Back to Journals » International Journal of Nanomedicine » Volume 11
Novel electrosprayed nanospherules for enhanced aqueous solubility and oral bioavailability of poorly water-soluble fenofibrate
Authors Yousaf AM, Mustapha O ![]() , Kim D, Kim DS, Kim KS, Jin SG, Yong CS, Youn YS
, Kim D, Kim DS, Kim KS, Jin SG, Yong CS, Youn YS ![]() , Oh Y, Kim JO, Choi H
, Oh Y, Kim JO, Choi H
Received 1 October 2015
Accepted for publication 9 December 2015
Published 12 January 2016 Volume 2016:11 Pages 213—221
DOI https://doi.org/10.2147/IJN.S97496
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Abid Mehmood Yousaf,1,2 Omer Mustapha,1 Dong Wuk Kim,1 Dong Shik Kim,1 Kyeong Soo Kim,1 Sung Giu Jin,1 Chul Soon Yong,3 Yu Seok Youn,4 Yu-Kyoung Oh,5 Jong Oh Kim,3 Han-Gon Choi1
1College of Pharmacy and Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, Gyeonggi, South Korea; 2Faculty of Pharmacy, University of Central Punjab, Johar, Lahore, Pakistan; 3College of Pharmacy, Yeungnam University, Gyongsan, North Gyeongsang, 4School of Pharmacy, Sungkyunkwan University, Suwon, Gyeonggi, 5College of Pharmacy, Seoul National University, Seoul, South Korea
Purpose: The purpose of the present research was to develop a novel electrosprayed nanospherule providing the most optimized aqueous solubility and oral bioavailability for poorly water-soluble fenofibrate.
Methods: Numerous fenofibrate-loaded electrosprayed nanospherules were prepared with polyvinylpyrrolidone (PVP) and Labrafil® M 2125 as carriers using the electrospray technique, and the effect of the carriers on drug solubility and solvation was assessed. The solid state characterization of an optimized formulation was conducted by scanning electron microscopy, powder X-ray diffraction, differential scanning calorimetry, and Fourier transform infrared spectroscopic analyses. Oral bioavailability in rats was also evaluated for the formulation of an optimized nanospherule in comparison with free drug and a conventional fenofibrate-loaded solid dispersion.
Results: All of the electrosprayed nanospherule formulations had remarkably enhanced aqueous solubility and dissolution compared with free drug. Moreover, Labrafil M 2125, a surfactant, had a positive influence on the solubility and dissolution of the drug in the electrosprayed nanospherule. Increases were observed as the PVP/drug ratio increased to 4:1, but higher ratios gave no significant increases. In particular, an electrosprayed nanospherule composed of fenofibrate, PVP, and Labrafil M 2125 at the weight ratio of 1:4:0.5 resulted in a particle size of <200 nm with the drug present in the amorphous state. It demonstrated the highest solubility (32.51±2.41 µg/mL), an excellent dissolution (~85% in 10 minutes), and an oral bioavailability ~2.5-fold better than that of the free drug. It showed similar oral bioavailability compared to the conventional solid dispersion.
Conclusion: Electrosprayed nanospherules, which provide improved solubility and bioavailability, are promising drug delivery tools for oral administration of poorly water-soluble fenofibrate.
Keywords: fenofibrate, electrospray technique, nanospherule, enhanced bioavailability
Introduction
Fenofibrate, a Biopharmaceutics Classification System class II drug, is a therapeutic agent used for the treatment of hypercholesterolemia and hypertriglyceridemia.1,2 It is a very lipophilic entity (logP =5.24)3 that is practically insoluble in aqueous gastrointestinal fluids (<100 μg/mL);4 therefore, it exhibits very poor absorption5 and bioavailability after oral administration.1 Thus, an enhancement in bioavailability is dependent upon an improvement in aqueous solubility.
