Back to Journals » Cancer Management and Research » Volume 10
Novel endocrine therapeutic strategy in endometrial carcinoma targeting estrogen-related receptor α by XCT790 and siRNA
Authors Sun PM ![]() , Mao XD
, Mao XD ![]() , Gao M, Huang MM, Chen LL, Ruan GY
, Gao M, Huang MM, Chen LL, Ruan GY ![]() , Huang WY
, Huang WY ![]() , Braicu EI, Sehouli J
, Braicu EI, Sehouli J
Received 13 March 2018
Accepted for publication 8 May 2018
Published 10 August 2018 Volume 2018:10 Pages 2521—2535
DOI https://doi.org/10.2147/CMAR.S168043
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
PengMing Sun,1,* XiaoDan Mao,1,* Min Gao,2 MeiMei Huang,1 LiLi Chen,1 GuanYu Ruan,1 WeiYi Huang,1 Elena Ioana Braicu,3 Jalid Sehouli3
1Laboratory of Gynecologic Oncology, Fujian Provincial Maternity and Children’s Hospital, Affiliated Hospital of Fujian Medical University, Fuzhou 350001, People’s Republic of China; 2Department of Gynecology Oncology, Beijing Cancer Hospital, Beijing 100142, People’s Republic of China; 3Department of Gynecology, Campus Virchow Clinic, Charité Medical University Berlin, Berlin, Germany
*These authors contributed equally to this work
Purpose: To explore the targeted therapy of estrogen-related receptor α (ERRα) in endometrial cancer (EC) cells and its potential mechanisms.
Methods: The mRNA and protein expression levels of ERRα and estrogen receptor α (ERα) were detected by qPCR and Western blotting in RL-952, AN3-CA, HEC-1A, and HEC-1B EC cell lines. After treatment with the ERRα-specific antagonist XCT790 or infection with lentivirus-mediated small interfering RNA (siRNA) targeting the ERRα (siRNA-ERRα), cell proliferation and apoptosis were evaluated by MTS assay and flow cytometry. After treatment with siRNA-ERRα, the expression profiles of transcription factors (TFs) were analyzed by protein/DNA arrays in EC cells.
Results: The relative mRNA levels of ERRa in RL-952 (1±0.0831) and AN3-CA (1.162±0.0325) were significantly higher than those in HEC-1A (0.3081±0.0339) and HEC-1B (0.1119±0.0091) (P<0.05), and similar results were observed for ERRα protein levels. A higher ratio of ERa/ERRa was observed in ERα-positive RL-952 (10-fold) and ANC-3A (8.5-fold) cells, whereas a lower ratio was observed in ERα-negative HEC-1A (3.75-fold) and HEC-1B cells (0-fold). Both – exogenous XCT790 and endogenous siRNA-ERRa – can decrease the expression of ERRα, thereby inhibiting proliferation but promoting apoptosis in both ERα-positive and -negative EC cells. The XCT790 presented higher proliferation-inhibition and apoptosis rates in the ERα-positive than ERα-negative cells, whereas the siRNA-ERRα exhibited higher proliferation-inhibition and apoptosis rates in the ERα-negative than in ERα-positive cells. In total, 3 upregulated and 17 downregulated TFs were screened out by knocked-down expression of ERRα in all EC cells. Among them, the upregulated TFs organic cation transporter 3/4(Oct3/4), hepatic nuclear factor 4 (HNF4), HNF4 and chicken ovalbumin upstream TF (COUP-TF) as well as downregulated transcription factor EB (TFEB) were found to be statistically significant (P<0.05).
Conclusion: Targeting ERRα provides a promising novel endocrine therapeutic strategy.
Keywords: ERRα, XCT790, siRNA, endocrine target therapy, endometrial cancer cells
Introduction
In 2018, >63,230 estimated new cases of endometrial cancer (EC) were diagnosed and ~11,350 estimated patients died due to uterine carcinomas in the United States.1 The incidence of EC has been slowly, but stably, increasing over the past two decades in China, with an estimated 63,400 new cases of EC and 21,800 estimated deaths in 2015.2 In most cases, endometrial carcinoma can be diagnosed in the early stages (67%). In this case, the disease is confined to the uterus, usually presents with bleeding, and has a 5-year survival rate of 95% reported in the USA.3 In contrast, if the disease is identified in more advanced stages, the 5-year survival rate is only 17%.4
Endocrine therapy is one of the treatment options for EC.5 Estrogen plays a predominant role in the proliferation and malignancy of estrogen-dependent tumors, such as breast cancer and EC.6 The biological activity of estrogen is known to be transduced by two classic nuclear receptors – estrogen receptor (ER) α and β.7 At present, hormone therapies such as medroxyprogesterone and megestrol usually aim to block estrogen and ER binding in breast cancer treatment.8,9 However, the mechanism by which endocrine therapy blocks estrogen and its receptors is still uncertain in EC. Estrogen-related receptor α (ERRα) was earlier identified by screening a kidney cDNA library using the sequence of the ERα DNA-binding domain (ERα-DBD) as a probe.8 ERRα and ERα have a very high similarity in the amino acid sequence of their DBDs and ligand-binding domains (LBDs).9,10 However, ERRα is not activated by any known natural estrogens or physiological ligands and is, therefore, classified as an orphan nuclear receptor.11 ERRα is expressed in a wide variety of tissues, and its high expression correlates with poor prognoses in breast,12–14 colon15 and prostatic cancers.16 In our previous study, we demonstrated that ERRα overexpression was an independent marker of poor prognosis in ovarian cancer.17 In EC, we found a positive correlation between ERRα mRNA expression and the International Federation of Gynecology and Obstetrics (FIGO) stage of myometrial invasion.18 Furthermore, ERRα overexpression inhibited cell proliferation in ERα-responsive Ishikawa cells, but stimulated cell proliferation in ERα-nonresponsive HEC-1A cells.19 These findings indicate that the function of ERRα depends on the level of ERRα expression as well as the function of ERα. Thus, ERRα may be a potential target for endocrine therapy of hormone-related cancers.
The synthetic molecule XCT790 is an ERRα-specific ligand and acts as an antagonist.20 Recent studies have reported the anticancer effect of XCT790 in breast cancer through ERRα targeting via different mechanisms.14 It is interesting that ERRα activity was shown to be elevated in tumors that are either human epidermal growth factor receptor-2 (HER2)-positive or ER-negative or those that contain P53 mutations.21,22 Moreover, studies showed that XCT790 was efficacious in both ERα-positive and ERα-negative breast cancer cells.23,24 XCT790 may be used as an exogenous agent to downregulate ERRα in EC cells. Small interfering RNA (siRNA) – a symmetric or asymmetric double-stranded RNA measuring ~20 bp – have long been used as a molecular tool to regulate the expression of the gene of interest in basic research.25 siRNAs are currently used as biotherapeutics to silence disease-causing genes, and siRNA-mediated gene therapy is a promising strategy for transient inhibition of the expression of genes involved in the development of breast cancer.26 Given its significant inhibitory and silencing effect on target genes, siRNA has become an invaluable tool for the downregulation of ERRα in endometrial carcinoma cells. Endocrine therapy for the treatment of EC may not be as effective as in breast cancer. Therefore, we aimed to demonstrate a novel endocrine therapy strategy by endogenous and exogenous downregulation of ERRα in endometrial carcinoma cells.
Materials and methods
Relative real-time quantitative polymerase chain reaction analysis
Total RNA was extracted by the TRIzol reagent (Thermo Fisher, Los Angeles, CA, USA). Briefly, 1 µg DNase I-treated RNA samples was reverse transcribed to complementary DNA (cDNA) using a reverse transcription kit (Promega, Madison, WI, USA). The polymerase chain reaction (PCR) primer sets used were as follows: ERRα sense 5′-ACC GAG AGA TTG TGG TCA CCA-3′, anti-sense 5′-CAT CCA CAC GCT CTG CAG TACT-3′ (101 bp); glyceraldehyde 3-phosphate dehydrogenase (GADPH, control) sense 5′-GCA CCG TCA AGG CTG AGA AC-3′, anti-sense 5′-TGG TGA AGA CGC CAG TGGA-3′ (138 bp); and ERα sense 5′-CCA ATT ATG AGA TGG ACT GTG GGT A-3′, anti-sense 5′-CTT GGG AAT GGT GCA GTG TAA TCT A-3′ (99 bp). The PCR products were electrophorized on 2% agarose gel and stained with ethidium bromide. The relative level of ERRα mRNA expression was quantified by the image-intensity ratio of the target gene mRNA against GAPDH mRNA. The target gene mRNA level was quantified using the comparative method (2−ΔΔCT method) and normalized to GAPDH expression.
Western blot analysis
Cell lysates were prepared by lysis/extraction reagent (Sigma-Aldrich, St. Louis, MO, USA). Proteins were quantified by a bicinchoninic acid (BCA) protein assay reagent kit (Pierce, Rockford, IL, USA). Protein samples (50 µg) were electrophorized and transblotted. Blotted membranes were incubated with the anti-human ERRα rabbit monoclonal antibody (1:300 dilution; Abcam, San Francesco, CA, USA) overnight at 4°C and β-Actin mouse monoclonal antibody(1:1000 dilution; Beyotime, Shanghai, China). An enhanced chemiluminescence (ECL) detection system (Thermo Fisher) was used to visualize the bands.
