Back to Journals » International Journal of Nanomedicine » Volume 11
Novel in situ self-assembly nanoparticles for formulating a poorly water-soluble drug in oral solid granules, improving stability, palatability, and bioavailability
Authors Guo S, Pham K, Li D, Penzak S, Dong X
Received 16 November 2015
Accepted for publication 14 January 2016
Published 7 April 2016 Volume 2016:11 Pages 1451—1460
DOI https://doi.org/10.2147/IJN.S100621
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Shujie Guo,1 Kevin Pham,2 Diana Li,2 Scott R Penzak,3 Xiaowei Dong2
1State Key Laboratory of Medical Genomics, Shanghai Key Laboratory of Hypertension, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 2Department of Pharmaceutical Sciences, 3Department of Pharmacotherapy, University of North Texas Health Science Center, Fort Worth, TX, USA
Purpose: The purpose of this study was to develop a novel lipid-based nanotechnology to formulate poorly water-soluble drugs in oral solid granules to improve stability, palatability, and bioavailability.
Materials and methods: In one method, we prepared ritonavir (RTV) nanoparticles (NPs) by a microemulsion-precursor method and then converted the RTV NPs to solid granules by wet granulation to produce RTV NP-containing granules. In the other innovative method, we did not use water in the formulation preparation, and discovered novel in situ self-assembly nanoparticles (ISNPs). We prepared RTV ISNP granules that did not initially contain NPs, but spontaneously produced RTV ISNPs when the granules were introduced to water with gentle agitation. We fully characterized these RTV nanoformulations. We also used rats to test the bioavailability of RTV ISNP granules. Finally, an Astree electronic tongue was used to assess the taste of the RTV ISNP granules.
Results: RTV NP-containing granules only had about 1% drug loading of RTV in the solid granules. In contrast, RTV ISNP granules achieved over 16% drug loading and were stable at room temperature over 24 weeks. RTV ISNPs had particle size between 160 nm and 300 nm with narrow size distribution. RTV ISNPs were stable in simulated gastric fluid for 2 hours and in simulated intestinal fluid for another 6 hours. The data from the electronic tongue showed that the RTV ISNP granules were similar in taste to blank ISNP granules, but were much different from RTV solution. RTV ISNP granules increased RTV bioavailability over 2.5-fold compared to RTV solution.
Conclusion: We successfully discovered and developed novel ISNPs to manufacture RTV ISNP granules that were reconstitutable, stable, and palatable, and improved RTV bioavailability. The novel ISNP nanotechnology is a platform to manufacture oral solid dosage forms for poorly water-soluble drugs, especially for pediatric formulation development.
Keywords: ritonavir, pediatric formulation, taste, pharmacokinetics, lipid formulation, drug loading
Introduction
Poorly water-soluble drug candidates are prevalent in drug-development pipelines. For oral administration, low solubility could lead to low bioavailability of drugs.1–3 Lipid-based nanoparticles (NPs), which take advantage of NPs as well as lipid-based formulations, have been studied as a means of improving drug solubility and bioavailability.4–6 They are prepared from biodegradable and biocompatible lipids and surfactants, reducing the likelihood of excipient-safety concerns.7–9 However, lipid-based NPs and formulations are generally prepared as liquid forms. Considering oral solid dosage forms are preferred for stability, manufacturing, storage, and transportation, lipid-based NPs have limited applications for oral medications.7,10 To address the issue, research efforts were directed toward converting liquid formulations into solid forms. However, low drug loading in the final solid forms was often obtained, as great amounts of solid excipients had to be added to remove water and/or achieve good flow properties.6,11–15
Potent combination antiretroviral therapy has converted human immunodeficiency virus (HIV) infection from an acute, rapidly lethal illness into a chronic, manageable disease.16–18 Nonetheless, lifelong adherence to oral medication regimens is crucial to ensure virologic and immunologic response. However, most of lifesaving HIV medications are available as oral capsule and tablet formulations, which pose a potential barrier to medication adherence for patients who are not able to ingest those dosage forms; such patients typically include pediatric and geriatric individuals.19 HIV protease inhibitors were discovered in the mid 1990s and are key components of combination antiretroviral therapy.17,20 Currently available protease inhibitors are substrates for the enzyme CYP3A, which reduces the systemic availability of these agents. To address the problem, the HIV protease inhibitor ritonavir (RTV) is widely coadministered to boost concentrations of other protease inhibitors by inhibiting their metabolism through CYP3A.21,22 Currently, Norvir® is the only RTV oral solution commercially available. Because of its low solubility in water, RTV is formulated in high concentrations of propylene glycol (<27%) and ethanol (>43%) in Norvir, causing adverse events and unpalatable taste. To improve palatability, RTV solution is often mixed with chocolate milk and consumed within 1 hour.23 In addition, due to poor stability, RTV solution has only a 6-month shelf life at room temperature, which increases the cost of the drug. Indeed, heat-stable 25 mg RTV sprinkle was highlighted as an urgently needed formulation in the World Health Organization’s list of missing drugs and formulations for pediatric HIV treatment.24
The objective of this work was to develop a novel nanotechnology platform that could formulate poorly water-soluble drugs into oral solid dosage forms and also improve their bioavailability. The nanoformulation should be flexible for dosing, stable at room temperature, and palatable for administration to patients with swallowing difficulty. For these purposes, RTV was chosen as the model drug for the studies reported herein. We initiated the NP development using a previously published microemulsion-precursor method,25 and then converted NPs to solid granules by wet granulation. We refer to this nanoformulation as RTV NP-containing granules (Figure 1A). However, the drug loading was too low to meet the oral dosage requirement (normally >25 mg per dose). As a breakthrough, we prepared the formulation using a new procedure that did not include water in the preparation, and discovered novel in situ self-assembly NPs (ISNPs). We successfully utilized the novel ISNP nanotechnology to manufacture RTV ISNP granules with high drug loading and a minimum 6-month shelf life at room temperature. RTV ISNP granules did not contain NPs; however, they produced RTV-loaded ISNPs when introduced in liquids (eg, water or the fluid in the gastrointestinal tract) (Figure 1B). Moreover, RTV ISNP granules had comparable taste with the placebo ISNP granules, as evaluated by using an Astree electronic tongue (e-tongue; Alpha MOS, Toulouse, France). Collectively, the novel ISNP nanotechnology could be a technology platform to formulate poorly water-soluble drugs for flexible oral solid dosage forms with acceptable palatability.
