (89%) (Figures 8: C1–C3 and D1–D3). As a result, these findings indicate that compound 5f forms a stable and well-defined complex with the 5-LOX target, exhibiting a binding pattern comparable to that of the native ligand arachidonic acid. These results support the potential of compound 5f as a promising anti-inflammatory agent through competitive inhibition of 5-LOX, warranting further biological evaluation.
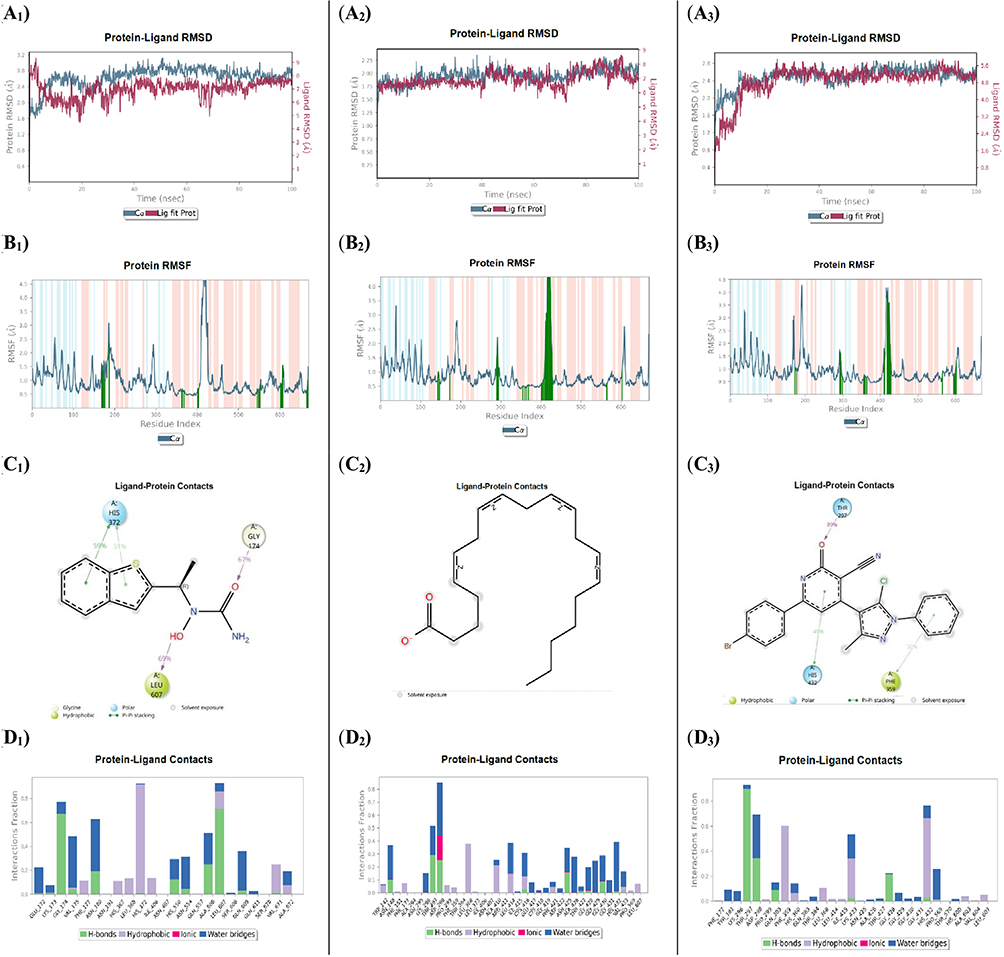 |
Figure 8 Molecular dynamics (MD) simulation analysis of compound 5f in comparison with the native ligand arachidonic acid and the reference drug zileuton, each bound to the 5-LOX active site. (zileuton A1, arachidonic acid A2, and compound 5f A3) Root Mean Square Deviation (RMSD) plots for the 5-LOX protein (gray) and the respective ligands (red), illustrating the structural stability of the complexes over the simulation period. (zileuton B1, arachidonic acid B2, and compound 5f B3) Root Mean Square Fluctuation (RMSF) plots of 5-LOX residues, highlighting regions of local flexibility influenced by ligand binding. (zileuton C1, arachidonic acid C2, and compound 5f C3) Two-dimensional interaction maps depicting key ligand–protein interactions were consistently maintained throughout the simulation. (zileuton D1, arachidonic acid D2, and compound 5f D3) Histograms showing the frequency and nature of ligand–protein interactions, offering insights into the stability and durability of binding over time. |
Comparative MD Simulation of iNOS Complexes with Compound 5m and the Native Ligand I58
During the molecular dynamics (MD) simulation, the Cα atom fluctuations of the iNOS protein complexed with compound 5m and the native ligand I58 were maintained within acceptable limits. Specifically, the 5m–iNOS complex exhibited fluctuations up to 6.4 Å from an initial value of 3.2 Å, while the I58–iNOS complex showed deviations reaching 4.20 Å from a starting point of 3.00 Å, both remaining within the normal dynamic range. Ligand RMSD analysis revealed that I58 achieved equilibrium after the first 10 ns, stabilizing at approximately 5.4 Å from an initial value of 3.8 Å. Similarly, compound 5m displayed a steady RMSD profile, fluctuating within 4.2 Å from an initial 2.4 Å, throughout the simulation period (Figure 9: A1–A2). These results collectively indicate stable complex formation for both ligands with the iNOS protein over the course of the simulation. The RMSF analysis showed that the I58 ligand-maintained contacts with 18 residues throughout the simulation: Cys194, Gln257, Arg260, Tyr341, Pro344, Ala345, Val346, Phe363, Asn364, Gly365, Trp366, Tyr367, Met368, Glu371, Ile372, Arg375, Asp376, and Arg382.46 Compound 5m, in comparison, shared 9 interacting residues with I58, namely: Gly194, Gln257, Val346, Phe363, Trp366, Tyr367, Met368, Glu371, and Arg375. These overlapping interactions suggest an approximate binding orientation and mechanism between the novel compound 5m and the native ligand I58 (Figure 9: B1–B2). Further insight from the ligand–protein contact histograms revealed that the I58 ligand exhibited a high-frequency hydrogen bond interaction with Met368, exceeding an 80% interaction fraction, along with a water-bridged hydrogen bond with Asp376 (40%), consistent with docking predictions.46 For compound 5m, two prominent interactions were observed:1 hydrogen bonding with Trp366 (64%), and2 π–π* stacking interactions with Trp188 (44%) and Phe363 (82%) (Figures 9: C1–C2 and D1–D2). These findings support the strong affinity of compound 5m for the iNOS active site, maintaining stable interactions throughout the simulation. The results highlight its potential as a nitric oxide synthase (iNOS) inhibitor, suggesting its promise in reducing NO levels and alleviating inflammation, which merits further biological validation.