Cell culture and treatment with XCT790
Human endometrial carcinoma cell lines RL-952, AN3-CA, HEC-1A, and HEC-1B were purchased from the American Type Culture Collection (ATCC) and maintained in our laboratory. All cells used were the most common in vitro EC cell models. Moreover, the expressions of ERα in RL-952 and AN3-CA were positive but the ERα expressions were negative in HEC-1A and HEC-1B, which created conditions for exploring the relevance of ERα and ERRα. RL-952 was cultured in DMEM/F12 medium supplemented with 0.005 mg/mL insulin and 10% fetal bovine serum (FBS) and AN3-CA was cultured by Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% FBS. HEC-1A and HEC-1B were cultured in DMEM high-glucose medium, respectively, supplemented with 10% FBS at 37°C in a 5% CO2 atmosphere. All culture media were purchased from Thermo Fisher Scientific. Cells treated with XCT790 (Sigma-Aldrich, St. Louis, MO, USA) were transferred in phenol red-free medium (Thermo Fisher) containing 1% serum-replacement-2 (Sigma-Aldrich) for 24 h. In regard to the XCT790 concentrations affecting endometrial stromal cells,27 EC cells were treated with 1 or 10 µM XCT790 (dimethyl sulfoxide [DMSO], soluble; Sigma-Aldrich) following incubation14 or DMSO (control) for 24 or 48 h in this study.
Lentivirus-mediated siRNA construction and infection
A lentivirus-mediated siRNA named siRNA-ERRα was purchased from Genechem (Shanghai, People’s Republic of China), whose specificity and knockdown effect had already been confirmed and guaranteed by the manufacturers. The siRNA target sequence selected in the ERRa gene (ESRA; GenBank accession NM_001282451.1) was as follows: siRNA-ERRα GAG CGA GAG GAG TAT GTT CTA. Stem-loop oligonucleotides were synthesized and cloned into a lentivirus-based vector carrying the green fluorescent protein (GFP) gene (GV115; Genechem). A universal sequence (PSC-NC: TTC TCC GAA CGT GTC ACG T) – NC – was used as a negative control for RNA interference, whereas cells without treatment were used as a blank. Lentivirus particles were prepared as described previously for a siRNA target sequence. The lentiviral vector constructs carrying siRNA-ERRα and NC were used to infect four EC cells at multiplicities of infection (MOI) of 100. After 72 h of infection, GFP expression was detected to calculate the infection efficiency. Cells were harvested when the infection efficiency was ≥90%.
3-(4,5-Dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) analysis
RL-952, AN3-CA, HEC-1A, and HEC-1B cells were seeded at a density of 1×104 cells/well in 96-well plates and cultured in a complete medium for 24 h. Cells were transferred to phenol red-free medium containing 1% serum-replacement-2 and incubated for an additional 24 h. Following incubation, cells were treated with 10 µM XCT790 for 0, 24, 48, 72, and 96 h. The media was removed, and fresh media added to each well along with 20 µL MTS reagent. Following a 2 h incubation, the absorbance of each well was measured at 490 nm with a microplate reader (Stat FAX 2100; Los Angeles, CA, USA). The experiment was repeated triplicate, with four replicates for each treatment.
Apoptosis analysis via flow cytometry
For flow cytometric analysis, all cells with XCT790 treatment or siRNA treatment were seeded into six-well plates. When the cells reached 80% confluence, EDTA-free trypsin was added, and the cells were harvested. After centrifugation, cell pellets were washed twice with pre-cooled PBS. Cells were resuspended in a buffer at 106 cells/mL. Cells were stained with the Annexin-V-FLUOS or 7-AAD staining kit (BD, New York, NY, USA) according to the manufacturer’s instructions. The proportions of apoptotic cells were measured using a FACS Canto II flow cytometer (BD) and analyzed with Diva software (BD). All experiments were conducted in triplicate.
Protein/DNA array analysis
The protein/DNA array analysis was conducted by KangChen Bio-tech Inc. (Shanghai, People’s Republic of China). The array analysis procedure was undertaken according to the manufacturer’s instructions. Briefly, 106/mL endometrial cancer cells were seeded in 25 cm2 cell-culture flasks (Corning, Lowell, CA, USA). Nuclear proteins were extracted using an NE-PER Nuclear Protein Extraction Kit (Pierce, Rockford, IL, USA) and quantified with a BCA protein assay kit (Beyotime, Haimen, People’s Republic of China). Biotin-labeled DNA-binding probes were mixed with nuclear extract to form DNA/protein complexes, which were then passed through spin columns to remove unbound probes. The eluted bound probes were hybridized to a membrane which contained an array of 345 transcription factor (TF) consensus binding sequences (Spin Column version, Panomics, Freemont, CA, USA). After being washed, the DNA/protein array was incubated with horseradish peroxidase (HRP)-conjugated streptavidin solution (Pierce, Rockford, IL, USA) and visualized by using Horseradish Peroxidase (HRP) Substrate Working Solution (Millipore, Billerica, MA, USA). Images of the chemiluminescent signal were captured using Syngene GBox Imaging System (Cambridge, UK) and quantitated with MeV software. For data analysis, we retained significant changes (fold change ≥2.0) in every experiment.