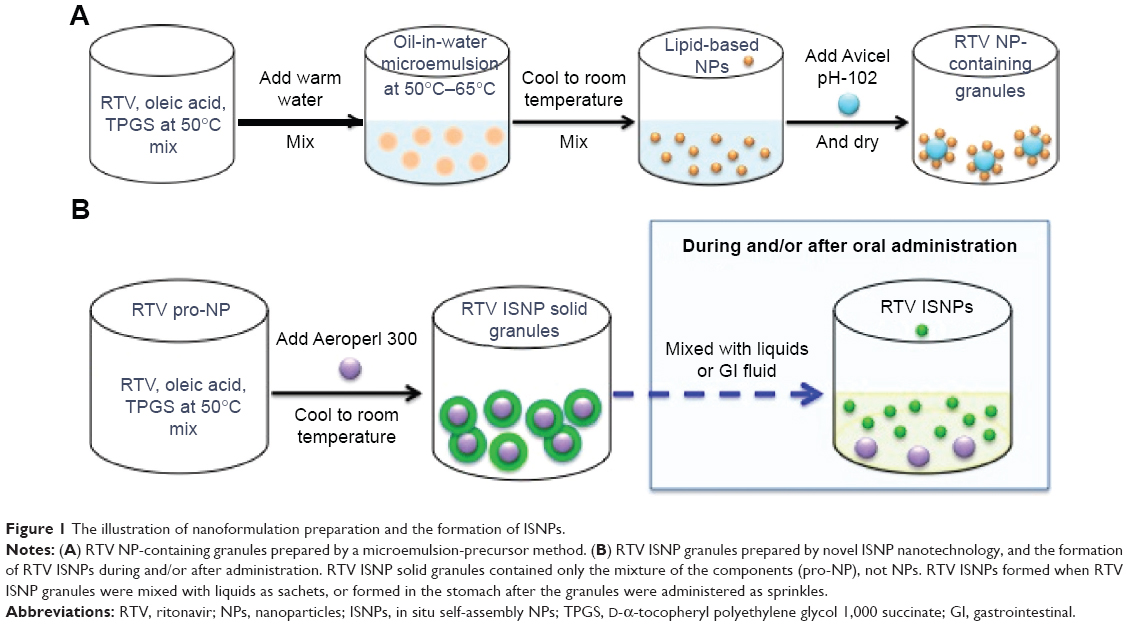 | Figure 1 The illustration of nanoformulation preparation and the formation of ISNPs. |
Materials and methods
Materials
RTV was purchased from USP Reference Standards (Rockville, MD, USA). d-α-Tocopheryl polyethylene glycol 1,000 succinate (TPGS) was provided as a gift from BASF SE (Ludwigshafen, Germany). Avicel PH 102 was provided as a gift from FMC BioPolymer (Philadelphia, PA, USA). Aeroperl 300 Pharma was provided as a gift from Evonik Industries (Essen, Germany). NaCl, KH2PO4 and NaOH were purchased from Sigma-Aldrich (St Louis, MO, USA). Oleic oil, high-performance liquid chromatography (HPLC)-grade acetonitrile, and methanol were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Quantitative measurement of RTV using HPLC
RTV concentration was measured using an Alliance 2695 Separation Module, equipped with a model 2487 wavelength ultraviolet detector (Waters, Milford, MA, USA). The column used was an Inertsil ODS-3 (150×4.6 mm internal diameter, 3 μm particle size; GL Sciences Inc, Tokyo, Japan). The mobile phase consisted of acetonitrile (solvent A) and 0.1% trifluoroacetic acid (solvent B). The isotopic program was 70% A and 30% B at a flow rate of 0.5 mL/min. The injection volume was 20 μL. RTV was monitored at a wavelength of 210 nm. The HPLC method was validated in terms of linearity, accuracy, precision, and limitation of detection.
Solubility measurement in simulated gastric fluid and simulated intestinal fluid
Solubility was determined in simulated gastric fluid (SGF; pH 1.2) and simulated intestinal fluid (SIF; pH 6.8).26 An excess of RTV was added into 5 mL of a medium and stirred for 48 hours at 37°C. Then, 1 mL of the suspension was centrifuged at 14,000 rpm for 10 minutes at room temperature, and the supernatant was taken. This procedure was repeated twice to completely separate RTV powder and dissolved RTV. After the last centrifugation, 150 μL of the supernatant was measured by HPLC. The experiment was performed in triplicate.