Nanotechnology is an elegant science with the capability to resolve aqueous solubility, dissolution, and absorption problems of poorly water-soluble active pharmaceutical agents.6,7 A pharmaceutical nanoparticle is a particle having a diameter <1,000 nm.8,9 Nanoparticles can be fabricated using polymeric compounds.6 Natural polymers confer outstanding protection, effectiveness, biocompatibility, and biodegradation characteristics; accordingly, they are very popular matrices for pharmaceutical nanoparticle formation. There are several methods to develop pharmaceutical nanoparticles, such as solvent evaporation, emulsification, spray drying, phase inversion, self-assembly, and solvent displacement.10–13 Electrospraying is a promising technique for the preparation of pharmaceutical nanoparticles.14,15 In the preparation of a nanospherule via this technique, all of the constituents are comprehensively dissolved and the final transparent solution is subjected to electrospraying. The resulting nanospherule is a spherical nanoparticle containing evenly scattered drug molecules in the polymeric matrix.8,16
The electrospraying approach has been successfully used to produce polymeric nanoparticles for several drug classes, such as antibiotics,17–19 anti-inflammatories,20,21 and hormones,22–24 among others. In this technique, a syringe is filled with a transparent solution of drug and other excipients such as polymeric components. Then, the loaded syringe is secured with the specified holder in the instrument. The pump, used for regulating the flow rate of the feed, is positioned so that it touches the open end of the plunger. An electrode (usually anode) is attached to the capillary nozzle of the syringe, while the other electrode (usually cathode) is attached to the product-collecting metallic sheet. At a specific high voltage, a Taylor cone is formed at the tip of the capillary nozzle, and the flowing liquid atomizes into minute droplets bearing the same charges on their surfaces; therefore, maximal scatter is achieved as a result of Coulombic repulsion. These charged droplets are directed to the collector due to gravity and Coulombic attraction. Evaporation takes place during the journey from ejector to collector.25
In this study, to develop a novel electrosprayed nanospherule conferring the most optimized aqueous solubility, dissolution, and oral bioavailability to poorly water-soluble fenofibrate, numerous fenofibrate-loaded electrosprayed nanospherules were prepared with polyvinylpyrrolidone (PVP) and Labrafil M 2125 as carriers using the electrospraying technique. Then, the effect of the carriers on the solubility and dissolution of the drug present in them was assessed. The solid state characterization of an optimized formulation was conducted by powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), scanning electron microscopy (SEM), and Fourier transform infrared (FTIR) spectroscopic analyses. Oral bioavailability in rats was evaluated in comparison with free drug and a conventional fenofibrate-loaded solid dispersion for the formulation of an optimized preparation.
Materials and methods
Materials
Fenofibrate and fenofibric acid were provided by Hanmi Pharmaceutical Co (Suwon, South Korea). PVP K30 was procured from BASF (Ludwigshafen, Germany). Labrafil M 2125 was purchased from Gattefosse (Saint Priest Cedex, Lyon, France). Clofibric acid was obtained from Sigma-Aldrich (St Louis, MO, USA). All other chemical substances were of reagent grade.
Preparation of nanospherules
Each nanospherule formulation was prepared using ESR 100 electrospraying equipment (NanoNC, Seoul, South Korea). All the components, including nonmicronized fenofibrate, were completely dissolved in dichloromethane. This clear solution was filled in a gas-tight glass syringe (Hamilton Co, Reno, NV, USA) equipped with a polytetrafluoroethylene plunger tip (Figure 1). Subsequently, electrospraying was performed at a flow rate of 0.5 mL/h under an optimized voltage of 8.5 kV. The dried product was carefully collected and preserved in dry microtubes.