Statistical analysis
All values were reported as mean±SD of three independent experiments, otherwise specified. An independent sample t-test and analysis of variance (ANOVA) were used to compare the parametric data using SPSS 19.0 software. A value of P<0.05 was considered statistically significant.
Results
Expression of ERRα and ERα in different EC cells
Real-time PCR (RT-PCR) analysis revealed the relative mRNA expression of ERRa in RL-952, AN3-CA, HEC-1A, and HEC-1B to be 1±0.0831, 1.162±0.0325, 0.3081±0.0339, and 0.1119±0.0091, respectively (Figure 1A). A similar tendency was observed for ERRα protein expression in these EC cells. The semiquantitative ERRα protein expression in RL-952, AN3-CA, HEC-1A, and HEC-1B was 1±0.0513, 1.1434±0.0419, 0.3968±0.0367, and 0.2632±0.0243, respectively (Figure 1B). The relative expression of classic ERα in RL-952 (1.0±0.0687) and AN3-CA (0.984±0.0521) was significantly higher than that in HEC-1A (0.115±0.0468) (P<0.05). Moreover, negative ERa expression was observed in HEC-1B (Figure 1C). In line with the previous reports, our result confirmed that ERa expression was positive in RL-952 and AN3-CA cells, whereas it was weak in HEC-1A and negative in HEC-1B cells.28 The ratio of ERa/ERRa was ~10.00-fold and 8.47-fold in RL-952 and AN3-CA, respectively, whereas it was significantly lower in HEC-1A (~3.75-fold) and HEC-1B (~0) cells (Figure 1D).
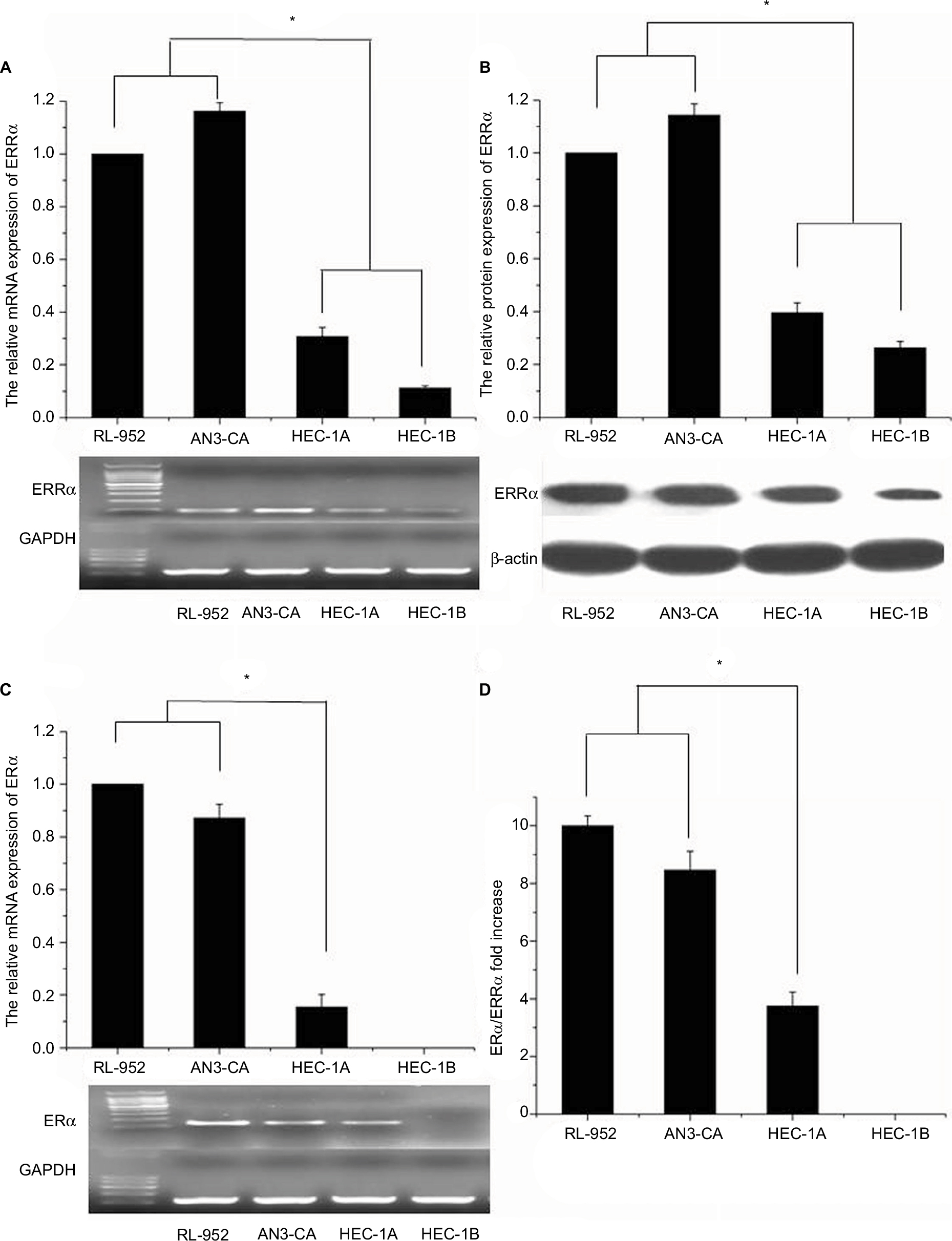 | Figure 1 Expression of ERRα and ERα in EC cell lines. (A) mRNA expressions of ERRα in RL-952, AN3-CA, HEC-1A, and HEC-1B were detected by real-time quantitative PCR, and the PCR products were electrophorized on 2% agarose gel and stained with ethidium bromide (EB). There was higher expression of relative ERRα mRNA in ERα-positive RL-952 and AN3-CA cells than in ERα-nonfunctional HEC-1A and ERα-negative HEC-1B cells. (B) Protein expression of ERRα in four endometrial cancer cells was detected by Western blotting, and a similar trend to the mRNA expression was also observed in the protein expression of ERRα. (C) mRNA expression of ERα in all cells was detected by real-time quantitative PCR and electrophorized. (D) The relative ratio of ERα/ERRα mRNA expression in all cells was calculated. All experiments were repeated in triplicate. *Significant differences (P<0.05) are indicated. Abbreviation: EC, endometrial cancer. |
XCT790 treatment downregulated the expression of ERRα but not of ERα
Results of RT-PCR revealed that treatment of ERα-positive and -negative EC cells with different concentrations of XCT790 for 24 h resulted in the downregulation of the relative mRNA expression of ERRa in a dose-dependent manner. In comparison to the untreated cells and those treated with DMSO, cells treated with 10 and 1 µM XCT790 showed significantly lower levels of relative ERRa mRNA expression. Treatment with 10 µM XCT790 inhibited ERRa mRNA expression by 41.06% (0.7311±0.0212 vs 1.2404±0.0831, P<0.05) in RL-952 (Figure 2A), 40.73% (0.8525±0.0236 vs 1.4413±0.0325, P<0.05) in AN3-CA (Figure 2B), 22.76% (0.2692±0.0034 vs 0.3822±0.0339, P<0.05) in HEC-1A (Figure 2C), and 20.32% (0.1101±0.0046 vs 0.1388±0.0091 P<0.05) in HEC-1B (Figure 2D) cells, respectively, as compared to the untreated cells (Blank group). As the highest ERRa inhibition was achieved with 10 µM XCT790, we used this concentration to observe the time-effect of treatment with XCT790. ERα-positive and functional EC cells, RL-952 (Figure 2E), and ERα-positive but non-functional EC cells, HEC-1A (Figure 2F), were treated with 10 µM XCT790 for 24 and 48 h. We observed a time-dependent inhibition of ERRa expression following XCT790 treatment. Although the inhibitory effect of ERRa expression treated with 10 µM XCT790 for 48 h was better than that treated for 24 h, the suitable time was still 24 h in consideration of the cell quantity declining sharply after 48 h. A sharp reduction in cell quantity would influence the content of available total RNA and protein directly, which would increase the difficulty of the experiment. Above all, the inhibitory effect of ERRa expression treated with 10 µM XCT790 for 24 h was sufficiently obvious. A similar result was observed for ERRα protein expression when RL-952 (Figure 3B), AN3-CA (Figure 3C), HEC-1A (Figure 3D), and HEC-1B (Figure 3E) were treated with 10 μM XCT790 for 24 h. In line with previous reports, XCT790 failed to exert any effect on ERa expression in all EC cells at both time points (Figure 3A).
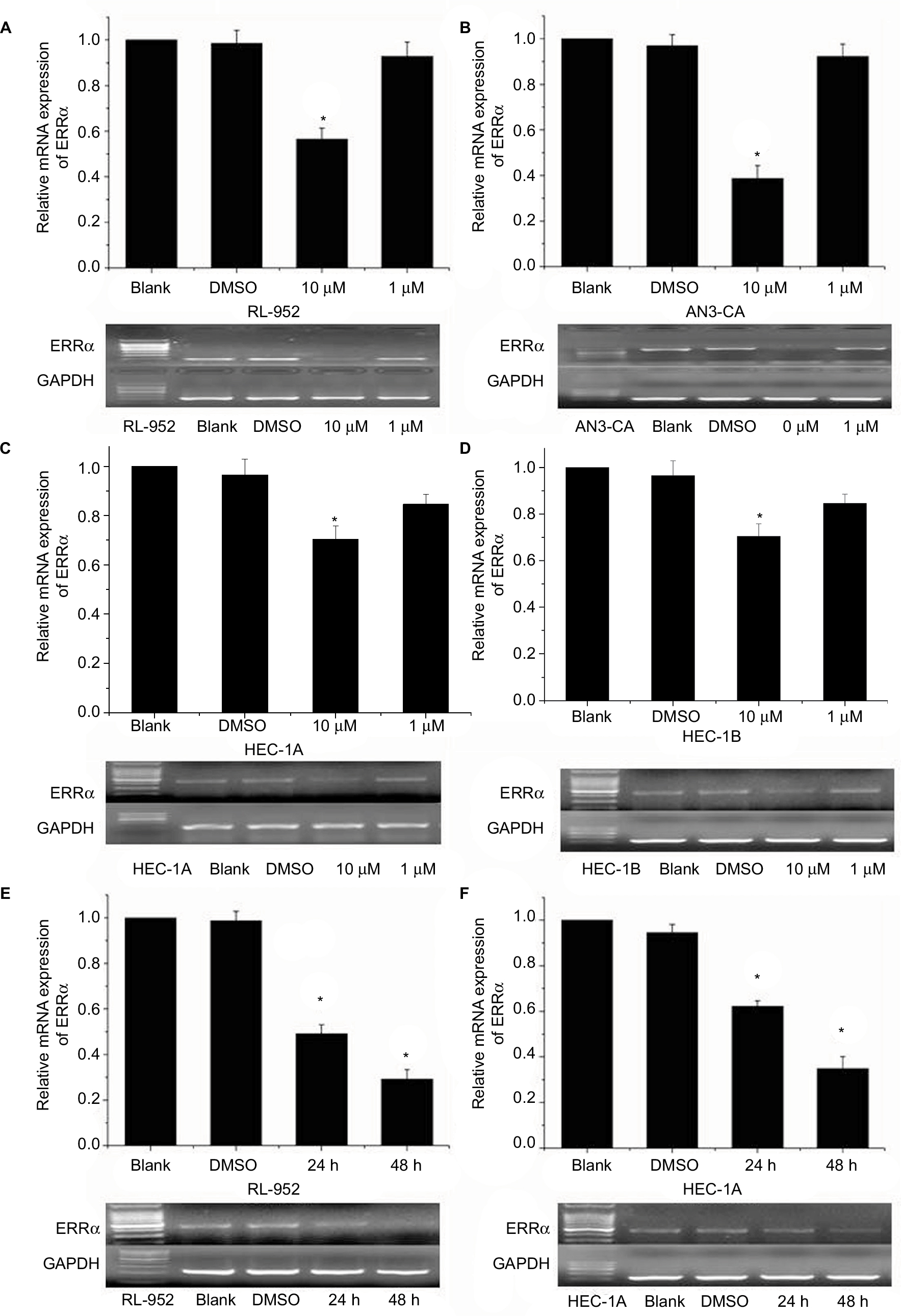 | Figure 2 Relative ERRα mRNA expression in EC cells treated with different concentrations of XCT790. Relative mRNA expression of ERRα was analyzed in RL-952 (A), AN3-CA (B), HEC-1A (C), and HEC-1B (D) EC cell lines treated with XCT790. After treatment with 10 or 1 µM XCT790 (XCT group), the expressions of ERRα mRNA were compared with the cells treated with 0.1% DMSO (DMSO group) or untreated cells (blank group). The RL-952 (E) and HEC-1A (F) cells treated with 10 µM XCT790 for 24 or 48 h were also analyzed. Compared to the DMSO or blank group, the ERRα mRNA in cells treated with 10 µM XCT790 for 24 h and 48 h showed a downregulation of ERRα induced by XCT790. All experiments were conducted in triplicate. *Significant differences (P<0.05) are indicated. Abbreviation: EC, endometrial cancer. |