Preparation of RTV NP-containing solid granules
NP preparation by a microemulsion-precursor method has been described previously.25 The procedure to prepare RTV NP-containing solid granules is shown in Figure 1A. To prepare NPs suitable for RTV, we used oleic acid as a lipid and TPGS as a surfactant. Briefly, 600 μg of RTV, 2 mg of oleic acid, and 4 mg of TPGS were weighed and mixed in a glass vial for 10 minutes at 50°C. One milliliter of water was added to the mixture, stirred for 20 minutes at 50°C, and then cooled to room temperature to form the NP suspension. Avicel PH-102 (700 mg) was added to the NP suspension to prepare wet granules by wet granulation using a mortal and pestle. The wet granules were dried in a vacuum oven at 37°C overnight to obtain the solid granules.
Preparation of RTV ISNP granules
We prepared RTV ISNP granules by using our novel ISNP nanotechnology, as shown in Figure 1B. Briefly, 100 mg of RTV, 100 mg of oleic acid, and 200 mg of TPGS were mixed in a glass vial for 10 minutes at 50°C. We named the melted mixture “pro-ISNP”. Aeroperl 300 (110 mg) was added to the pro-NP, mixed, and cooled to room temperature to obtain RTV ISNP granules.
Particle-size and zeta-potential measurement
To measure the particle size of RTV NPs, 10 μL of RTV NPs was diluted with 1 mL Milli-Q water and measured by dynamic light scattering using a Delsa Nano HC particle analyzer (Beckman Coulter Inc, Brea, CA, USA). To measure particle size from RTV ISNPs, 10 mg RTV ISNP granules was reconstituted with 1 mL of Milli-Q water and centrifuged at 14,000 rpm for 5 minutes to obtain RTV ISNPs presenting in the supernatant. The particle size of RTV ISNPs was measured by the particle analyzer. Polydispersity index (PI) indicated size distribution. Zeta potentials of RTV NPs and RTV ISNPs were measured by the particle analyzer.
Differential scanning calorimetry measurement
Differential scanning calorimetry (DSC) analysis was performed using a DSC 400 (PerkinElmer Inc, Waltham, MA, USA) to determine the physical state of RTV in RTV ISNP granules. Placebo granules and RTV powder were used as controls. The heating curves were recorded at a scan rate of 10°C/min from 20°C to 250°C.
Determination of drug-loading and -entrapment efficiency
To analyze drug loading for RTV NPs, 100 μL of RTV NPs was mixed with 1 mL of methanol for HPLC measurement. For RTV ISNP granules, 5 mg of RTV ISNP granules was dissolved in 1.5 mL of methanol and mixed by a vortex mixer for 1 minute. The mixture was sonicated for 5 minutes and then centrifuged at 14,000 rpm for 5 minutes. The supernatant was removed and RTV quantitated using HPLC. Drug loading was calculated as follows:
|
|
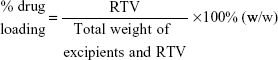
To measure entrapment-efficiency percentage (EE%), 10 mg of RTV ISNP granules were mixed with 1 mL of Milli-Q water and centrifuged to remove the solid excipient and obtain the supernatant that contained RTV ISNPs, which was mixed with methanol to measure total RTV in RTV ISNPs by HPLC. Free RTV was separated from either RTV NPs or RTV ISNPs (the supernatant) by a Microcon Y-100 (EMD Millipore, Billerica, MA, USA) by centrifugation at 14,000 rpm for 5 minutes at 4°C. The RTV NPs or RTV ISNPs in the insert of the Microcon were collected and mixed with 1,000 μL of methanol to measure the entrapped RTV by HPLC, as described earlier. EE was calculated as follows:
|
|

Short-term stability of RTV ISNPs in simulated physiological condition
To evaluate change in particle size in the gastrointestinal tract, blank ISNP granules and RTV ISNP granules were incubated in SGF and SIF, respectively. Briefly, 30 mg of granules was added to 24 mL of SGF with shaking at 150 rpm at 37°C. After 2 hours, 0.98 mL of 2 M KH2PO4 and 5.34 mL of 0.5 N NaOH were added to the SGF to adjust the pH to 6.8 (SIF). At each given time interval, 1.5 mL aliquots were collected and centrifuged at 14,000 rpm for 10 minutes. The supernatant was measured for particle size. Stability of ISNPs was evaluated in terms of particle size and distribution (n=3).
Long-term stability of RTV ISNP granules at room temperature
Three independent batches of RTV ISNPs were prepared and stored at room temperature for 24 weeks. At predetermined time intervals, samples were taken and measured for drug-loading percentage, EE%, particle size, PI, and zeta potential, as described earlier. The degradation of RTV was monitored according to the appearance of extra peaks on HPLC chromatograms.
In vitro release/dissolution studies
Release of RTV NPs and dissolution of RTV ISNP granules were performed in SGF and SIF. Briefly, 500 μL of RTV NPs and 2 mg of RTV ISNP granules were added to 5 mL of SGF to maintain sink conditions and shaken at 150 rpm at 37°C for 2 hours. Then, 0.98 mL of 2 M KH2PO4 and 5.34 mL of 0.5 N NaOH were added to adjust the pH value to 6.8 (SIF), and the study was continued for another 6 hours. At predetermined time points, sample aliquots were collected. Free RTV was separated from RTV NPs or RTV ISNPs using the Microcon and measured by HPLC, as described earlier.