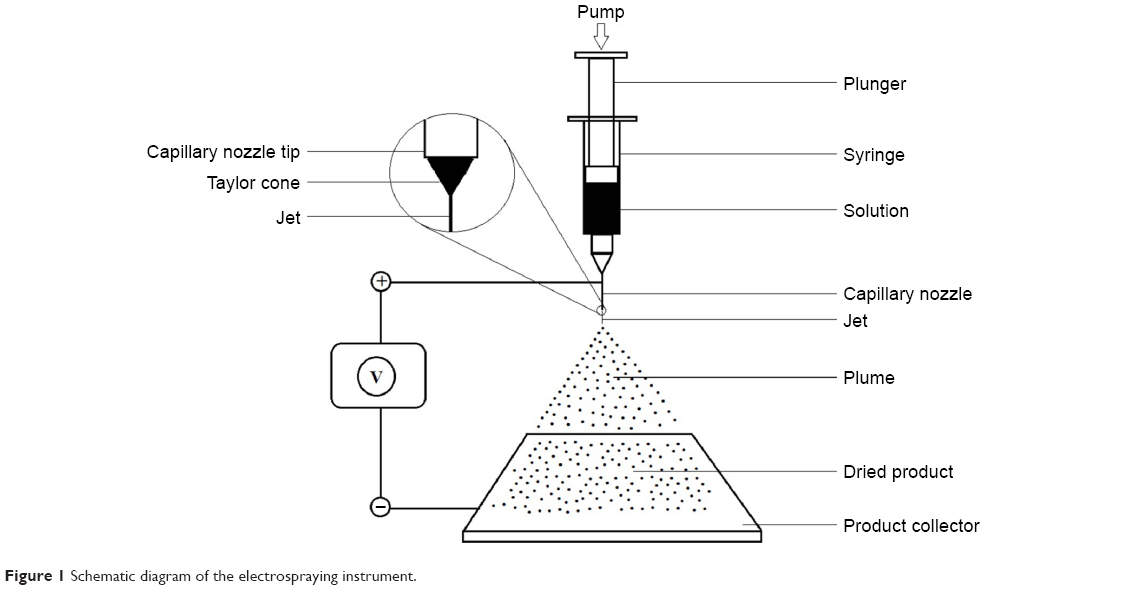 | Figure 1 Schematic diagram of the electrospraying instrument. |
Aqueous solubility test
An ample quantity of each nanospherule formulation (~10 mg) was added to 500 μL of distilled water in a 2 mL microtube and vortex-mixed (~1 minute). Subsequently, the tubes were agitated at 100 rpm for 7 days in a water bath at 25°C. Then, after vortex-mixing again, centrifugation was performed at 10,000× g for 10 minutes (Smart 15; Hanil Science Industrial Co, Gangneung, South Korea). A 300 μL aliquot of supernatant was diluted with 300 μL of acetonitrile in another clean microtube. The diluent was filtered through a syringe filter (0.45 μm, number 6789–1304; Whatmann Co, Shrewsbury, MA, USA) and the concentration of fenofibrate in the filtrate (50 μL) was quantified by high-performance liquid chromatography (HPLC) (Agilent 1260 Infinity; Agilent Technologies, Santa Clara, CA, USA). The HPLC system was equipped with a Capcell Pak C18 column (4.6 mm inner diameter ×250 mm, 5 μm particle size; Shiseido, Tokyo, Japan). The column temperature was set at 30°C. The mobile phase, consisting of acetonitrile and 0.1% (v/v) aqueous phosphoric acid at a volume ratio of 75:25, was eluted at a flow rate of 2 mL/minutes. The eluent was monitored at 286 nm for the quantification of fenofibrate.26
Dissolution test
The dissolution test was performed using the basket apparatus (Vision® Classic 6™; Hanson Research Co, Los Angeles, CA, USA). Each nanospherule formulation, equivalent to 15 mg of fenofibrate, was properly enclosed in a dialysis bag (Spectra/Por® dialysis membrane, molecular weight cutoff: 3,500 Da; Spectrum Laboratories, Rancho Dominguez, CA, USA) and the loaded bag was placed in the basket. The basket was immersed in 500 mL of 2% (w/v) aqueous Tween 8027,28 at 37°C±0.5°C. The rotation speed of the basket was set to 100 rpm. At preset time intervals, 1 mL of the dissolution medium was sampled, filtered (0.45 μm), and analyzed using the HPLC method described earlier.26 After each sampling, the medium was replenished by the addition of 1 mL of fresh dissolution medium.