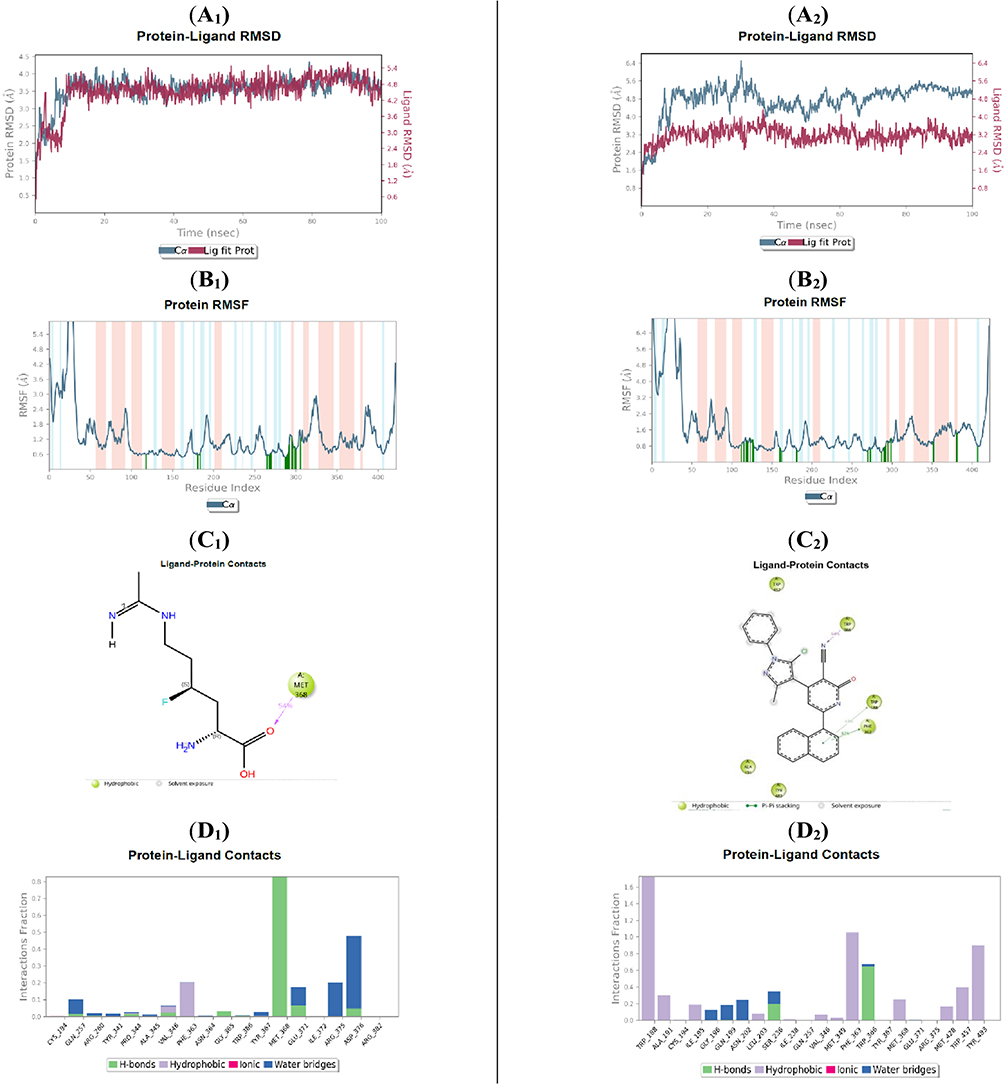 |
Figure 9 Molecular dynamics (MD) simulation analysis of compound 5m in comparison with the native ligand I58, each bound to the iNOS active site. (native ligand I58 A1, compound 5m A2) Root Mean Square Deviation (RMSD) plots for the iNOS protein (gray) and the respective ligands (red), illustrating the structural stability of the complexes over the simulation period. (native ligand I58 B1, compound 5m B2) Root Mean Square Fluctuation (RMSF) plots of iNOS residues, highlighting regions of local flexibility influenced by ligand binding. (native ligand I58 C1, compound 5m C2) Two-dimensional interaction maps depicting key ligand–protein interactions were consistently maintained throughout the simulation. (native ligand I58 D1, compound 5m D2) Histograms showing the frequency and nature of ligand–protein interactions, offering insights into the stability and durability of binding over time. |
Structure Activity Relationship
The structure-activity relationship (SAR) analysis of the synthesized pyrazole/3-cyano-2-pyridinone derivatives 5a–n reveals that variations in the aryl moiety attached to the pyrazole/3-cyano-2-pyridinone derivatives significantly influence their anti-inflammatory profiles across multiple targets (Figure 10). Additionally, bulky groups, electron-donating, and electron-withdrawing groups have an impact on their anti-inflammatory potency through modulation of hydrogen bonding, lipophilicity, and receptor binding. In terms of inhibition of NO overproduction, compounds bearing electron-donating or bulky lipophilic groups showed superior activity and iNOS modulation. Notably, compound 5g (Ar = 4-hydroxyphenyl) demonstrated the highest potency in inhibiting NO over-release, attributed to the electron-donating OH group, which enhances hydrogen bonding with polar active sites, affecting cellular levels of NO. Compound 5m (Ar = 1-naphthyl) ranks second due to its lipophilic conjugated π-system, which favors π-π stacking or binding to lipophilic active sites. In compound 5f (Ar = 4-bromophenyl), the presence of halogen enhances lipophilicity and increases binding affinity to hydrophobic sites, regarding the inhibition and selectivity of COX-2. Compound 5k (Ar = 3-pyridyl) displayed the highest selectivity as a COX-2 inhibitor (SI = 19.6), likely attributable to its interaction with polar residues within the COX-2 active site. Conversely, compounds 5g (Ar = 4-hydroxyphenyl) and 5m (Ar = 1-naphthyl) showed remarkable COX-2 inhibition; however, their selectivity for COX-2 declined, potentially due to concurrent COX-1 