Taste evaluation by e-tongue
The taste of RTV ISNP granules was evaluated with the Astree e-tongue. Sensor set 2, including ZZ, AB, GA, BB, CA, DA, and JE sensors, was equipped with the instrument for taste evaluation. The analysis procedures were conducted according to the manufacturer’s recommendations. Conditioning, calibration, and diagnostic tests were conducted before sample testing. For sample preparation, 500 mg of RTV ISNP granules was put into 100 mL of water and stirred for 20 minutes, which was equivalent to 80 μg/mL of RTV in the sample. According to the manufacturer’s recommendations, sensors can be used with up to 40% ethanol. To match the RTV concentration in the sample of RTV ISNP granules, we dissolved RTV powder into 35% ethanol to produce 80 μg/mL of RTV solution as a control. Oleic acid was dissolved in 35% ethanol and TPGS dissolved in water to corresponding concentrations equivalent to those in the RTV ISNP granules. Blank ISNP granules were also tested as a placebo. The data were collected and analyzed according to principal-component analysis. Each sample was measured in triplicate.
In vivo pharmacokinetic studies in rats
Three Sprague Dawley rats (males, 276–300 g; Charles River Laboratories, Wilmington, MA, USA) used in the study received standard care in compliance with an approved protocol by the Institutional Animal Care and Use Committee at the University of North Texas Health Science Center. All procedures were followed according to the Guide for the Care and Use of Laboratory Animals and referred to the Animal Welfare Act. Prior to conduction of the experiment, the rats were kept on a 12-hour light/dark cycle with unlimited access to food and water. RTV ISNP granules and RTV oral solution were administered to the rats by oral gavage at 10 mg/kg of RTV. After administration, 250 μL of blood was collected through the tail vein at 0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, and 24 hours, and centrifuged at 3,400 rpm for 5 minutes at 4°C to obtain plasma. Plasma was harvested and stored at −20°C until further analysis by an LC–mass spectrometry method previously reported.27
Plasma concentrations of RTV were analyzed by noncompartmental methods using Phoenix WinNonlin pharmacokinetic software (version 6.4; Certara, St Louis, MO, USA). Maximum plasma concentration (Cmax) and time to reach Cmax (Tmax) were obtained by direct inspection of the plasma concentration–time profile. The area under the plasma concentration–time curve from time 0 to the last measured sample (AUC0–last) was determined using the log-linear trapezoidal rule.
Statistical analysis
Data are presented as mean ± standard deviation. Continuous variables were compared by either Student’s t-test or an analysis of variance followed by the Student–Newman–Keuls post hoc test with SPSS software version 11.5 (SPSS Inc, Chicago, IL, USA). P<0.05 was considered statistically significant.
Results
HPLC method for measurement of RTV concentration
We developed and validated an HPLC method to measure RTV concentration in this study. In this HPLC method, RTV had a linear range from 0.39 to 100 μg/mL (R2=0.999). The limitation of detection was 0.1 μg/mL. Accuracy ranged from 99.7% to 99.9% for concentrations of 7.6, 38.1, and 76.1 μg/mL of RTV. Intraday precision was evaluated at three different concentration levels. Intermediate precision was evaluated in the same solutions on different days. Values show good precision of the method, with relative standard deviation lower than 2%. These data confirm that our HPLC method was valid and accurate to measure RTV concentration.
Solubility of RTV in SGF and SIF
RTV is known to be a water-insoluble drug; however, we did not find solubility data for RTV in SGF and SIF in the literature. To characterize the solubility of RTV in simulated physiological conditions and to design release/dissolution studies, we measured the saturated solubility of RTV in SGF and SIF. The saturated solubility of RTV in SGF and SIF was found to be 287.9±1.3 μg/mL and 1.5±0.1 μg/mL, respectively (n=3).
RTV NPs and RTV NP-containing granules
With our composition, we successfully prepared RTV NPs with particle size <200 nm and narrow size distribution (PI <0.3) using a previously reported microemulsion-precursor method (Figure 2). Physicochemical properties of the RTV NPs are summarized in Table 1. It is noteworthy that the theoretical drug loading of RTV NPs (4.8%) in Table 1 was calculated without water. If we included water in the preparation to represent the drug loading for the final batch of RTV NPs, the drug loading would be 0.06%; that means there is only 0.06 mg of RTV in 100 mg of RTV NP suspension. After we added Avicel PH-102 as a solid absorption agent to convert RTV NPs into a solid form, we ended up with about 1% drug loading in the final RTV NP-containing granules.