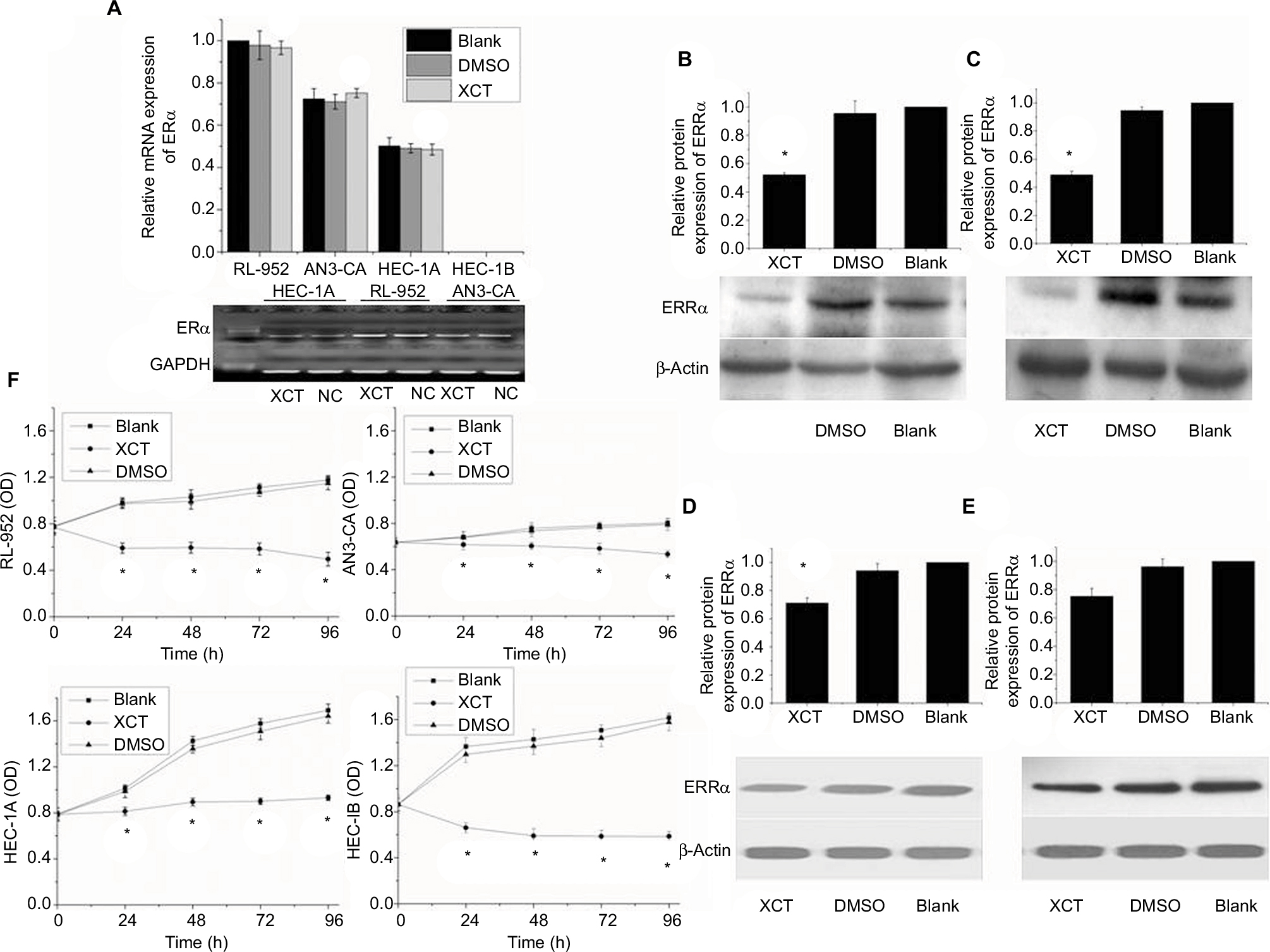 | Figure 3 XCT790 effect on the expression of ERα and ERRα and cellular proliferation in EC cells. (A) mRNA expression of ERα in all endometrial cancer cells treated with 10 µM XCT790 for 24 h was analyzed by real-time quantitative PCR and electrophorized on 2% agarose gel and stained with EB. XCT790 had no effect on the expression of ERα. Protein expression of ERRα in RL-952 (B), AN3-CA (C), HEC-1A (D), and HEC-1B (E) cells treated with 10 µM XCT790 for 24 h was detected by Western blotting. Comparing the cells treated with 0.1% DMSO or untreated cells (DMSO or blank group, respectively), ERRα protein expression had a similar trend to the mRNA expression. (F) The growth rates of all EC cells with different treatment were analyzed for 24, 48, 72, and 96 h by using an MTS assay. XCT790 suppressed cell proliferation effectively. All experiments were conducted in triplicate. *Significant differences (P<0.05) are indicated. Abbreviations: EC, endometrial cancer; PCR, polymerase chain reaction; EB, ethidium bromide; MTS, 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium. |
XCT790 inhibited cell proliferation
RL-952, AN3-CA, HEC-1A, and HEC-1B cells were treated with 10 µM XCT790 for 24, 48, 72, and 96 h. The growth inhibition ratio at 0, 24, 48, 72, and 96 h was as follows: RL-952 – 2.832%±1.330%, 39.670%±0.2.868%, 40.710%±1.435%, 43.660%±6.036%, and 54.247%±6.300%, respectively; AN3-CA – −0.052%±0.020%, 9.328%±0.379%, 17.708%±0.386%, 25.278%±1.438%, and 33.121%±0.800%, respectively; HEC-1A – 0.212%±0.020%, 20.661%±3.837%, 32.654%±0.378%, 43.271%±0.249%, and 44.736%±0.135%, respectively; and HEC-1B–2.373%±0.292%, 49.191%±0.504%, 57.747%±3.065%, 59.427%±0.314%, and 62.500%±1.398%, respectively. MTS assay revealed that XCT790 reduced cell proliferation by downregulating ERRα at 24 h (P<0.05; Figure 3F).
siRNA-mediated downregulation of ERRα expression
Results from RT-PCR and Western blot analysis showed that the relative expression of ERRa was downregulated by siRNA in RL-952, AN3-CA, HEC-1A, and HEC-1B (P<0.05). The relative expression of ERRa was decreased in RL-952 (0.7014±0.0172 vs 1.1646±0.0187, P<0.05), AN3-CA (0.8990±0.0185 vs 1.3624±0.0069, P<0.05), HEC-1A (0.0929±0.0036 vs 0.5405±0.0379 P<0.05), and HEC-1B (0.0975±0.0041 vs 0.3562±0.0010 P<0.05) as compared with the untransfected cells (negative control [NC]). The inhibition ratios were 39.77%, 34.01%, 82.82%, and 72.63% for RL-952, AN3-CA, HEC-1A, and HEC-1B, respectively (Figure 4A). Similar results were observed for ERRα protein expression in RL-52 (Figure 4B), AN3-CA (Figure 4C), HEC-1A (Figure 4D), and HEC-1B (Figure 4E).
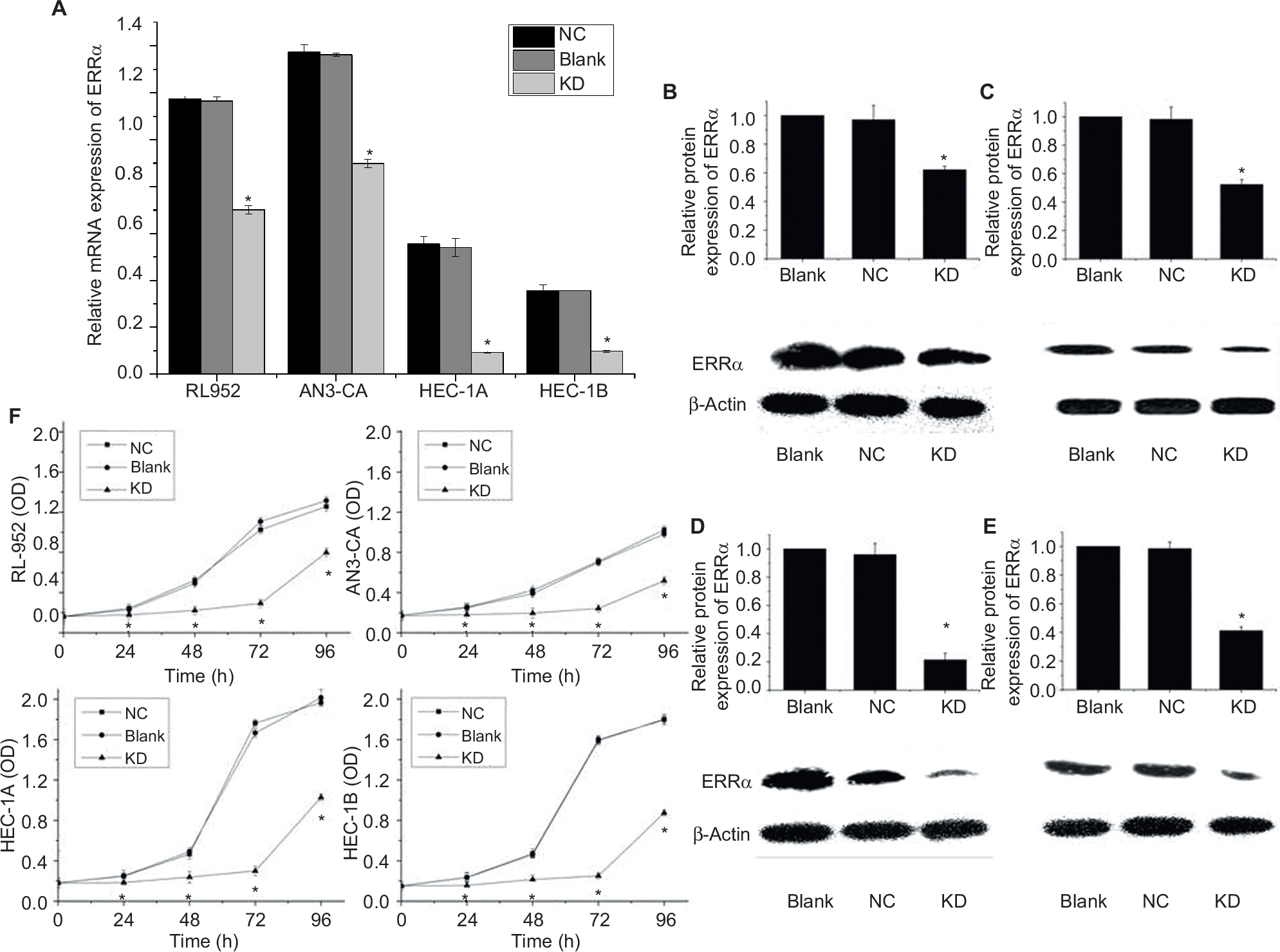 | Figure 4 siRNA targeting downregulates ERRα expression and suppresses EC cell proliferation. (A) mRNA expression of ERRα in four endometrial cancer cells infected by siRNA specific for (siRNA-ERRα) and named as the knockdown (KD) group was analyzed by real-time PCR. The ERRα mRNA levels of all EC cells infected with siRNA-ERRα obviously decreased as compared with the blank cells or cells infected with nonspecific siRNA (the negative control [NC] group). Protein expression of ERRα in RL-952 (B), AN3-CA (C), HEC-1A (D), and HEC-1B (E) cells infected with siRNA-ERRα for 72 h was detected by Western blotting in comparison with cells infected with nonspecific siRNA or cells without treatment. ERRα protein expression showed a similar trend to the mRNA expression. (F) Growth rates of all EC cells were analyzed for 24, 48, 72, and 96 h using an MTS assay. siRNA-ERRα suppressed cellular proliferation effectively. All experiments were conducted in triplicate. *Significant differences (P<0.05) are indicated. Abbreviations: EC, endometrial cancer; PCR, polymerase chain reaction; MTS, 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium. |
Cell proliferation inhibition induced by siRNA
RL-952, AN3-CA, HEC-1A, and HEC-1B were cultured in medium, followed by infection with siRNA- ERRα for 0, 24, 48, 72, and 96 h. The growth inhibition ratio at 0, 24, 48, 72, and 96 h were as follows: RL-952 – 2.652%±0.569%, 48.15%±0.368%, 57.18%±0.512%, 85.70%±0.635%, and 62.22%±0.579%, respectively; AN3-CA – −1.234%±0.077%, 28.46%±0.312%, 36.21%±0.791%, 44.34%±0.789%, and 30.12%±0.875%, respectively; HEC-1A – −0.068%±0.047%, 51.36%±0.321%, 52.51%±0.048%, 82.01%±0.042%, and 49.00%±0.020%, respectively; and HEC-1B – −0.172%±0.189%, 38.28%±0.199%, 58.12%±0.257%, 67.46%±0.236%, and 53.46%±0.346%, respectively. MTS assay showed that siRNA transfection resulted in the inhibition of cell proliferation through the downregulation of ERRα expression in a time-dependent manner (Figure 4F).