inhibition. The presence of the bulky halogen in compound 5f (Ar = 4-bromophenyl) was anticipated to restrict orientation, resulting in decreased COX-2 affinity and selectivity. Consequently, the selectivity for COX-2 is improved by the incorporation of polar groups (such as OH and pyridyl N) and by the presence of planar hydrophobic moieties that facilitate the interactions with the COX-2 active site. The inhibition of PGE2 correlates strongly with the inhibition of COX-2. Compound 5g (Ar = 4-hydroxyphenyl) exhibited the highest inhibition, followed by 5k (Ar = 3-pyridyl), indicating that hydroxylated and heteroaryl rings favor the suppression of prostaglandin synthesis. In TNF-α inhibition, 5m (Ar = 1-naphthyl) was the most effective, while compound 5f (Ar = 4-bromophenyl) ranks second in reducing TNF-α levels. Conversely, compounds 5g (Ar = 4-hydroxyphenyl) and 5k (Ar = 3-pyridyl) were the least active, indicating that bulkier, hydrophobic aromatic systems are more favorable for TNF-α suppression. Evaluation of lipoxygenase enzymes revealed that 5f (4-bromophenyl) demonstrated significant inhibitory activity against both 5-LOX and 15-LOX enzymes. This dual activity may be associated with the optimal size and lipophilicity of the halogen, which favors hydrophobic interactions with 5-/15-LOX active sites. Whereas compounds 5g (Ar = 4-hydroxyphenyl) and 5m (Ar = 1-naphthyl) exhibited weaker inhibitory activity against 5-/15-LOX. Hence, bulkier and lipophilic aryl groups (such as Br or naphthyl) are expected to be essential for fitting with the hydrophobic pocket in LOX active sites. In the context of direct iNOS enzymatic inhibition, compound 5m (Ar = 1-naphthyl) demonstrated the highest inhibitory activity, followed closely by 5f (4-bromophenyl). This implies that lipophilic non-polar aromatic rings positively influence enzyme interaction. Nonetheless, compound 5g (Ar = 4-hydroxyphenyl) displayed the lowest inhibitory activity against iNOS; however, it significantly suppressed NO production, indicating an indirect effect on NO levels rather than direct enzymatic inhibition.
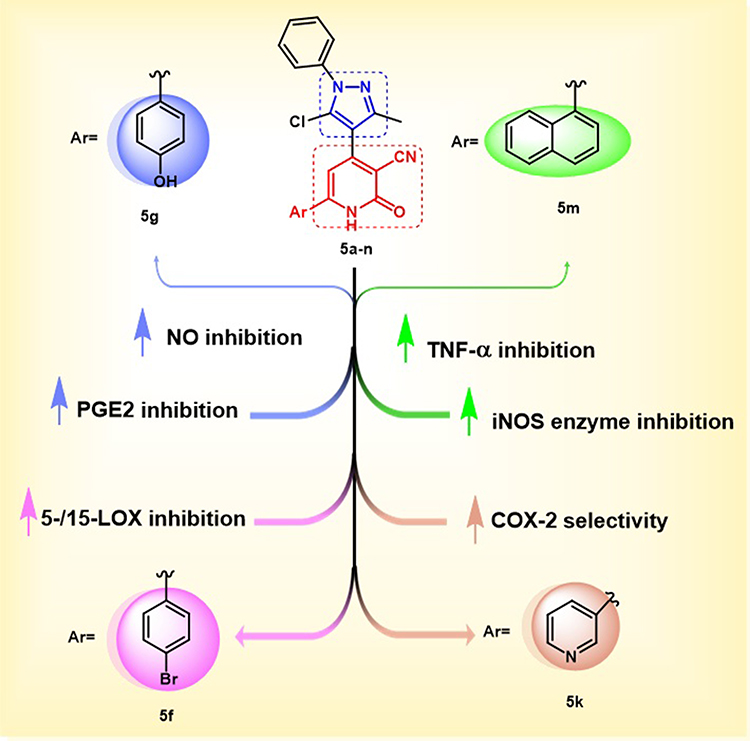 |
Figure 10 Structure-activity relationship (SAR) for the newly synthesized pyrazole/3-cyano-2-pyridinone derivatives 5a-n. |
Overall, the SAR indicates that electron-donating (eg., OH), halogen, or bulky aromatic substitutions on the pyrazole/3-cyano-2-pyridinone core can significantly tune the anti-inflammatory activity depending on the specific molecular target, with 4-hydroxyphenyl and 1-naphthyl groups showing a broad and potent profile across most targets.