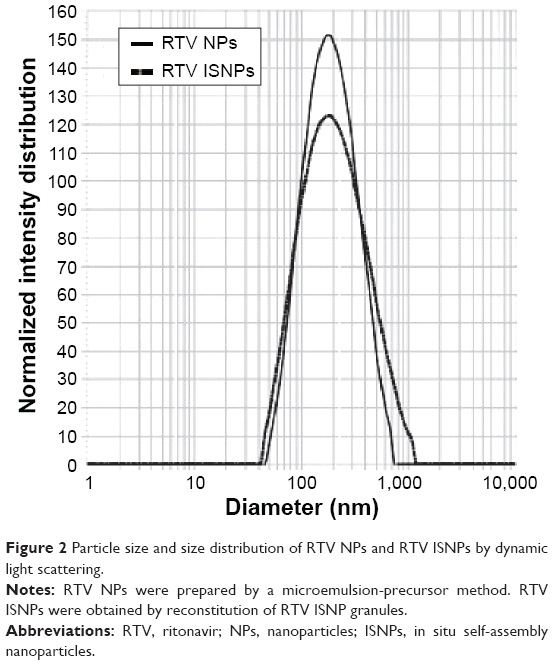 | Figure 2 Particle size and size distribution of RTV NPs and RTV ISNPs by dynamic light scattering. |
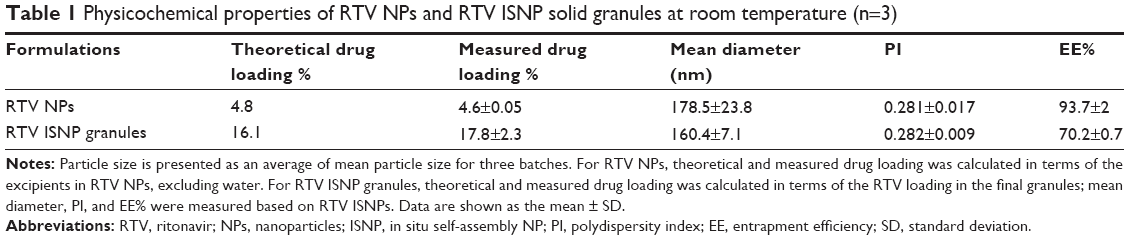 | Table 1 Physicochemical properties of RTV NPs and RTV ISNP solid granules at room temperature (n=3) |
We measured the release of RTV from RTV NPs. As shown in Figure 3, 35% of RTV quickly released when we put RTV NPs in SGF, and was maintained for 2 hours in SGF. After we changed SGF to SIF, dissolved RTV decreased to 5% within 1 hour, and then remained at 5% for another 4 hours in SIF.
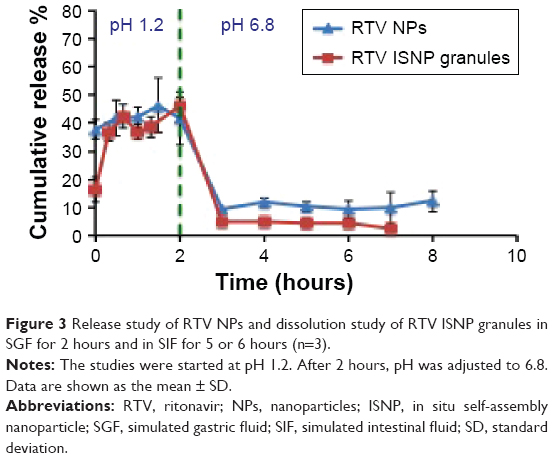 | Figure 3 Release study of RTV NPs and dissolution study of RTV ISNP granules in SGF for 2 hours and in SIF for 5 or 6 hours (n=3). |
RTV ISNPs and the granules
Characterization of RTV ISNPs from the granules
After we reconstituted RTV ISNP granules with water and obtained RTV ISNPs, we fully characterized the physicochemical properties of RTV ISNPs (Table 1). RTV ISNPs had similar particle size and size distribution to RTV NPs (Figure 2). RTV ISNPs were monodispersed with a narrow size distribution (PI <0.3). Since we removed water from the preparation of the granules, we were able to load 16.1% of RTV in the solid granules. At this high drug loading, over 70% of RTV was entrapped inside the ISNPs.
To understand the short-term stability of particle size for RTV ISNPs in physiological conditions, we measured the particle size of RTV ISNPs over time in SGF and SIF (Figure 4). Both blank ISNPs and RTV ISNPs showed comparable particle size in SGF for 2 hours (P>0.05). After SGF was changed to SIF, particle size of blank ISNPs slightly decreased from 2 hours to 3 hours and then remained consistent over the rest of the study. In contrast, RTV ISNPs slightly increased in particle size upon pH change, but remained the same size for the remaining 6 hours in SIF.
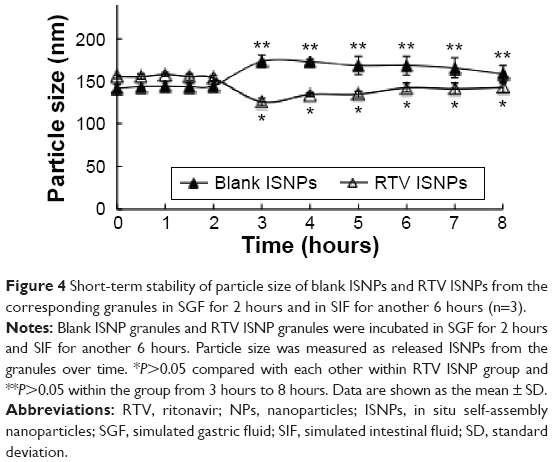 | Figure 4 Short-term stability of particle size of blank ISNPs and RTV ISNPs from the corresponding granules in SGF for 2 hours and in SIF for another 6 hours (n=3). |
We measured the dissolution of RTV ISNP granules in SIF and SGF (Figure 3). About 15% of RTV quickly released initially when we put RTV ISNP granules into SGF, and then the release was maintained in SGF for 2 hours. After SGF was changed to SIF, dissolved RTV decreased to about 10% and maintained this concentration for another 5 hours in SIF.
To evaluate the long-term stability of RTV ISNP granules, we stored RTV ISNP granules at room temperature for 24 weeks. Data for long-term stability are shown in Table 2. Over 24 weeks, we did not detect any degradation products based on HPLC measurements. Neither drug-loading percentage, EE%, particle size, nor zeta potential significantly changed (P>0.05). All together, these data demonstrate that RTV ISNP granules are stable at room temperature over 24 weeks.