PXRD analysis
The crystallinity of the samples was examined using a Rigaku X-ray diffractometer (D/MAX-2500 PC; Rigaku Corporation, Tokyo, Japan). The analysis was carried out using a Cu Kα1 monochromatic radiation source with 40 mA of current and 40 kV voltage. The X-ray diffraction patterns were recorded in the range of 10°C–80°C, with a 2θ scanning mode, a step size of 0.02°/second, and a scan speed of 5°/minute.29
DSC analysis
The thermograms of samples were recorded using a DSC (DSC Q20; TA Instruments, New Castle, DE, USA). Each sample was enclosed in an aluminum pan and scanned over a range of 30°C–130°C, at a heating rate of 10°C/minutes and a nitrogen flow of 25 mL/minutes.30
FTIR spectroscopic analysis
The FTIR spectroscopic analysis was executed to determine the potential interaction between the drug and the excipients. The FTIR spectra of samples were recorded using an FTIR spectrophotometer-430 (Shimadzu Corporation, Tokyo, Japan). Each sample, thoroughly triturated with potassium bromide and subsequently compressed to make a disk, was scanned over a range of 400–4,000 cm−1 with 2 cm−1 resolution.30
SEM analysis
The shape and surface attributes of free drug and nanospherule were observed using a scanning electron microscope (S-4800; Hitachi Ltd., Tokyo, Japan). The samples, attached on the exposed side of double-sided adhesive tape affixed on a metallic disk, were coated with platinum using an EMI Teck Ion Sputter (K575K). Coating was continued for 4 minutes under a pressure of 8×10−3 mbar, 15 mA current, and 100% turbo speed with the intention of making the samples electrically conductive for imaging.31
Pharmacokinetic studies
Care of animals
Eighteen male Sprague Dawley rats (each 270–300 g) were placed under a controlled environment at 22°C–25°C, with 50%–60% relative humidity, and ad libitum access to food and water. Access to food was discontinued 36 hours prior to the start of the pharmacokinetic procedures because food intake greatly affected the oral absorption of fenofibrate;32 nevertheless, free access to drinking water was continued. The procedures for the animal studies were consistent with National Institutes of Health Policy and the Animal Welfare Act under the approval of the Institutional Animal Care and Use Committee at Hanyang University.
Dose administration and periodic blood sampling
Before compound administration, each fasted rat was surgically cannulized in the right carotid artery. Then, it was placed in a separate restraining box, in which it was sufficiently free to move and drink water. The cannulized rats were divided into three groups, each comprising six rats. Each treatment, whether the nonmicronized free drug, the fenofibrate-loaded solid dispersion, or the nanospherule formulation, was equivalent to 20 mg/kg fenofibrate and was enclosed in tiny, hard gelatin capsules (#9; Suheung Capsule Co, Seoul, Korea). The capsules were administered via the oral route to the corresponding rat.33 The electrosprayed nanospherules and the solid dispersion were composed of fenofibrate, PVP, and Labrafil M 2125 at a weight ratio of 1:4:0.5. This fenofibrate-loaded conventional solid dispersion was prepared by dissolving nonmicronized fenofibrate in ethanol and then spray-drying it with a Büchi B-290 mini spray dryer (Büchi Co, Flawil, Switzerland).11–13 Blood samples were collected at the following preset time intervals: 0.25, 0.5, 1, 1.5, 2, 4, 6, 10, 14, 24, and 36 hours using heparinized syringes.30 Plasma was immediately isolated from each blood sample (400 μL) by centrifugation (Hanil Science Industrial Co) at 10,000× g for 10 minutes. Subsequently, plasma samples were refrigerated at −20°C until further experimentation.34,35
Sample preparation