Physicochemical and Oral Bioavailability Assessment
The predicted physicochemical properties of the synthesized 3-cyano-2-pyridinone derivatives 5k, 5f, and 5m were assessed using the Swiss ADME online platform.73 The analysis yielded promising insights into the oral drug-likeness of these novel compounds. Lipophilicity, estimated using the atomistic, knowledge-based XLOGP3 model, is considered favorable for absorption when the value remains below 5.0074 compounds 5k and 5f satisfied this criterion, with XLOGP3 values of 3.21 and 4.97, respectively. However, compound 5m slightly exceeded the optimal range, displaying a value of 5.53. All compounds demonstrated molecular weights below the recommended threshold of 500 g/mol for orally administered drugs,75 indicating favorable molecular size (Table 3). In terms of polarity, optimal oral bioavailability is typically associated with a topological polar surface area (TPSA) of less than 130 Å2.47,76 All three compounds met this benchmark, with TPSA values of 87.36 Å2 for 5k and 74.47 Å2 for both 5f and 5m. Water solubility, evaluated via the ESOL Log S model (where values below 0 denote acceptable solubility), was within acceptable limits for all compounds, with Log S values of −4.68 (5k), −6.26 (5f), and −6.46 (5m). Regarding saturation, the fraction of sp3-hybridized carbons (Csp3) was found to be 0.05 for both 5k and 5f, and 0.04 for 5m, which is below the upper limit of 1.00, indicating an acceptable level of molecular unsaturation.77 All compounds exhibited limited molecular flexibility, as indicated by three rotatable bonds per molecule, which falls within the recommended threshold of fewer than nine. This restricted flexibility may contribute to improved pharmacokinetic stability and reduced off-target interactions (Figure 11). Lastly, all three compounds fully conformed to Lipinski’s Rule of Five, underscoring their potential as orally bioavailable drug candidates.
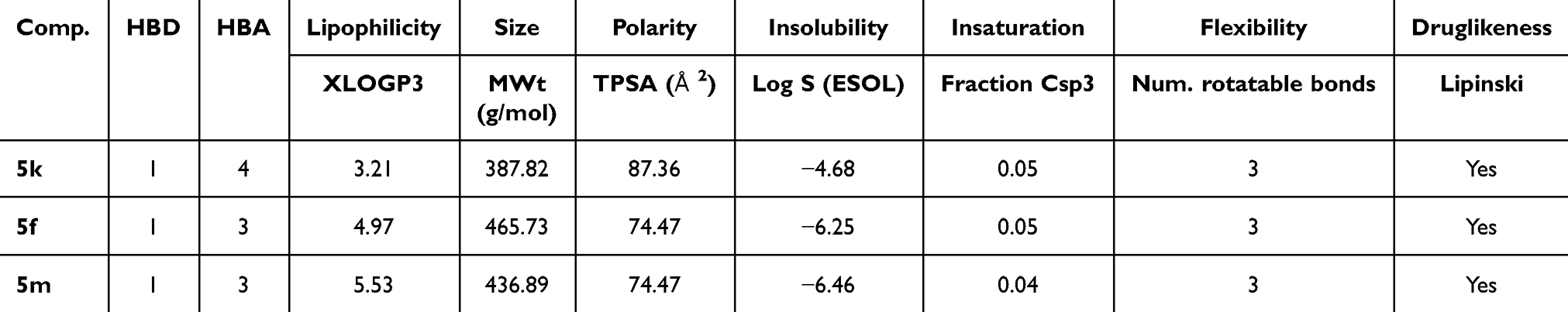 |
Table 3 Predicted ADME Profiles of the Novel 3-Cyano-2-Pyridinone Derivatives 5k, 5f, and 5m. The Assessment Encompasses Key Physicochemical Parameters and Drug-Likeness Descriptors Pertinent to Oral Bioavailability and Therapeutic Efficacy |
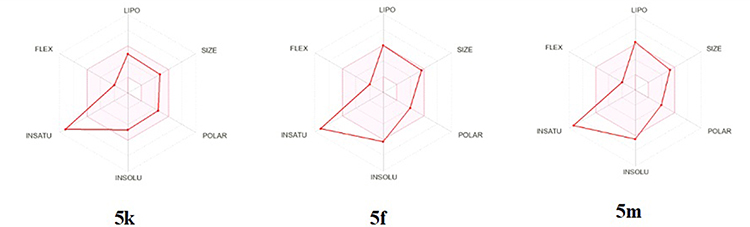 |
Figure 11 The oral bioavailability plot illustrates the drug-likeness and oral suitability of the compounds 5k, 5f, and 5m under investigation. The red-shaded region denotes areas associated with unfavorable pharmacokinetic characteristics, such as excessive molecular size, elevated lipophilicity, increased polarity, limited solubility, low degrees of unsaturation, and high molecular flexibility. |
Conclusion
In this study, novel pyrazole-3-cyano-2-pyridinone hybrids were synthesized and evaluated for their anti-inflammatory potential. The compounds demonstrated selective inhibition of COX-2 over COX-1, with compound 5k showing the highest COX-2 potency (IC50 = 0.92 μM) and selectivity index (SI = 19.6). Additionally, compounds 5f and 5g exhibited significant inhibition of 5-LOX and 15-LOX enzymes, with 5f showing IC50 values of 0.34 μM and 0.21 μM, respectively. The tested derivatives also effectively reduced pro-inflammatory mediators, including nitric oxide, PGE2, and TNF-α, in LPS-stimulated RAW 264.7 cells. These results confirm that hybridizing pyrazole and cyanopyridine scaffolds results in multi-target inhibitors with promising in vitro anti-inflammatory activity. In agreement with the in vitro biological evaluation, molecular docking analyses revealed that compounds 5k, 5f, and 5m displayed strong binding affinities toward the COX-2, 5-LOX, and iNOS enzymes, respectively, highlighting their potential as viable candidates for the treatment of inflammatory disorders. To investigate the stability of these ligand-target interactions under simulated physiological conditions, atomistic molecular dynamics (MD) simulations were carried out for 100 ns. The simulation results confirmed that all three compounds maintained stable conformations within their respective binding sites throughout the simulation period. Furthermore, pharmacokinetic predictions suggested that compounds 5k, 5f, and 5m exhibit favorable oral bioavailability and possess drug-like properties. This integrated in vitro and in silico evaluation supports their potential as promising lead compounds for further development as novel anti-inflammatory agents, warranting future studies including in vivo validation and optimization for therapeutic applications.