Change in cell apoptosis following treatment with XCT790 and siRNA
We examined apoptosis in RL-952, AN3-CA, HEC-1A, and HEC-1B EC cells treated with 10 µM XCT790 for 24 h. The apoptosis ratios of the treated cells as compared with the DMSO and blank groups were as follows: RL-952 – 69.61%±5.506% vs 9.452%±1.202% and 10.21%±1.675% (Figure 5A), AN3-CA – 77.55%±7.182% vs 11.55%±4.596% and 9.135%±2.241% (Figure 5B), HEC-1A – 24.43%±6.564% vs 5.233%±1.839% and 6.321%±2.042% (Figure 5C), and HEC-1B – 25.52%±3.674% vs 8.664%±1.474% and 10.01%±2.067% (Figure 5D). The apoptosis ratios for RL-952 and AN3-CA were noticeably higher than those in HEC-1A and HEC-1B (P<0.05). Furthermore, we examined apoptosis in the EC cells following siRNA treatment for 48 h when the fluorescence intensity of GFP was higher than 90%. The results showed that the apoptosis ratios of the four EC cells, as compared with the NC and blank groups, were as follows: RL-952 –17.52%±1.056% vs 2.814%±1.026% and 0.2031%±0.0652% (Figure 6A), AN3-CA–19.43%±1.613% vs 3.272%±1.692% and 3.265%±1.582% (Figure 6B), HEC-1A – 25.51%±2.949% vs 4.031%±1.219% and 4.127%±1.608% (Figure 6C), and HEC-1B – 27.87%±5.877% vs 5.173%±1.635% and 2.434%±0.5712% (Figure 6D). The apoptosis ratios for RL-952 and AN3-CA were noticeably lower than those for HEC-1A and HEC-1B (P<0.05).
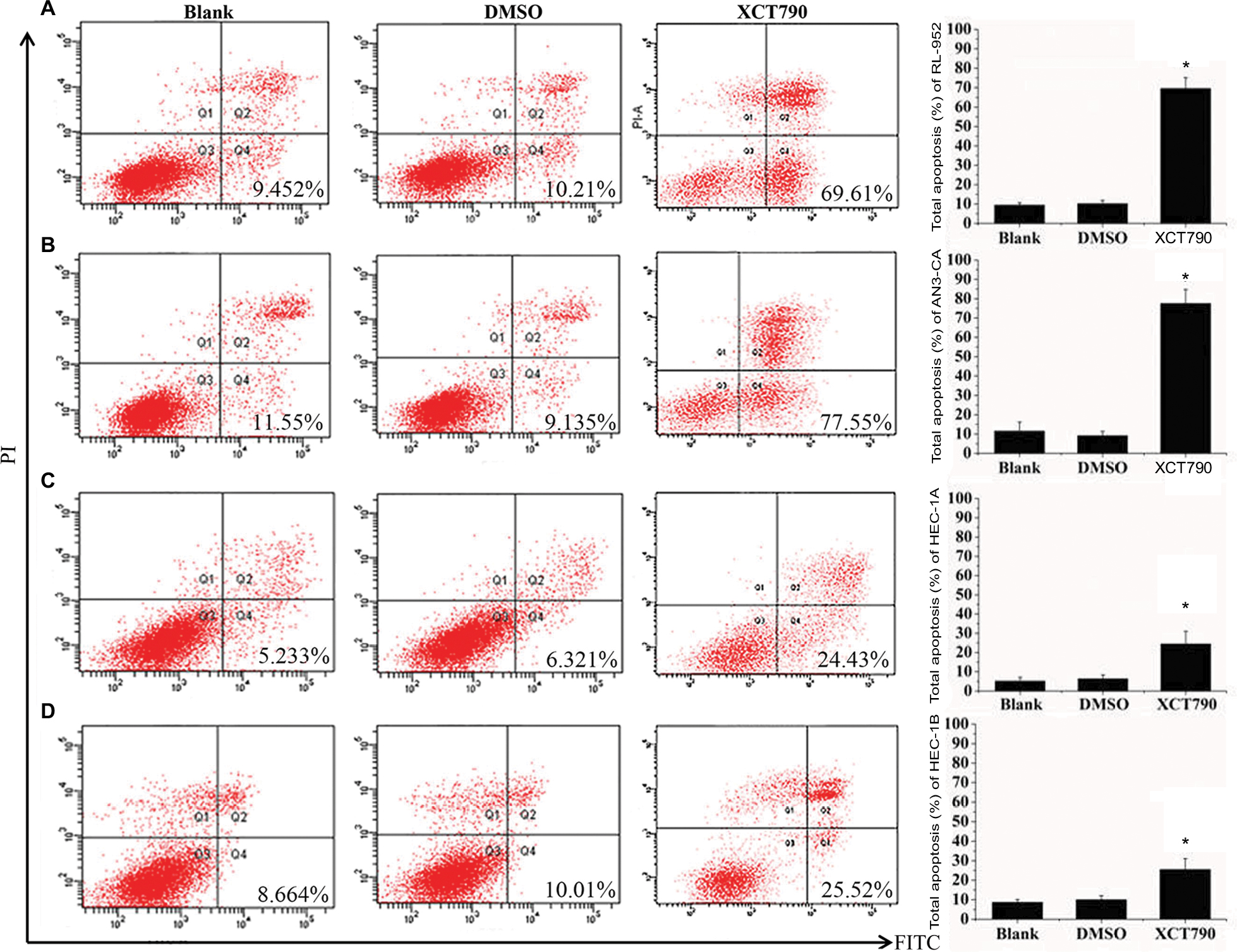 | Figure 5 Downregulation of ERRα by XCT790 induces cell apoptosis. The apoptosis rates of EC cell lines RL-952 (A), AN3-CA (B), HEC-1A (C), and HEC-1B (D) induced by XCT790 were analyzed by flow cytometry using an Annexin-V/FITC Kit. Cells treated with 0.1% DMSO (the solvent of XCT790) were called the DMSO group and those treated with 10 µM XCT790 for 24 h were called the XCT group. Downregulation of ERRα by treatment with XCT790 led to a significant increase in the cellular apoptosis as compared to the blank or DMSO group. All experiments were conducted in triplicate. *Significant differences (P<0.05) are indicated. Abbreviations: FITC, fluorescein-5-isothiocyanate; PI, propidium iodide; EC, endometrial cancer. |
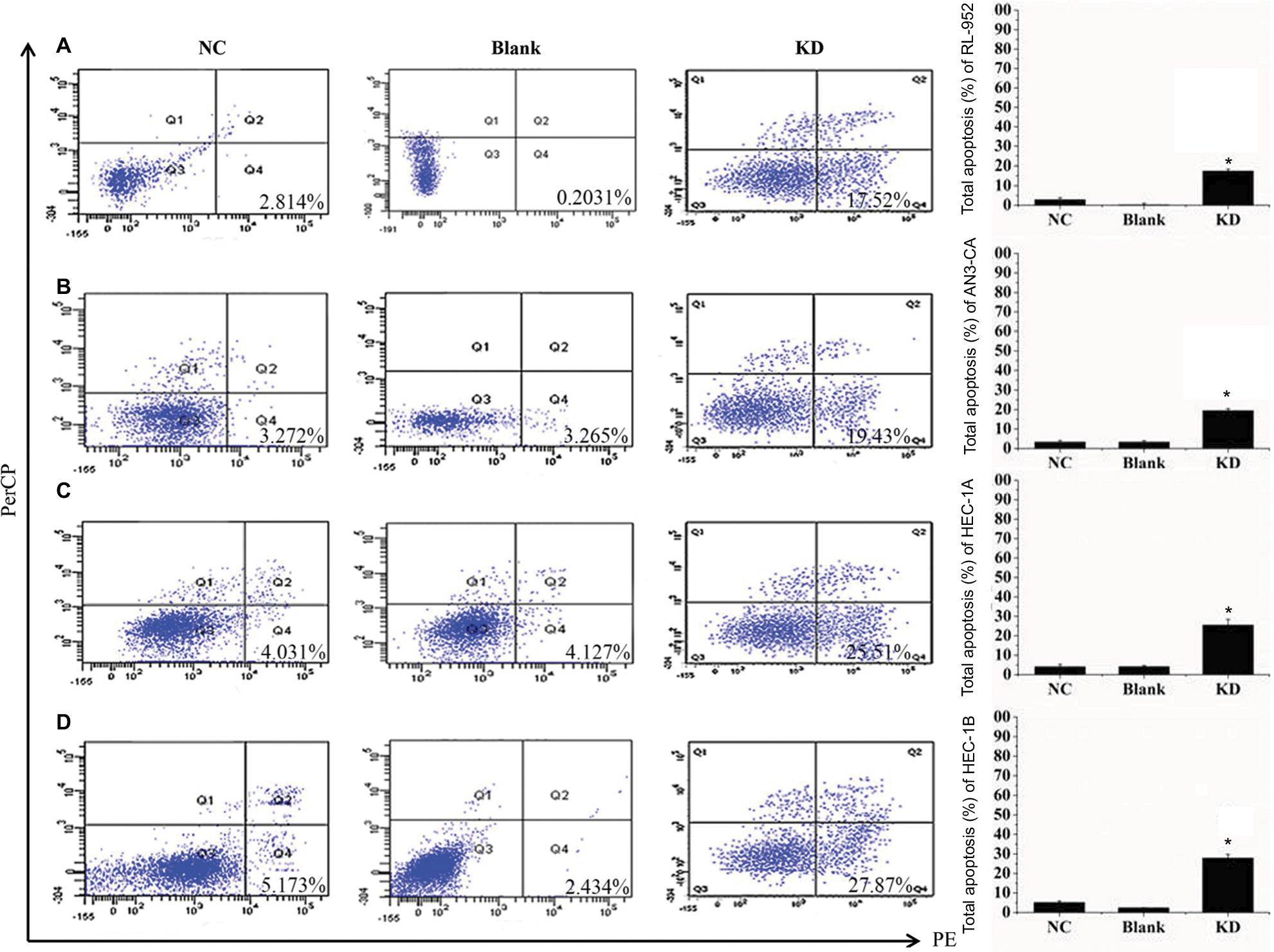 | Figure 6 Downregulation of ERRα by siRNA induces cell apoptosis. The apoptosis rates of four endometrial cancer cells RL-952 (A), AN3-CA (B), HEC-1A (C), and HEC-1B (D) infected with siRNA targeted on ERRα (siRNA-ERRα) were analyzed by flow cytometry using an Annexin-V 7AAD Kit. Untreated EC cells were designated as the blank group, the cells infected with nonspecific siRNA as the negative control (NC) group, and the cells infected with siRNA-ERRα for 72 h as the KD group. Compared to the blank or NC group, the downregulation of ERRα by infection with siRNA-ERRα led to a significant increase in cellular apoptosis. All experiments were conducted in triplicate. *Significant differences (P<0.05) are indicated. Abbreviation: KD, knockdown. |
Downregulating ERRα by siRNA alters TF profiles in EC cell lines
To discuss the potential molecular mechanism of endocrine therapy targeting on ERRα in EC, the TFs in RL-952, AN3-CA, and HEC-1A cells were analyzed by high-throughput protein/DNA array analysis and compared with cells infected with ERRα-siRNA (Figure 7A). Compared with the RL-952 cells, 117 TFs showed upregulated activities and 34 TFs showed downregulated activities in RL-952 cells infected with ERRα-siRNA. Similarly, 12 TFs were upregulated, and 247 TFs were downregulated in ANC-3A vs ANC-3A-ERRα-siRNA; 95 TFs were upregulated and 91 TFs were downregulated in HEC-1A vs HEC-1A-ERRα-siRNA (Figure 7B). In general, three TFs analyzed by protein/DNA array were found to be at least 2-fold higher, whereas 17 TFs were at least 2-fold lower in KD group cells as compared to NC group cells (Figure 7C; Table 1). According to the results, there were four TFs whose P-values were below 0.05 including three upregulated TFs and one downregulated TF. Transcription factor EB (TFEB) was the only downregulated TF induced by ERRα knockdown that was related to the mTOR pathway and affected ERRα via the peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α). Moreover, the organic cation transporter 3/4 (Oct3/4) was an upregulated TF which participated in the Wnt pathway by combining with β-catenin (β-cat), and inversely regulated T-cell factor/lymphoid enhancer factor (TCF/LEF). Hepatic nuclear factor 4 (HNF-4) could modulate PGC-1α to indirectly affect ERRα (Figure 7D).