Separated plasma samples were acidified by the addition of 200 μL of 1 M HCl. Subsequently, 40 μL of clofibric acid solution, at a concentration of 400 μg/mL in acetonitrile, was added and mixed. Then, liquid–liquid extraction was performed twice with 2 mL of diethyl ether by vigorous, thorough mixing for 5 minutes and centrifuging at 10,000× g for 10 minutes (Hanil Science Industrial Co). The transparent supernatant was evaporated in another clean microtube at 40°C. Samples were reconstituted in 200 μL of acetonitrile. Each reconstituted sample was placed in a small-volume HPLC vial and subjected to HPLC analysis.35
HPLC analysis
Fenofibrate is a prodrug. After oral administration, it is completely metabolized to fenofibric acid by biological esterases. Thus, an evaluation of the bioavailability of fenofibrate is dependent upon the measurement of fenofibric acid in the blood stream.36–38 A 100 μL aliquot of the prepared sample was analyzed for fenofibric acid content. An Agilent 1260 Infinity HPLC system (Agilent Technologies) was used for this purpose. A Capcell Pak C18 column (4.6 mm inner diameter ×250 mm, 5 μm particle size), with temperature set to 30°C, was used for reverse-phase HPLC analysis. The mobile phase consisted of acetonitrile and 0.1% (v/v) phosphoric acid (54:46, v/v). The flow rate of the mobile phase was 2 mL/minutes. Fenofibric acid quantification was accomplished at 285 nm.35
Results and discussion
Electrospraying is a promising technique for the preparation of pharmaceutical polymeric nanoparticles.14,15,40 In this study, fenofibrate, PVP, and Labrafil M 2125 were entirely dissolved in volatile dichloromethane (boiling point: 39.6°C) and subjected to electrospraying via the solvent evaporation method, leading to the novel fenofibrate-loaded nanospherules. PVP and Labrafil M 2125 were selected as the most suitable excipients owing to their excellent fenofibrate solubilization ability35,40 and their complete solubility in dichloromethane to form an absolutely clear solution for electrospraying.
To determine the effect of Labrafil M 2125 concentration on the aqueous solubility and dissolution of fenofibrate, nanospherule formulations I–VI (Table 1) were prepared with gradually increasing amounts of Labrafil M 2125 while maintaining the fenofibrate/PVP ratio constant at 1:2. All of the electrosprayed nanospherule formulations had remarkably enhanced aqueous solubility and dissolution compared with free drug. With increasing amounts of Labrafil M 2125, solubility (Figure 2A) and dissolution (Figure 2B) were enhanced further. Labrafil M 2125, a surfactant, improved the drug solubility and dissolution due to its high solubilization ability.35 Among the nanospherule formulations I–VI, formulation VI, containing fenofibrate/PVP/Labrafil M 2125 at the weight ratio of 1:2:0.5, showed the highest apparent aqueous solubility (21.23±2.54 μg/mL) and dissolution (~70% at 1 hour) of fenofibrate. Therefore, this formulation was selected for further study.
 | Table 1 Compositions of electrosprayed nanospherules |
 | Figure 2 Effect of Labrafil M 2125 on the aqueous solubility (A) and the dissolution (B) of drug in the electrosprayed nanospherule. |
Next, the influence of the quantity of polymeric matrix on the solubility and dissolution of the drug were investigated. The nanospherule formulations VII–XI (Table 1) were prepared with varying amounts of PVP while keeping the fenofibrate/Labrafil M 2125 ratio fixed at 1:0.5. As the PVP/drug ratio increased to 4:1, the solubility (Figure 3A) and dissolution (Figure 3B) increased. Further increasing the quantity of PVP in formulations IX, X, and XI did not significantly change the solubility or dissolution. Thus, on the basis of the highest apparent solubility (32.51±2.41 μg/mL) and dissolution (~97% in 20 minutes) behavior of the drug, the formulation IX, containing fenofibrate/PVP/Labrafil M 2125 at the optimized weight ratio of 1:4:0.5, was chosen as an optimal formulation of electrosprayed nanospherule.