Acknowledgments
The Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, Saudi Arabia, has funded this project under grant no. (RG-15-140-43). The authors, therefore, acknowledge with thanks the DSR for technical and financial support.
Author Contributions
Bahgat R. M. Hussein: Writing original draft, Resources, Methodology, Formal analysis, Data curation, Visualization, Conceptualization. Ibrahim M. Salem: Writing original draft, Resources, Methodology, Software, Data curation. Hossameldin A. Aziz: Visualization, Resources, Methodology, Investigation, Formal analysis. Omar Alshazly: Writing review & editing, Project administration. Heba A. Hofny; Conceptualization and writing original draft. Aliaa M. Mohassab: Visualization, Resources, Methodology, Investigation, Formal analysis Zuhier A. Awan: Writing review & editing, Visualization, Funding, Software, Resources, Formal analysis, Data curation. Tarek S. Ibrahim: Writing – review & editing, Writing – original draft, Project administration, Methodology, Formal analysis, Conceptualization. Elsayed M. Mahmoud: Writing – original draft, Resources, Methodology, Formal analysis, Writing review & editing, Visualization, Supervision. Stefan Bräse: Writing review & editing, Visualization, Software, Resources, Formal analysis, Data curation. Mamdouh F.A. Mohamed: Writing original draft, Resources, Methodology, Formal analysis, Data curation, Writing review & editing, Visualization, Supervision, Conceptualization. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The Deanship of Scientific Research (DSR) at King Abdulaziz University (KAU), Jeddah, Saudi Arabia has funded this project, under grant No. (RG-15-140-43).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Medzhitov R. The spectrum of inflammatory responses. Science. 2021;374(6571):1070–26. doi:10.1126/science.abi5200
2. Kotas ME, Medzhitov RJC. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160(5):816–827. doi:10.1016/j.cell.2015.02.010
3. Hayallah AM, Shaaban MA, Abdelrahman MH, et al. Microwave assisted synthesis, biological assessment and in-silico studies of some new 5, 5′-(phenylmethylene) bisuracil and dihydropyrido [2, 3-d: 6, 5-d’] dipyrimidine derivatives as selective COX-2 inhibitors with potential anti-inflammatory activities. J. Mol. Struct. 2025;1331:141504. doi:10.1016/j.molstruc.2025.141504
4. Chimenti M, Triggianese P, Conigliaro P, et al. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Disease. 2015;6(9):e1887–e. doi:10.1038/cddis.2015.246
5. Mahmoud AM, Alfadl EMA, Ahmed AR, et al. Disclosing the impact of metformin and methotrexate in adjuvant arthritis in female rats: molecular docking and biochemical insights on visfatin. Naunyn Schmiedebergs Arch Pharmacol. 2025:1–12. doi:10.1007/s00210-025-03823-7
6. Ramos GP, Papadakis KA, editors.. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clinic Proceedings. Elsevier; 2019.
7. Korniluk A, Koper O, Kemona H, Dymicka-Piekarska V. From inflammation to cancer. Irish J Med Sci. 2017;186(1):57–62. doi:10.1007/s11845-016-1464-0
8. Ahmed AH, Mohamed MF, Allam RM, et al. Design, synthesis, and molecular docking of novel pyrazole-chalcone analogs of lonazolac as 5-LOX, iNOS and tubulin polymerization inhibitors with potential anticancer and anti-inflammatory activities. Bioorg Chem. 2022;129:106171. doi:10.1016/j.bioorg.2022.106171
9. Yao C, Bjop NSJ. Prostaglandin-cytokine crosstalk in chronic inflammation. Br Jo Pharmacol. 2019;176(3):337–354. doi:10.1111/bph.14530
10. Sasaki F, Yokomizo T. The leukotriene receptors as therapeutic targets of inflammatory diseases. Int Immunol. 2019;31(9):607–615. doi:10.1093/intimm/dxz044
11. Tripathi P, Tripathi P, Kashyap L, Singh VJFI. The role of nitric oxide in inflammatory reactions. FEMS Immunol Med Microbiol. 2007;51(3):443–452. doi:10.1111/j.1574-695x.2007.00329.x
12. Umare V, Pradhan V, Nadkar M, et al. Effect of proinflammatory cytokines (IL-6, TNF-α, and IL-1β) on clinical manifestations in Indian SLE patients. Mediators of Inflammation. 2014;2014(1):385297. doi:10.1155/2014/385297
13. Giménez-Bastida JA, Gonzalez-Sarrias A, Laparra-Llopis JM, Schneider C, Espín JC. Targeting mammalian 5-lipoxygenase by dietary phenolics as an anti-inflammatory mechanism: a systematic review. Int J Mol Sci. 2021;22(15):7937. doi:10.3390/ijms22157937
14. Aziz MA, Salem IM, Al-Awadh MA, et al. Exploration of anti-inflammatory activity of pyrazolo [3, 4-d] pyrimidine/1, 2, 4-oxadiazole hybrids as COX-2, 5-LOX and NO release inhibitors: design, synthesis, in silico and in vivo studies. Bioorg Chem. 2025;156:108181. doi:10.1016/j.bioorg.2025.108181