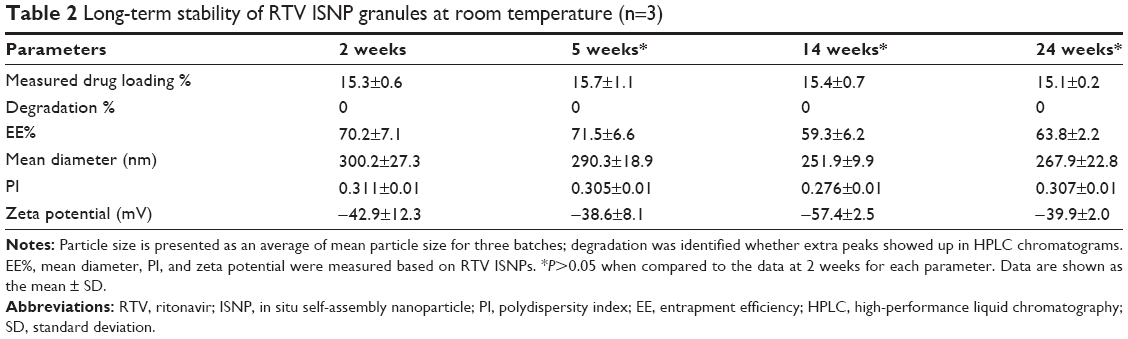 | Table 2 Long-term stability of RTV ISNP granules at room temperature (n=3) |
To evaluate the physical state of RTV in RTV ISNP granules, we utilized DSC to measure the granules. As shown in Figure 5, RTV existed in an amorphous state, as we did not observe the melting peak of RTV in RTV ISNP granules.
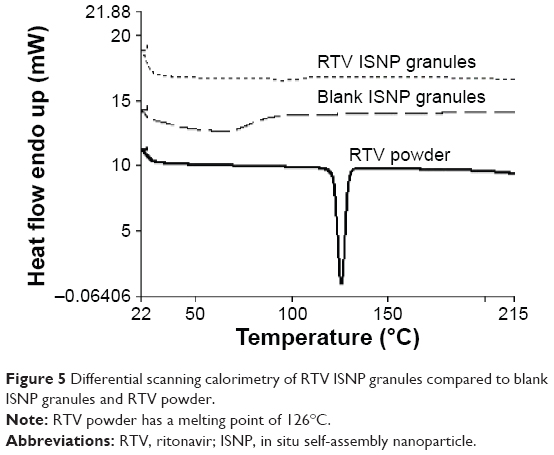 | Figure 5 Differential scanning calorimetry of RTV ISNP granules compared to blank ISNP granules and RTV powder. |
Taste of RTV ISNP granules
Taste is an important element for pediatric formulation development. We used the Astree e-tongue to assess the taste of RTV ISNP granules. As shown in Figure 6, the cluster of RTV ISNP granules was very close (almost in the same position) to the cluster of blank ISNP granules and TPGS, but far from the cluster of RTV solution on the principal-component analysis map, demonstrating that RTV ISNP granules have a taste that is similar to blank ISNP granules and TPGS.
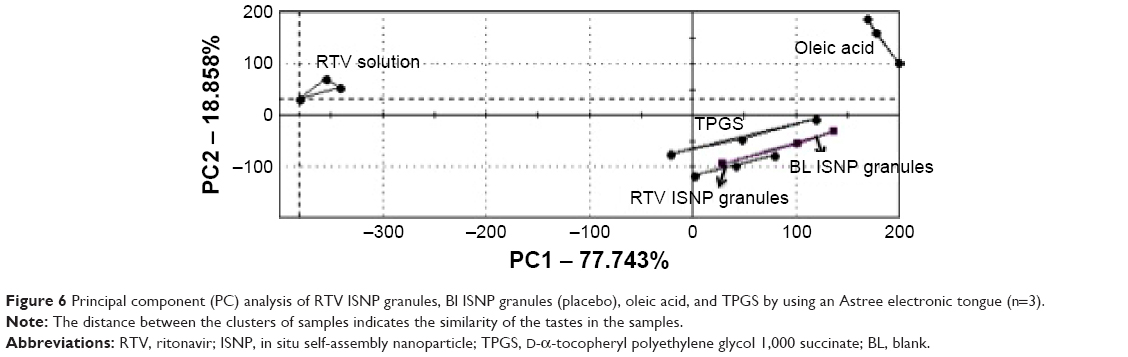 | Figure 6 Principal component (PC) analysis of RTV ISNP granules, Bl ISNP granules (placebo), oleic acid, and TPGS by using an Astree electronic tongue (n=3). |
Pharmacokinetics of RTV ISNP granules
We explored the pharmacokinetics of RTV ISNP granules in rats. Pharmacokinetic parameter values are shown in Table 3, and RTV versus time curves are shown in Figure 7. Compared to RTV oral solution, RTV ISNP granules increased AUC0–last 2.3-fold and Cmax 2.3-fold. Tmax was not significantly different between the formulations.