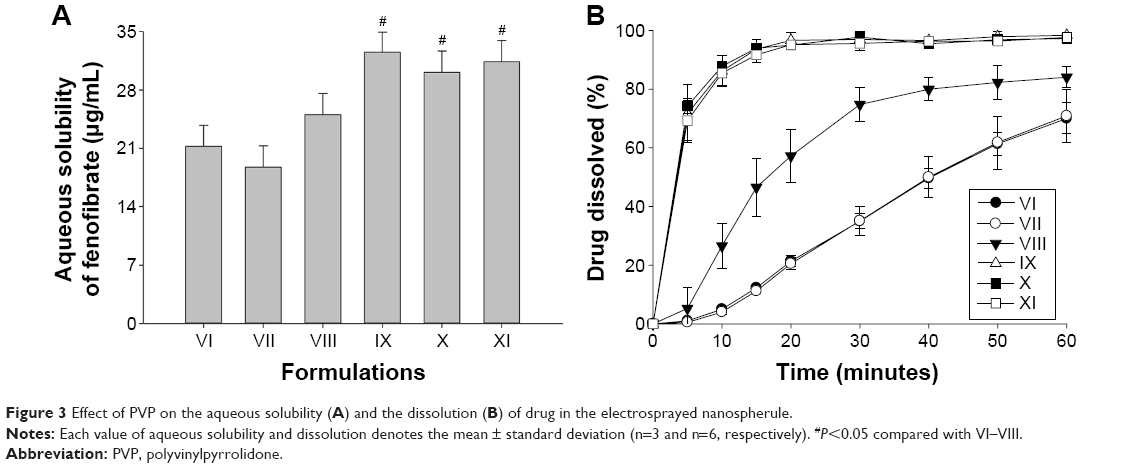 | Figure 3 Effect of PVP on the aqueous solubility (A) and the dissolution (B) of drug in the electrosprayed nanospherule. |
The X-ray diffraction patterns of the samples are displayed in Figure 4A. The free drug pattern was characterized by sharp spikes, representing its typical crystalline nature (Figure 4A-a). The diffraction for PVP did not show any peaks, suggesting that it was amorphous in nature (Figure 4A-b). The main distinguishing peaks attributed to crystalline fenofibrate were also seen in the physical mixture (Figure 4A-c). However, the drug was present in the amorphous state in the electrosprayed nanospherule (Figure 4A-d).11,12
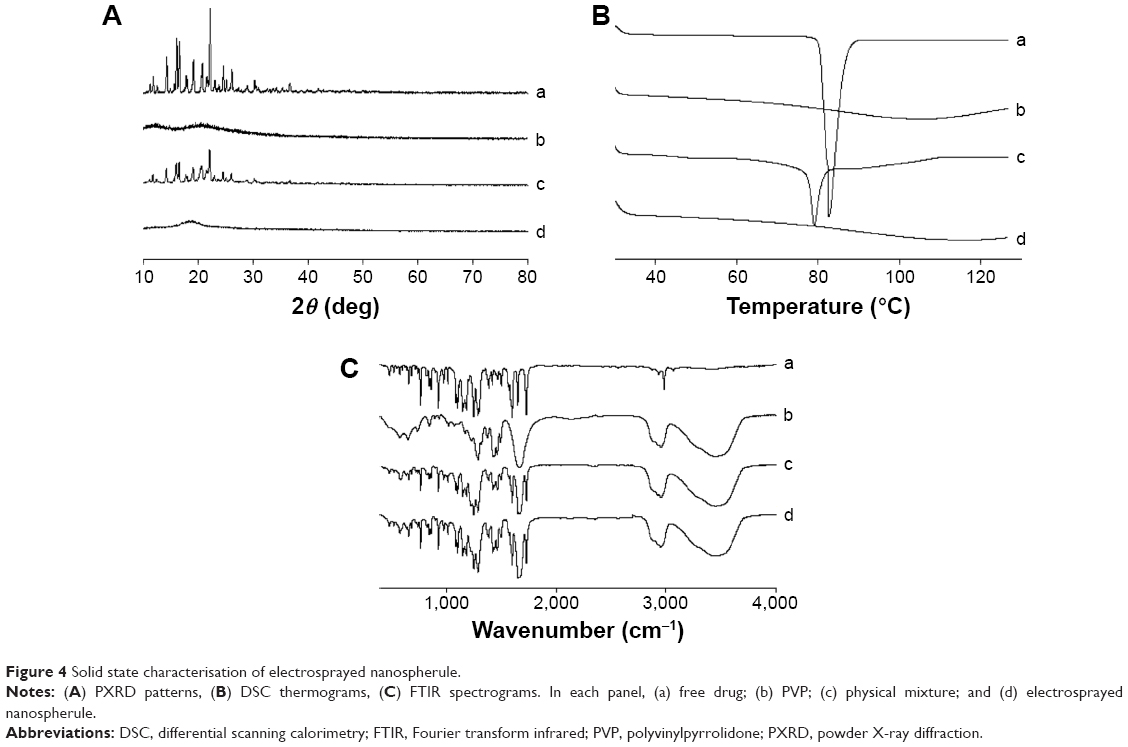 | Figure 4 Solid state characterisation of electrosprayed nanospherule. |
Figure 4B shows the thermal properties of the samples. The free drug gave a sharp endothermic peak at the melting point of fenofibrate at ~81.5°C (Figure 4B-a). This confirmed that the drug was typically crystalline in nature. PVP did not generate any endothermic peaks (Figure 4B-b); however, a broad downward curve was observed due to moisture evaporation.41 Fenofibrate also generated an endothermic peak (Figure 4B-c) in the physical mixture, suggesting that the drug was present in the crystalline form in the physical mixture. On the other hand, the drug was completely transformed into the amorphous state in the nanospherule (Figure 4B-d).12,13
Figure 4C reveals the FTIR spectra of the samples. The free drug produced the chief distinguishing spikes at approximately 1,729, 1,600, 1,180, 1,145, 925, 765, 525, and 480 cm−1 (Figure 4C-a). These distinguishing spikes also appeared in the spectra of the physical mixture (Figure 4C-c) and electrosprayed nanospherule (Figure 4C-d). Moreover, the peak pattern in the spectrum of the nanospherule formulation was exactly the same as that in the spectrum of the physical mixture. There was no shift of the chief distinguishing spikes; therefore, the drug had no covalent interaction with the excipients in the nanospherule formulation.11–13