15. Tziona P, Theodosis-Nobelos P, Rekka A. Medicinal Chemistry approaches of controlling gastrointestinal side effects of non-steroidal anti-inflammatory drugs. Endogenous protective mechanisms and drug design. Med Chem. 2017;13(5):408–420. doi:10.2174/1573406413666170209123433
16. Ahmadi M, Bekeschus S, Weltmann K-D, von Woedtke T, Wende K. Non-steroidal anti-inflammatory drugs: recent advances in the use of synthetic COX-2 inhibitors. RSC Med Chem. 2022;13(5):471–496. doi:10.1039/d1md00280e
17. Asghar W, Jamali FJI. The effect of COX-2-selective meloxicam on the myocardial, vascular and renal risks: a systematic review. Inflammopharmacology. 2015;23(1):1–16. doi:10.1007/s10787-014-0225-9
18. Wang B, Wu L, Chen J, et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Signal Transduct Target Ther. 2021;6(1):94. doi:10.1038/s41392-020-00443-w
19. Cinelli MA, Do HT, Miley GP, Silverman RB. Inducible nitric oxide synthase: regulation, structure, and inhibition. Med Res Rev. 2020;40(1):158–189. doi:10.1002/med.21599
20. Koeberle A, Werz O. Multi-target approach for natural products in inflammation. Drug Dis Today. 2014;19(12):1871–1882. doi:10.1016/j.drudis.2014.08.006
21. Ghanim AM, Rezq S, Ibrahim TS, Romero DG, Kothayer HJE. Novel 1, 2, 4-triazine-quinoline hybrids: the privileged scaffolds as potent multi-target inhibitors of LPS-induced inflammatory response via dual COX-2 and 15-LOX inhibition. Eur J Med Chem. 2021;219(219):113457. doi:10.1016/j.ejmech.2021.113457
22. Mantzanidou M, Pontiki E, Hadjipavlou-Litina DJM. Pyrazoles and pyrazolines as anti-inflammatory agents. Molecules. 2021;26(11):3439. doi:10.3390/molecules26113439
23. Dannhardt G, Laufer S. Structural approaches to explain the selectivity of COX-2 inhibitors: is there a common pharmacophore? Curr Med Chem. 2000;7(11):1101–1112. doi:10.2174/0929867003374237
24. Ibrahim TS, Salem IM, Mostafa SM, et al. Design, synthesis, and pharmacological evaluation of novel and selective COX-2 inhibitors based on bumetanide scaffold. Bioorg Chem. 2020;100:103878. doi:10.1016/j.bioorg.2020.103878
25. Ragab MA, Eldehna WM, Nocentini A, et al. 4-(5-Amino-pyrazol-1-yl) benzenesulfonamide derivatives as novel multi-target anti-inflammatory agents endowed with inhibitory activity against COX-2, 5-LOX and carbonic anhydrase: design, synthesis, and biological assessments. Eur J of Med Chem. 2023;250:115180. doi:10.1016/j.ejmech.2023.115180
26. Yildirim M, Poyraz S, MJMCR E. Recent advances on biologically active coumarin-based hybrid compounds. 2023;32(4):617–642. doi:10.1007/s00044-023-03025-x
27. Dallob A, Hawkey CJ, Greenberg H, et al. Characterization of etoricoxib, a novel, selective COX-2 inhibitor. J Clin Pharmacol. 2003;43(6):573–585. doi:10.1177/0091270003253703
28. Cochrane DJ, Jarvis B, Keating GM. Etoricoxib. Drugs. 2002;62(18):2637–2651. doi:10.2165/00003495-200262180-00006
29. Alsayed SSR, Elshemy HAH, Abdelgawad MA, Abdel-Latif MS, Abdellatif KRA. Design, synthesis and biological screening of some novel celecoxib and etoricoxib analogs with promising COX-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg Chem. 2017;70:173–183. doi:10.1016/j.bioorg.2016.12.008
30. Kumar N, Chauhan A, Drabu S. Synthesis of cyanopyridine and pyrimidine analogues as new anti-inflammatory and antimicrobial agents. Biomedicine & pharmacotherapy = Biomédecine & pharmacothérapie. 2011;65(5):375–380. doi:10.1016/j.biopha.2011.04.023
31. Atla SR, Nagireddy NR, Yejella RP. Anti-inflammatory, analgesic and antimicrobial activity studies of novel 4, 6-disubstituted-2-amino-3-cyanopyridines. 2014;1(1):47–57. doi:10.1248/cpb.59.1079
32. Hussein BR, Mohammed HH, Ahmed EA, Alshazly O, Mohamed MF, Omran O. Design, synthesis, and anti-breast cancer activity evaluation of novel 3-cyanopyridine derivatives as PIM-1 inhibitors. Mol. Divers. 2025;29(3):2565–2584. doi:10.1007/s11030-024-11010-8
33. Hussein BR, El-Saghier SM, Allam RM, Mohamed MF, Amer A. An efficient methodological approach for synthesis of selenopyridines: generation, reactions, anticancer activity, EGFR inhibitory activity and molecular docking studies. Mol. Divers. 2025;29(1):519–534. doi:10.1007/s11030-024-10872-2
34. Hussein BR, Keesing A, Pendleton J. Multicomponent reaction for synthesis of novel 2-tosyloxyphenylpyridines. J Med Entomol. 2019;56(4):1420–1425. doi:10.1002/jhet.2649
35. A-E-BA G, Khodairy A, Moustafa HM, Hussein BR, Farghaly MM, MOJPCJ A. Synthesis, in vitro antibacterial and in vivo anti-inflammatory activity of some new pyridines. 2017;51(8):652–660. doi:10.1007/s11094-017-1670-8
36. Ghattas AEBA, Khodairy A, Moustafa HM, BRJJoHC H. New heterocyclic compounds derived from 4, 6-diamino-3-cyano-2-methylthiopyridine and their biological activity. Indian Pediatrics. 2017;54(2):879–888. doi:10.1002/jhet.2649