 | Table 3 Pharmacokinetic parameters for RTV after oral administration of RTV ISNP granules and RTV oral solution in rats (n=3) |
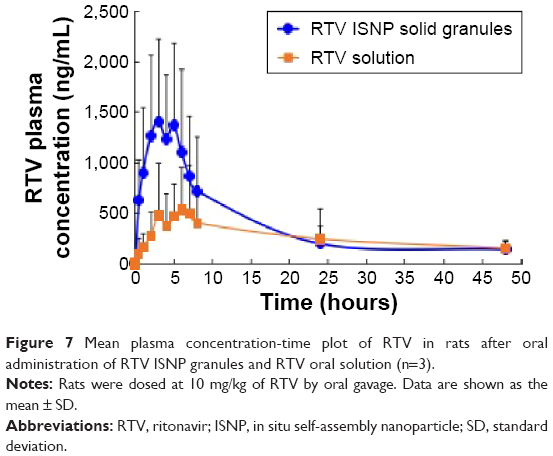 | Figure 7 Mean plasma concentration-time plot of RTV in rats after oral administration of RTV ISNP granules and RTV oral solution (n=3). |
Discussion
Lipid formulations, such as micelles, emulsions, and microemulsions, represent commonly used technologies to formulate poorly water-soluble drugs. As liquids in nature, they are primarily filled into soft gel capsules. Stability and manufacturing difficulties are major limitations when using these technologies.7 Lipid NPs, such as solid lipid NPs, liposomes, and nanocapsules, combine the advantages of lipid formulations and NPs, and offer alternative technologies for poorly water-soluble drugs. To prepare lipid-based NPs, water is added to NP preparation to disperse materials and form NPs. As a result, NPs are in a suspension that compromises the stability of lipid-based NPs, making them poor candidates for long-term storage. Solid absorption agents were added to lipid formulations or lipid-based NPs to convert them into solid forms, avoiding the use of soft gel capsules. Also, several approaches, including freeze-drying,12 extrusion,6 and spray-drying5 have been used to convert lipid-based NPs into solid forms. Until now, the key shortcoming of these approaches was low drug loading (<0.1%), which cannot satisfy dose requirements in solid dosage forms for oral administration. We have prepared paclitaxel NPs and doxorubicin NPs using a microemulsion-precursor method.25 Using the same method, we prepared RTV NPs, and then converted them to solid granules by adding a solid absorption agent (Figure 1A). Although we increased drug loading compared to that previously reported, we still only finished the solid granules with about 1% drug loading. The RTV oral human dose is at least 100 mg per day. Therefore, 1% drug loading is not sufficient to manufacture clinically relevant RTV formulations.
In this study, we discovered a novel ISNP nanotechnology (Figure 1B). To solve the issue of low drug loading, we completely removed water from the NP preparation and prepared pro-NP by simply mixing oleic acid and TPGS, which are both approved by the US Food and Drug Administration for oral administration. After we added water into the pro-NP preparation via gentle shaking, we detected ISNPs in the supernatant. This result was unexpected, as dispersing by water is an essential step in the microemulsion-precursor method. Therefore, the ISNP nanotechnology is novel and completely different from the microemulsion-precursor method. The ISNPs can form at different pH values and different temperatures. As demonstrated by DSC, RTV dissolved in oleic acid and TPGS when we prepared pro-NP. While oleic acid and TPGS form ISNPs in water, RTV was able to be entrapped into the ISNPs and formed stable RTV ISNPs. During the measurement of particle size, we diluted RTV ISNP granules over 1,000 times with water. Interestingly, with this extremely high dilution, we still detected ISNPs with high drug loading and EE% (Table 1). They were also stable in pH 1.2 and pH 6.8 over 8 hours (Figure 4). They had similar physicochemical properties with RTV NPs in terms of particle size, size distribution (Table 1), and release profiles (Figure 3). Therefore, we infer that RTV ISNPs and RTV NPs are very likely a similar type of NPs. Unlike self-emulsifying microemulsions and nanoemulsions,10 RTV ISNPs were stable for changes of environment, RTV was kept inside the ISNPs over the period of the release study, and there was no RTV precipitation after RTV ISNPs were diluted over 1,000 times. With this novel ISNP nanotechnology, we achieved over 16% drug loading of RTV in RTV ISNP granules, which could provide a dose of about 100 mg per administration in humans. Importantly, the manufacturing process of RTV ISNP granules is simple and scalable (Figure 1B).
RTV is a weak acid, and has high solubility in SGF and low solubility in SIF. Therefore, free RTV that released from NPs or ISNPs within 2 hours in SGF precipitated out when we changed SGF to SIF. Superior to RTV NPs, RTV ISNPs kept more RTV in NPs after changing the pH to 6.8 (Figure 3). Interestingly, although only 55% of RTV released from RTV ISNP granules in 2 hours in SGF in the in vitro dissolution study (Figure 3), RTV ISNP granules showed a twofold increase upon systemic exposure compared to RTV solution, demonstrating high oral absorption in vivo (Figure 7). Bile salts and enzymes could interact with RTV ISNPs, destroy RTV ISNP, and consequently release RTV into the gastrointestinal tract. Moreover, RTV ISNPs could enter the circulation through the lymphatic system. We are planning detailed studies to further understand mechanisms related to the oral absorption of RTV ISNP granules.
The major shortcomings of commercially available RTV oral solution are unpleasant taste and the need of a cool chain for storage and transportation. By using solid dosage forms, we produced RTV ISNP granules that were stable at room temperature for at least 24 weeks. The granules are reconstitutable and flexible, meaning they can be poured into liquids prior to administration as sachets or can be added to soft foods as sprinkles. Moreover, their taste is comparable to blank ISNPs (placebo), according to the taste evaluation by the e-tongue (Figure 6). Controlling RTV release from the ISNPs and encapsulating RTV inside the ISNPs may be the mechanisms by which the taste of RTV is masked by the ISNP granules.
Conclusion
We discovered a novel ISNP nanotechnology. RTV ISNP granules were successfully manufactured using the novel nanotechnology. RTV ISNP granules have improved taste, stability, and bioavailability. The granules are reconstitutable and flexible, and thus they are especially suitable for pediatric formulations. This novel ISNP nanotechnology has great potential for widespread applications to formulate poorly water-soluble drugs for oral solid dosage forms.