The SEM outcomes of the study of fenofibrate powder and electrosprayed nanospherule are presented in Figure 5. The free drug showed crystals of irregular shape and surface (Figure 5A). The electrosprayed nanospherule appeared as tiny spherical nanoparticles (Figure 5B). The SEM image indicated that these electrosprayed nanospherules had a particle size <200 nm. In general, a pharmaceutical nanoparticle is a particle having a diameter <1,000 nm.8,9
 | Figure 5 SEM images. |
Figure 6 shows the mean fenofibric acid concentration–time profiles after oral administration of the free drug, the solid dispersion, and the electrosprayed nanospherule; each dose was equivalent to 20 mg/kg fenofibrate. The electrosprayed nanospherule and the solid dispersion were prepared with fenofibrate, PVP, and Labrafil M 2125 at the weight ratio of 1:4:0.5 and dissolved in dichloromethane and ethanol, respectively. Fenofibrate, a prodrug, is completely biotransformed after oral administration to fenofibric acid, an active metabolite, by blood and tissue esterases. There is no fenofibrate detectable in plasma following oral administration; therefore, a bioavailability assessment relies upon quantification of its metabolite.36–38 The nanospherule and solid dispersion gave higher mean plasma concentrations of fenofibric acid than did the free drug at all time points, with particularly significant differences from 1 to 12 hours (P<0.05). Moreover, the plasma titers of fenofibric acid in the nanospherule group were not significantly different from those in the solid dispersion group, even if the former showed lower values than did the latter. The pharmacokinetic parameters are displayed in Table 2. The area under the curve (AUC) and maximum plasma concentration (Cmax) of the nanospherule and solid dispersion was remarkably improved as compared to the AUC and Cmax of the free drug (P<0.05). Similar to the plasma concentration, there were no significant differences in AUC and Cmax values between the nanospherule and the solid dispersion. The AUC values of the free drug and the novel nanospherule formulation were 17.72±1.52 and 41.01±10.60 h μg/mL, respectively. Similarly, the Cmax values of the free drug and