37. Yoon S-H, Cho D-Y, S-r C, et al. Synthesis and biological evaluation of salicylic acid analogues of celecoxib as a new class of selective cyclooxygenase-1 inhibitor. Biol. Pharm. Bull. 2021;44(9):1230–1238. doi:10.1248/bpb.b20-00991
38. Choi Y-J, Jung JI, Bae J, Lee JK, Kim EJ. Evaluating the anti-osteoarthritis potential of standardized boswellia serrata gum resin extract in alleviating knee joint pathology and inflammation in osteoarthritis-induced models. Int J Mol Sci. 2024;25(6):3218. doi:10.3390/ijms25063218
39. Zhou W, Dai Q, Su N, Liu Z, Hu J, Medicine T. IGF2BP2dependent STIM1 inhibition protects against LPSinduced pneumonia in vitro by alleviating endoplasmic reticulum stress and the inflammatory response. Exp Therap med. 2023;26(6):575. doi:10.3892/etm.2023.12273
40. Tang S, Bhatia B, Maldonado CJ, et al. Evidence that arachidonate 15-lipoxygenase 2 is a negative cell cycle regulator in normal prostate epithelial cells. J Biolog Chem. 2002;277(18):16189–16201. doi:10.1074/jbc.m111936200
41. ElBakary NM, Hagag SA, Ismail MA, El-Sayed WM. New thiophene derivative augments the antitumor activity of γ-irradiation against colorectal cancer in mice via anti-inflammatory and pro-apoptotic pathways. Dis Oncol. 2022;13(1):119. doi:10.1007/s12672-022-00583-1
42. Dreaden EC, Mwakwari SC, Sodji QH, Oyelere AK, El-Sayed MA. Tamoxifen− poly (ethylene glycol)− thiol gold nanoparticle conjugates: enhanced potency and selective delivery for breast cancer treatment. Bioconjug Chem. 2009;20(12):2247–2253. doi:10.1021/bc9002212
43. Lucido MJ, Orlando BJ, Vecchio AJ, Malkowski MGJB. Crystal structure of aspirin-acetylated human cyclooxygenase-2: insight into the formation of products with reversed stereochemistry. Biochemistry. 2016;55(8):1226–1238. doi:10.1021/acs.biochem.5b01378
44. Gilbert NC, Rui Z, Neau DB, et al. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 2012;26(8):3222. doi:10.1096/fj.12-205286
45. Hallinan EA, Kramer SW, Houdek SC, et al. 4-Fluorinated L-lysine analogs as selective i-NOS inhibitors: methodology for introducing fluorine into the lysine side chain. Organic & Biomol Chem. 2003;1(20):3527–3534. doi:10.1039/b307563j
46. Ahmed AKM, Beshr EA, Salem IM, et al. Novel Chalcone Candidates as Potential in Vitro and in Vivo Anti-Inflammatory Agents: Synthesis, in-silico Docking, Multitarget Bio Evaluation and Molecular Dynamic Simulation.2025.108540 10.1016/j.bioorg.2025.108540
47. Salem IM, El-Sabbagh OI, Mostafa SM, et al. Exploitation of Novel Pyrazolo [3, 4-D] Pyrimidine Scaffold Tethered to Thiazole as Potential EGFR/HER2 Dual Kinase Inhibitor to Overcome Lapatinib Resistant Breast Cancer: Design, Synthesis, in silico Docking and Molecular Dynamic Simulation.2025.108671 10.1016/j.bioorg.2025.108671
48. Wizard PP, Epik S, Prime L, Glide SJNY, LLC NY. 2021;34. doi: 10.1515/9783868599527-018.
49. Sørensen J, Demir Ö, Swift RV, Feher VA, Amaro RE. Molecular docking to flexible targets. In: Molecular Modeling of Proteins. Springer; 2014:445–469.
50. AM AE-H, Lotfallah AH, Nemr MT, et al. Novel di-aryl chalcone derived pyrazole linked to methane sulfonyl pharmacophore as potent selective COX-2 inhibitors; design, synthesis, molecular modeling, in vitro and in vivo anti-inflammatory activities. Future Med Chem. 2025;17(15):1849–1865. doi:10.1080/17568919.2025.2545171
51. Mohamed MF, Salem IM, Fouad A, et al. Synthesis and Apoptotic Induction of Sulfonamide-Based Chalcone Hybrids as First-in-Class Dual Histone Deacetylasecarbonic Anhydrase Inhibitors with Potential Anti-Tubulin Activity.2025.108694 10.1016/j.bioorg.2025.108694
52. Abdel-Rahman IM, Abdel-Aziz M, Hassan HA, et al. Unveiling ortho-phenolic Mannich ciprofloxacin-chalcone conjugates with potential antineoplastic activity: synthesis, mechanistic study, in silico docking and molecular dynamic simulation. Eur J Med Chem. 2025;296:117891. doi:10.1016/j.ejmech.2025.117891
53. Alyahyawy OY, Munshi RM, Badr-Eldin SM, et al. Reprofiling lamivudine as an antibiofilm and anti-pathogenic agent against Pseudomonas aeruginosa. AMB Express. 2025;15(1):33. doi:10.1186/s13568-025-01835-3
54. Khafagy ES, Saqr AA, Almutairy BK, et al. Repurposing nitroimidazoles: a new frontier in combatting bacterial virulence and quorum sensing via in silico, in vitro, and in vivo insights. Drug Develop Res. 2025;86(3):e70101. doi:10.1002/ddr.70101
55. Lundberg JO, Weitzberg EJC. Nitric oxide signaling in health and disease. Cell. 2022;185(16):2853–2878. doi:10.1016/j.cell.2022.06.010
56. Gantner BN, LaFond KM, Bonini MG. Nitric oxide in cellular adaptation and disease. Redox Bio. 2020;34:101550. doi:10.1016/j.redox.2020.101550
57. Yang T, Zelikin AN, Chandrawati RJAS. Progress and promise of nitric oxide-releasing platforms. Adv Sci. 2018;5(6):1701043. doi:10.1002/advs.201701043