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in either drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Bodmeier R, Chen HG, Paeratakul O. A novel approach to the oral delivery of micro- or nanoparticles. Pharm Res. 1989;6(5):413–417. | ||
De Jaeghere F, Allémann E, Kubel F, et al. Oral bioavailability of a poorly water soluble HIV-1 protease inhibitor incorporated into pH-sensitive particles: effect of the particle size and nutritional state. J Control Release. 2000;68(2):291–298. | ||
Bourzac K. Nanotechnology: carrying drugs. Nature. 2012;491(7425):S58–S60. | ||
Pathak K, Raghuvanshi S. Oral bioavailability: issues and solutions via nanoformulations. Clin Pharmacokinet. 2015;54(4):325–357. | ||
Li FQ, Yan C, Bi J, et al. A novel spray-dried nanoparticles-in-microparticles system for formulating scopolamine hydrobromide into orally disintegrating tablets. Int J Nanomedicine. 2011;6:897–904. | ||
Pinto JF, Müller RH. Pellets as carriers of solid lipid nanoparticles (SLN) for oral administration of drugs. Pharmazie. 1999;54:506–509. | ||
Das S, Chaudhury A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech. 2011;12(1):62–76. | ||
Bernkop-Schnürch A. Nanocarrier systems for oral drug delivery: do we really need them? Eur J Pharm Sci. 2013;49(2):272–277. | ||
Mehnert W. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47(2–3):165–196. | ||
Khan AW, Kotta S, Ansari SH, Sharma RK, Ali J. Potentials and challenges in self-nanoemulsifying drug delivery systems. Expert Opin Drug Deliv. 2012;9(10):1305–1317. | ||
Balakrishnan P, Lee BJ, Oh DH, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm. 2009;72(3):539–545. | ||
Ohshima H, Miyagishima A, Kurita T, et al. Freeze-dried nifedipine-lipid nanoparticles with long-term nano-dispersion stability after reconstitution. Int J Pharm. 2009;377(1–2):180–184. | ||
Taha EI, Al-Suwayeh SA, Anwer MK. Preparation, in vitro and in vivo evaluation of solid-state self-nanoemulsifying drug delivery system (SNEDDS) of vitamin A acetate. J Drug Target. 2009;17(6):468–473. | ||
Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech. 2009;10(1):183–192. | ||
Friedrich RB, Fontana MC, Bastos MO, Pohlmann AR, Guterres SS, Beck RC. Drying polymeric drug-loaded nanocapsules: the wet granulation process as a promising approach. J Nanosci Nanotechnol. 2010;10(1):616–621. | ||
Antonelli G, Turriziani O. Antiviral therapy: old and current issues. Int J Antimicrob Agents. 2012;40(2):95–102. | ||
Durand CM, Flexner C. HIV cure: knocking on the door. Clin Pharmacol Ther. 2013;93(5):382–384. | ||
Farmer P, Léandre F, Mukherjee J, Gupta R, Tarter L, Kim JY. Community-based treatment of advanced HIV disease: introducing DOT-HAART (directly observed therapy with highly active antiretroviral therapy). Bull World Health Organ. 2001;79(12):1145–1151. | ||
Duggan JM, Akpanudo B, Shukla V, Gutterson G, Eitniear L, Sahloff EG. Alternative antiretroviral therapy formulations for patients unable to swallow solid oral dosage forms. Am J Health Syst Pharm. 2015;72(18):1555–1565. | ||
Becker S. Atazanavir: improving the HIV protease inhibitor class. Expert Rev Anti Infect Ther. 2003;1(3):403–413. | ||
Chokephaibulkit K, Nuntarukchaikul M, Phongsamart W, et al. Once- versus twice-daily lopinavir/ritonavir tablets in virologically suppressed, HIV-infected, treatment-experienced children: comparative pharmacokinetics and virological outcome after switching to once-daily lopinavir/ritonavir. J Antimicrob Chemother. 2012;67(12):2927–2931. | ||
Colombo S, Buclin T, Cavassini M, et al. Population pharmacokinetics of atazanavir in patients with human immunodeficiency virus infection. Antimicrob Agents Chemother. 2006;50(11):3801–3808. | ||
Toronto HIV Clinic. Oral antiretroviral administration: information on crushing and liquid drug formulations. 2014. Available from: http://www.hivclinic.ca/main/drugs_extra_files/Crushing%20and%20Liquid%20ARV%20Formulations.pdf. Accessed February 2, 2016. | ||
World Health Organization. Item 4: Updated list of missing drug formulations for HIV treatment. 2011. Available from: http://www.who.int/selection_medicines/committees/expert/18/policy/policy4/en. Accessed February 2, 2016. | ||
Dong X, Mattingly CA, Tseng M, Cho M, Adams VR, Mumper RJ. Development of new lipid-based paclitaxel nanoparticles using sequential simplex optimization. Eur J Pharm Biopharm. 2009;72(1):9–17. | ||
Stippler E, Kopp S, Dressman J. Comparison of US Pharmacopeia simulated intestinal fluid TS (without pancreatin) and phosphate standard buffer pH 6.8, TS of the International Pharmacopoeia with respect to their use in in vitro dissolution testing. Dissolut Technol. 2004;5:6–10. | ||
Damaramadugu R, Inamadugu J, Kanneti R, Polagani S, Ponneri V. Simultaneous determination of ritonavir and lopinavir in human plasma after protein precipitation and LC-MS-MS. Chromatographia. 2010;71(9–10):815–824. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.