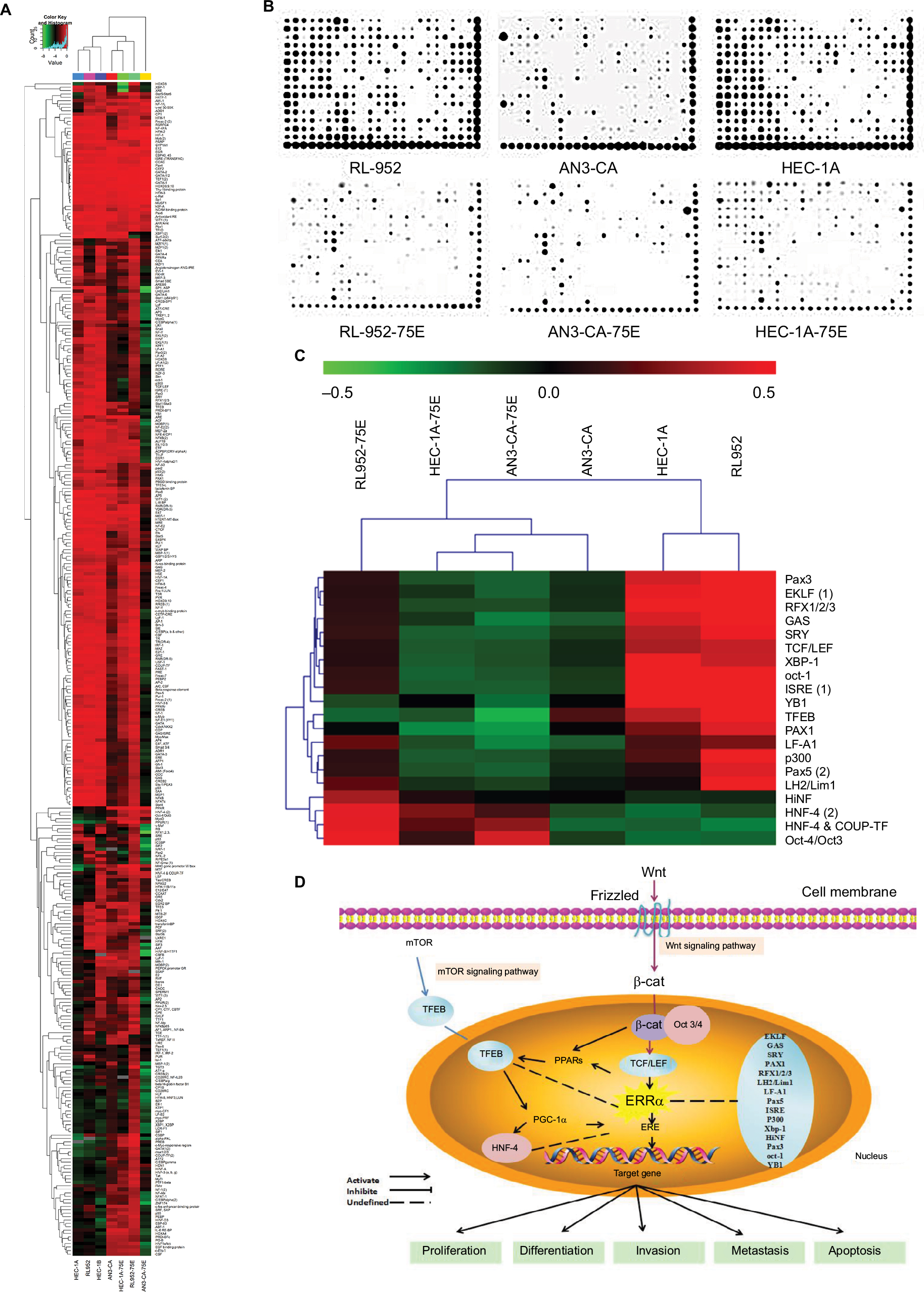 | Figure 7 Analysis of transcription factors by protein/DNA array. (A) Clustering analysis based on 345 TFs was undertaken in the KD and NC groups of RL-952, AN3-CA, HEC-1A, and HEC-1B. (B) Reading from the images of protein/DNA arrays, the plots of different TFs show the relative activity-level clusters of the NC and KD groups in RL-952, AN3-CA, HEC-1A, and HEC-1B. (C) Analyzed by MeV software, clustering analysis based on 345 TFs-chips between the NC and KD group revealed that the 20 TFs show the same change trend of transcriptional activities in the four RL-952, AN3-CA, HEC-1A, and HEC-1B EC cells. (D) The function of different TFs may be mediated by ERRα in the cellular nucleus as presented by the schematic diagram. Downregulation of ERRα then induced inhibition of TFEB which was mediated by PGC-1α and involved the mTOR signaling pathway. In addition, mediated by TCF, the downregulation of ERRα could cause Oct3/4 upregulation and participated in the Wnt signaling pathway. Abbreviations: TFs, transcription factors; KD, knockdown; NC, negative control; EC, endometrial cancer. |
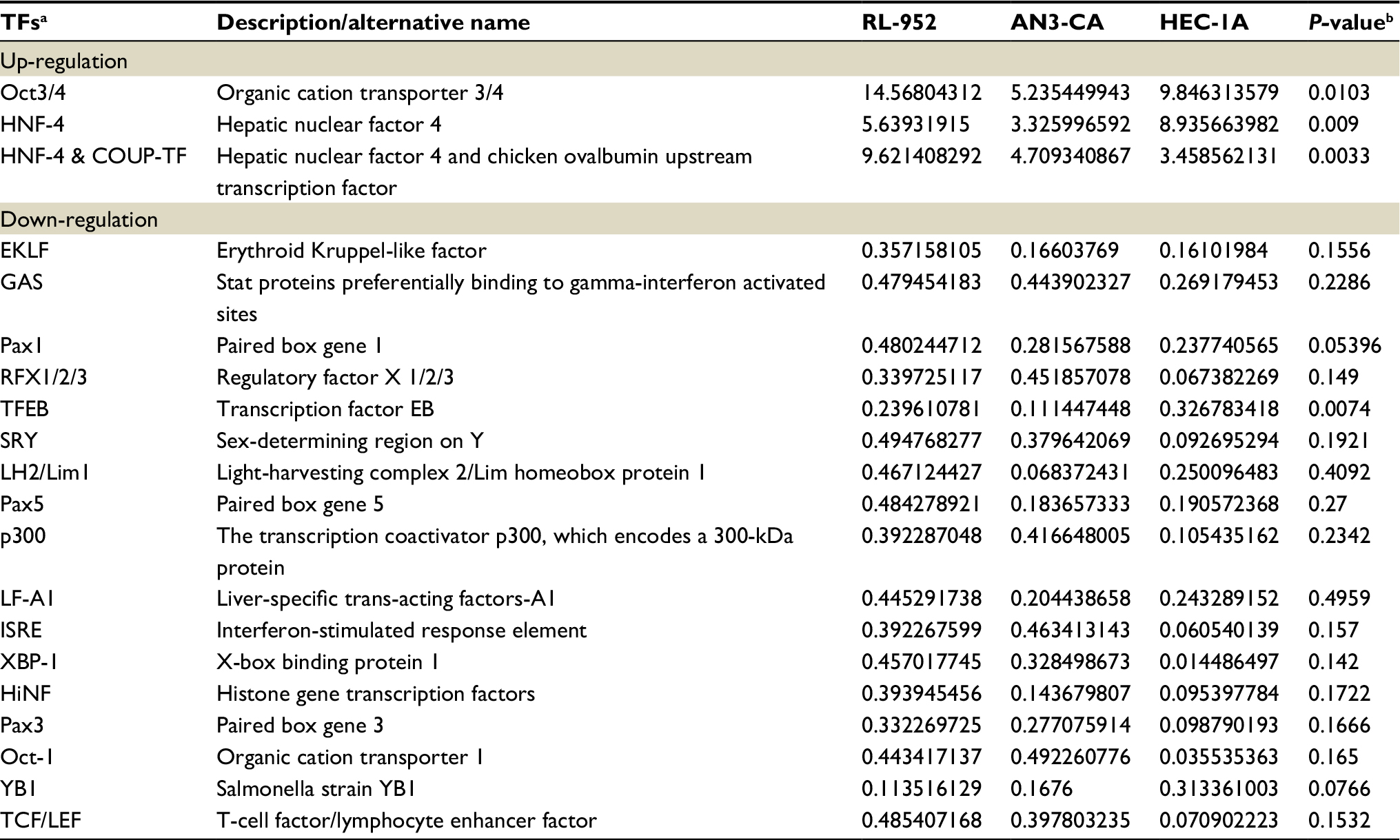 | Table 1 ERRα siRNA-related transcription factors Notes: aTFs named are cited from combo array annotation. bP-value of standardized regression coefficient. Abbreviation: TFs, transcription factors. |
Discussion
The development of EC is a complex process involving multiple factors and stages. This process is mediated via different signaling pathways that are composed of different oncogenes, tumor suppressor genes, and transcriptional signals. ERRα may participate in the tumorigenesis of estrogen-related tumors by interfering with ER signaling.29 Yang et al reported the regulatory effect of ERRα on the human lactoferrin gene promoter that is mediated via ERα in the EC cell line RL-952.30 This was the first study to describe the potential role of ERRα in EC cells in vitro. In subsequent studies, Gao et al compared the in vivo expression of ERα and ERRα in human endometrial carcinoma and the normal endometrial tissue.18 ERRα may be an indicator of poor prognoses in EC, similarly to that observed in breast,12–14 ovarian,17 colon,15 and prostatic cancers.16 The upregulation of ERRα may be related to tumor growth and progression in ECs.31 Thus, ERRα may be a candidate prognostic factor and potential target for the treatment of EC with specific pathological types.31
Although recent studies have improved our understanding of the role of orphan nuclear receptors in cancer, the function of ERRα remains unknown. It is well known that ERα and ERβ are inactive in the absence of hormonal signals, whereas ERRα displays a constitutive transcriptional activity as a repressor and activator.29 Previous studies have reported extensive cross talk between ERRα and ERα.17–19 In our previous work, the cross talk between ERRα and ERα at the transcription level was confirmed by using stable transfection experiments.18 ERRα overexpression repressed the mRNA level of ERα but not of ERβ. However, the mechanism involved in this adverse regulation is unclear. Moreover, ERRα overexpression was associated with a proliferative mechanism in ERα-nonresponsive EC cells in HEC-1A but not in ERα-positive Ishikawa cells.18 Therefore, the regulation of ERRα expression by estrogen and estrogenic response may vary depending upon the type and ERα responsiveness of cells. To study the effect of ERRα targeting better, we choose RL-952, AN3-CA, HEC-1A, and HEC-1B cells as in vitro models for types 1 and 2 EC. The expression of ERRα and ERα in these EC cells was determined by quantitative PCR (qPCR). Our experiments demonstrated the absolute expression of ERRα to be significantly higher in RL-952 and AN3-CA, which are ERα-positive EC cells, as compared to HEC-1A and HEC-1B cells (ER-negative and/or ERα-nonresponsive). Moreover, we observed an association between the expression of ERRα and ERα. The relative ratio of ER/ERRα in ERα-positive type 1 EC cells was higher than that in the ERα-negative type 2 EC cells. To downregulate the expression of ERRα, we used the exogenous drug XCT790 as well as the endogenous siRNA against ERRα (ERRα-siRNA). The synthetic molecule XCT790 has been identified as an ERRα-specific antagonist.14 XCT790 is thought to