58. Keshet R, Erez A. Arginine and the metabolic regulation of nitric oxide synthesis in cancer. Dis. Model. Mech. 2018;11(8):dmm033332. doi:10.1242/dmm.033332
59. Lee J, Bae EH, Ma SK, Kim SW. Altered nitric oxide system in cardiovascular and renal diseases. Chonnam Med J. 2016;52(2):81–90. doi:10.4068/cmj.2016.52.2.81
60. Yang Y, Yu T, Lian Y-J, Ma R, Yang S, Cho JY. Nitric oxide synthase inhibitors: a review of patents from 2011 to the present. Expert Opin. Ther. Pat. 2015;25(1):49–68. doi:10.1517/13543776.2014.979154
61. Bošković J, Dobričić V, Mihajlović M, Kotur-Stevuljević J, Čudina OJP. Synthesis, evaluation of enzyme inhibition and redox properties of potential dual COX-2 and 5-LOX inhibitors. Pharmaceuticals. 2023;16(4):549. doi:10.3390/ph16040549
62. Mohassab AM, Hassan HA, Abdelhamid D, et al. New quinoline/1, 2, 4-triazole hybrids as dual inhibitors of COX-2/5-LOX and inflammatory cytokines: design, synthesis, and docking study. 2021;1244:130948. doi:10.1016/j.molstruc.2021.130948
63. Abdellatif KR, Abdelall EK, Elshemy HA, Philoppes JN, Hassanein EH, Kahk NM. Design, synthesis, and pharmacological evaluation of novel and selective COX-2 inhibitors based on celecoxib scaffold supported with in vivo anti-inflammatory activity, ulcerogenic liability, ADME profiling and docking study. Bioorg Chem. 2022;120:105627. doi:10.1016/j.bioorg.2022.105627
64. Tsuge K, Inazumi T, Shimamoto A, Sugimoto Y. Molecular mechanisms underlying prostaglandinE2-exacerbated inflammation and immune diseases. Int Immunol. 2019;31(9):597–606. doi:10.1093/intimm/dxz021
65. Dawood DH, Nossier ES, Abdelhameed MF, Asaad GF, Abd El-Rahman SS. Design, synthesis, anti-inflammatory evaluation and molecular docking of novel thiophen-2-ylmethylene-based derivatives as potential TNF-α production inhibitors. Bioorg Chem. 2022;122:105726. doi:10.1016/j.bioorg.2022.105726
66. Su M, Zhou D, Huang J, Yang T, Zhou Q, Tan Y. Forsythiaside A exhibits anti-migration and anti-inflammation effects in rheumatoid arthritis in vitro model. Int. J. Rheum. Dis. 2024;27(1):e14976. doi:10.1111/1756-185x.14976
67. He Z, Tao D, Xiong J, et al. Phosphorylation of 5-LOX: the potential set-point of inflammation. Neurochem Res. 2020;45(10):2245–2257. doi:10.1007/s11064-020-03090-3
68. Çolakoğlu M, Tunçer S, Banerjee S. Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Proliferation. 2018;51(5):e12472. doi:10.1111/cpr.12472
69. Benkhelifa S, Rafa H, Belhadef S, et al. Aberrant up-regulation of iNOS/NO system is correlated with an increased abundance of Foxp3+ cells and reduced effector/memory cell markers expression during colorectal cancer: immunomodulatory effects of cetuximab combined with chemotherapy. Inflammopharmacology. 2019;27(4):685–700. doi:10.1007/s10787-019-00566-9
70. Liu Q, Chan S, Mahendran RJC. Nitric oxide induces cyclooxygenase expression and inhibits cell growth in colon cancer cell lines. Carcinogenesis. 2003;24(4):637–642. doi:10.1093/carcin/bgg014
71. Thuresson ED, Lakkides KM, Smith WL. Different catalytically competent arrangements of arachidonic acid within the cyclooxygenase active site of prostaglandin endoperoxide H synthase-1 lead to the formation of different oxygenated products. J Biolog Chem. 2000;275(12):8501–8507. doi:10.1074/jbc.275.12.8501
72. Hobbs AJ, Higgs A, SJ M. Inhibition of nitric oxide synthases as a potential therapeutic target. Ann Rev Pharmacol Toxicol. 1999;39(1):191–220. doi:10.1146/annurev.pharmtox.39.1.191
73. Roshdi M, Mohamed MF, Beshr EA, et al. Pharmaceuticals. design, synthesis, in silico docking, multitarget bioevaluation and molecular dynamic simulation of novel pyrazolo [3, 4-d] pyrimidinone derivatives as potential in vitro and in vivo anti-inflammatory agents. Pharmaceuticals. 2025;18(9):1326. doi:10.3390/ph18091326
74. Mannhold R, Poda GI, Ostermann C, Tetko IV. Calculation of molecular lipophilicity: state-of-the-art and comparison of log P methods on more than 96,000 compounds. J Pharm Sci. 2009;98(3):861–893. doi:10.1002/jps.21494
75. Salem IM, Mostafa SM, Salama I, El-Sabbagh OI, Hegazy WA, Ibrahim TS. Human dihydrofolate reductase inhibition effect of 1-Phenylpyrazolo [3, 4–d] pyrimidines: synthesis, antitumor evaluation and molecular modeling study. Bioorg Chem. 2022;129:106207. doi:10.1016/j.bioorg.2022.106207
76. Bytheway I, Darley MG, Popelier PLA. The calculation of polar surface area from first principles: an application of quantum chemical topology to drug design. ChemMedChem. 2008;3(3):445–453. doi:10.1002/cmdc.200700262
77. Petrauskas AA. ACD/Log P method description. Perspect Drug Discov Des. 2000;19(1):99–116. doi:10.1023/a:1008719622770
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.