downregulate the expression of the ERRα gene by inducing an actively repressive conformation.32 We found that XCT790 inhibited the expression of ERRα not only in the ERα-negative and/or ERα-nonresponsive EC cells HEC-1A and HEC-1B, but also in the ERα-positive and -responsive EC cell lines RL-952 and AN3-CA. XCT790 treatment resulted in the inhibition of cellular proliferation via ERRα downregulation in all four cell lines. These results are in agreement with those reported by Bianco et al,33 wherein XCT790 acted as an ERRα-specific antagonist and reduced cell proliferation in several tumor cell lines. In addition, the siRNA against ERRα decreased the expression of ERRα, inhibited cell proliferation, and induced cellular apoptosis in all EC cells.
The role of ERRα is entirely distinct between ERα-responsive and ERα-nonresponsive EC cells. The classic estrogen-ER signaling pathway is thought to be the most important factor that results in hormone-dependent endometrial carcinoma. In the presence of estradiol (E2), the estrogen–ER complex may directly or indirectly inhibit the expression of ERRα at transcriptional and/or translational level in ERα-responsive cells. This may partly explain the induction of hyperplasia and tumorigenesis of the endometrium with long-term estrogenic stimulation.34,35 However, the ERRα-mediated pathway may play an important role in ERα-independent endometrial carcinoma. ERRα may induce constitutive transactivation of estrogen-responsive genes, leading to an estrogenic effect. Therefore, we propose that the transformation of ERRα from the repressor to the activator may be key for the transformation of hormone-dependent type 1 endometrial carcinoma to hormone-independent type 2 endometrial carcinoma. It is interesting that XCT790 showed a stronger inhibitory effect against ERα-positive (high ERα/ERRα ratio) RL-952 and AN3-CA EC cells as compared to the ERα-negative (low ERα/ERRα ratio) HEC-1A and HEC-1B EC cells. However, the siRNA against ERRα was more effective in the ERα-negative cells than in the ERα-positive EC cells. Matsushima et al showed that ERRα silencing using endogenous siRNA resulted in an antitumor effect in ERα-independent HEC-1A and KLE EC cells but not in the ERα-dependent Ishikawa cells.36 Thus, the mechanism of exogenous XCT790 and endogenous siRNA downregulation of ERRα expression may involve multiple pathways.
To further discuss the potential mechanism involved, a high-throughput protein/DNA array was carried out. Four TFs, including three upregulating TFs and one downregulating TF, were screened out as statistically significant. We focused on TFEB because there was a positive correlation between TFEB and ERRα in our research. TFEB was recently identified as the master transcriptional regulator of autophagy and lysosomal biogenesis.37 TFEB can translocate to the nucleus following mitophagy induction to stimulate synthesis of new mitochondria via PGC-1α, because PGC-1α plays a role in mitochondrial biogenesis. TFEB can modulate PGC-1α and increase the mitochondrial content of cells.38 Moreover, TFEB activity is inhibited by mTOR phosphorylation.39 Therefore, it is obvious that TFEB was related to PGC-1α and might be involved in the mTOR signaling pathway. However, it is well known that PGC-1α was a coactivator of ERRα.40 Previous studies have reported that PGC-1α induced the expression of ERRα and interacted physically with ERRα.38 Therefore, we deduced that TFEB was associated with ERRα, which may be mediated by PGC-1α. On the other hand, Oct3/4 – an upregulated TF – is related to the self-renewal properties of stem cells.41,42 A study showed that Oct3/4 is downstream of the Wnt ligand, but upstream of the β-cat/TCF target genes. Oct3/4 expression promotes a dramatic decrease in β-cat. There is a feedback between the TCF/β-catenin and Oct3/4.42 TCF regulates Oct3/4 expression by binding directly to the Oct3/4 promoter and inhibiting Oct3/4 expression.43 Chang and McDonnell claimed ERRα was affected by the activity of TCF on several endogenous target genes, and the activation of β-cat-enhanced ERRα transcriptional activity.44 All of the observations suggested that there was some relationship between ERRα and Oct3/4. Thus, we speculate that ERRα downregulation induced Oct3/4 upregulation, which may be mediated by TCF and is involved in the Wnt signaling pathway.
In general, both XCT790 and siRNA showed proliferation-inhibitory effects against ERRα and induced cell apoptosis via downregulation of ERRα expression in ERα-positive and ERα-negative EC cells. Thus, it can be suggested that ERRα-specific inhibitors or synthetic ligands may be valuable for the treatment of both ERα-positive and ERα-negative ECs. Furthermore, the TFs screened out by using protein/DNA array analysis participated in modulating the expression of ERRα directly, or indirectly via different signaling pathways. The screening and analysis of TFs further clarified the potential mechanism of EC therapy targeting of ERRα. Future in vivo studies should explore the therapeutic applications of this strategy, and the TFs screened out need to be validated.
Conclusion
ERRα is the target of EC antitumor treatment. Above all, exogenous XCT790 was more effective in ER-positive EC antitumor treatment whereas endogenous siRNA was more suitable for ER-negative EC antitumor treatment. The potential mechanism may involve the mTOR and Wnt pathways mediated by TFEB, Oct3/4, and so on.
Acknowledgments
This work was partly sponsored by the Youth Foundation of Fujian Provincial Health and Family Planning Commission (2015-1-16), and the Natural Science Foundation of Fujian Province (grant nos. 2016J01428, 2016J01491, and 2017J01233).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. | ||
Makker V, Hensley ML, Zhou Q, Iasonos A, Aghajanian CA. Treatment of advanced or recurrent endometrial carcinoma with doxorubicin in patients progressing after paclitaxel/carboplatin: Memorial Sloan-Kettering Cancer Center experience from 1995 to 2009. Int J Gynecol Cancer. 2013;23(5):929–934. | ||
Ito K, Utsunomiya H, Niikura H, Yaegashi N, Sasano H. Inhibition of estrogen actions in human gynecological malignancies: new aspects of endocrine therapy for endometrial cancer and ovarian cancer. Mol Cell Endocrinol. 2011;340(2):161–167. | ||
Pasqualini JR. Estrogen sulfotransferases in breast and endometrial cancers. Ann N Y Acad Sci. 2009;1155:88–98. | ||
Levin ER. Membrane estrogen receptors signal to determine transcription factor function. Steroids. 2018;132:1–4. | ||
Shigeta H, Zuo W, Yang N, DiAugustine R, Teng CT. The mouse estrogen receptor-related orphan receptor alpha1:molecular cloning and estrogen responsiveness. J Mol Endocrinol. 1997;19(3):299–309. | ||
Suetsugi M, Su L, Karlsberg K, Yuan YC, Chen S. Flavone and isoflavone phytoestrogens are agonists of estrogen-related receptors. Mol Cancer Res. 2003;1(13):981–991. | ||
Greschik H, Wurtz JM, Sanglier S, et al. Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol Cell. 2002;9(2):303–313. | ||
Ranhotra HS. The orphan estrogen-related receptor alpha and metabolic regulation: new frontiers. J Recept Signal Transduct Res. 2015;35(6):565–568. | ||
Zhang L, Liu P, Chen H, et al. Characterization of a selective inverse agonist for estrogen related receptor alpha as a potential agent for breast cancer. Eur J Pharmacol. 2016;789:439–448. | ||
Park S, Chang CY, Safi R, et al. ERRα-regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep. 2016;15(2):323–335. | ||
Wu YM, Chen ZJ, Liu H, et al. Inhibition of ERRα suppresses epithelial mesenchymal transition of triple negative breast cancer cells by directly targeting fibronectin. Oncotarget. 2015;6(28):25588–25601. | ||
Bernatchez G, Giroux V, Lassalle T, Carpentier AC, Rivard N, Carrier JC. ERRα metabolic nuclear receptor controls growth of colon cancer cells. Carcinogenesis. 2013;34(10):2253–2261. | ||
Bonkhoff H. Estrogen receptor signaling in prostate cancer: implications for carcinogenesis and tumor progression. Prostate. 2018;78(1):2–10. | ||
Sun P, Sehouli J, Denkert C, et al. Expression of estrogen receptor-related receptors, a subfamily of orphan nuclear receptors, as new tumor biomarkers in ovarian cancer cells. J Mol Med (Berl). 2005;83(6):457–467. | ||
Gao M, Sun P, Wang J, Zhao D, Wei L. Expression of estrogen receptor–related receptor isoforms and clinical significance in endometrial adenocarcinoma. Int J Gynecol Cancer. 2006;16(2):827–833. | ||
Sun PM, Gao M, Wei LH, et al. An estrogen receptor alpha-dependent regulation of estrogen receptor-related receptor alpha in the proliferation of endometrial carcinoma cells. Int J Gynecol Cancer. 2006;16 Suppl 2:564–568. | ||
Chang CY, Kazmin D, Jasper JS, Kunder R, Zuercher WJ, McDonnell DP. The metabolic regulator ERRalpha, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell. 2011;20(4):500–510. | ||
Deblois G, Smith HW, Tam IS, et al. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat Commun. 2016;7:12156. | ||
Thewes V, Simon R, Schroeter P, et al. Reprogramming of the ERRα and ERα target gene landscape triggers tamoxifen resistance in breast cancer. Cancer Res. 2015;75(4):720–731. | ||
Lui K, Tamura T, Mori T, Zhou D, Chen S. MCF-7aro/ERE, a novel cell line for rapid screening of aromatase inhibitors, ERalpha ligands and ERRalpha ligands. Biochem Pharmacol. 2008;76(2):208–215. | ||
Wu YM, Chen ZJ, Jiang GM, Zhang KS. Inverse agonist of estrogen-related receptor α suppresses the growth of triple negative breast cancer cells through ROS generation and interaction with multiple cell signaling pathways. Oncotarget. 2016;7:12568–12581. | ||
Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457(7228):405–412. | ||
Zhou H, Wei J, Dai Q, et al. CaCO3/CaIP6 composite nanoparticles effectively deliver AKT1 small interfering RNA to inhibit human breast cancer growth. Int J Nanomedicine. 2015;10:4255–4266. | ||
Bombail V, Gibson DA, Collins F, MacPherson S, Critchley HO, Saunders PT. A role for the orphan nuclear receptor estrogen-related receptor alpha in endometrial stromal cell decidualization and expression of genes implicated in energy metabolism. J Clin Endocrinol Metab. 2010;95(10):E224–E228. | ||
Pekonen F, Nyman T, Rutanen EM. Human endometrial adenocarcinoma cell lines HEC 1B and KLE secrete insulin-like growth factor binding protein-1 and contain IGF-I receptors. Mol Cell Endocrinol. 1991;75(1):81–87. | ||
Mohideen-Abdul K, Tazibt K, Bourguet M, et al. Importance of the sequence-directed DNA shape for specific binding site recognition by the estrogen-related receptor. Front Endocrinol (Lausanne). 2017;8:140. | ||
Yang N, Shigeta H, Shi H, Teng CT. Estrogen-related receptor, hERR1, modulates estrogen receptor-mediated response of human lactoferrin gene promoter. J Biol Chem. 1996;271(10):5795–5804. | ||
Fujimoto J, Sato E. Clinical implication of estrogen-related receptor (ERR) expression in uterine endometrial cancers. J Steroid Biochem Mol Biol. 2009;116(1–2):71–75. | ||
Du Y, Song L, Zhang L, et al. The discovery of novel, potent ERR-alpha inverse agonists for the treatment of triple negative breast cancer. Eur J Med Chem. 2017;136:457–467. | ||
Bianco S, Lanvin O, Tribollet V, Macari C, North S, Vanacker JM. Modulating estrogen receptor-related receptor-alpha activity inhibits cell proliferation. J Biol Chem. 2009;284(35):23286–23292. | ||
Razavi P, Pike MC, Horn-Ross PL, Templeman C, Bernstein L, Ursin G. Long-term postmenopausal hormone therapy and endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2010;19(2):475–483. | ||
Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet. 2005;366(9484):491–505. | ||
Matsushima H, Mori T, Ito F, et al. Anti-tumor effect of estrogen-related receptor alpha knockdown on uterine endometrial cancer. Oncotarget. 2016;7(23):34131–34148. | ||
Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–1108. | ||
Ghosh A, Jana M, Modi K, et al. Activation of peroxisome proliferator-activated receptor α induces lysosomal biogenesis in brain cells: implications for lysosomal storage disorders. J Biol Chem. 2015;290(16):10309–10324. | ||
Marin Zapata PA, Beese CJ, Junger A, Dalmasso G, Brady NR, Hamacher-Brady A. Time course decomposition of cell heterogeneity in TFEB signaling states reveals homeostatic mechanisms restricting the magnitude and duration of TFEB responses to mTOR activity modulation. BMC C. 2016;16:355. | ||
Nsiah-Sefaa A, Brown EL, Russell AP, Foletta VC. New gene targets of PGC-1α and ERRα co-regulation in C2C12 myotubes. Mol Biol Rep. 2014;41(12):8009–8017. | ||
Karmakar S, Seshacharyulu P, Lakshmanan I, et al. hPaf1/PD2 interacts with OCT3/4 to promote self-renewal of ovarian cancer stem cells. Oncotarget. 2017;8(9):14806–14820. | ||
Abu-Remaileh M, Gerson A, Farago M, et al. Oct-3/4 regulates stem cell identity and cell fate decisions by modulating Wnt/β-catenin signalling. EMBO J. 2010;29(19):3236–3248. | ||
Ding VM, Ling L, Natarajan S, Yap MG, Cool SM, Choo AB. FGF-2 modulates Wnt signaling in undifferentiated hESC and iPS cells through activated PI3-K/GSK3beta signaling. J Cell Physiol. 2010;225(2):417–428. | ||
Chang CY, McDonnell DP. Molecular pathways: the metabolic regulator estrogen-related receptor α as a therapeutic target in cancer. Clin Cancer Res. 2012;18(22):6089–6095. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.