the novel nanospherule formulation were 2.36±0.34 and 5.12±1.61 μg/mL, respectively. On the other hand, the half-lives did not show significant variation from one another. With similar bioavailability as the conventional solid dispersion, the nanospherule improved bioavailability by ~2.5-fold as compared with the free drug. This enhancement might be attributed to the remarkable improvement in the aqueous solubility and dissolution resulting from particle size reduction and conversion of the crystalline drug to the amorphous state.14,15 Generally, improvement in aqueous solubility and bioavailability of poorly water-soluble drugs by conventional solid dispersion systems is due to wetting enhancement, particle size reduction, and/or a change of the crystalline drug to the amorphous form.11–13,32 Thus, in this study, the electrosprayed nanospherule may have improved the aqueous solubility and bioavailability of poorly water-soluble fenofibrate by the same mechanisms, except through wetting enhancement. On the other hand, unlike other microcapsules and solid dispersions, the electrosprayed nanospherule could be manufactured without heat, leading to production of more stable formulations compared to the former two. Moreover, the electrosprayed nanospherule might greatly enhance the aqueous solubility and oral bioavailability of various poorly water-soluble drugs compared to other ones due to the greater reduction in particle size, even if they present similar oral bioavailability, as in this study. Therefore, the novel well-established electrosprayed nanospherule system is a promising candidate to deliver poorly water-soluble drugs with remarkably improved oral bioavailability and excellent stability.
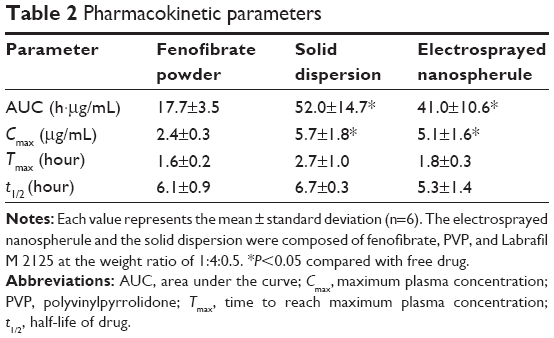 | Table 2 Pharmacokinetic parameters |
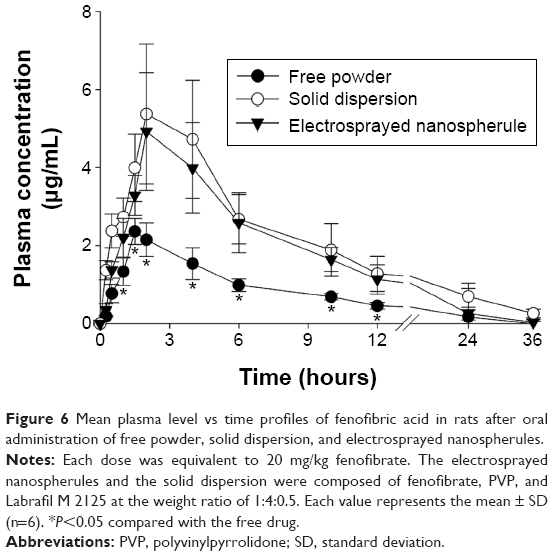 | Figure 6 Mean plasma level vs time profiles of fenofibric acid in rats after oral administration of free powder, solid dispersion, and electrosprayed nanospherules. |
Conclusion
The novel electrosprayed nanospherule, composed of fenofibrate, PVP, and Labrafil M 2125 at the weight ratio of 1:4:0.5, had a particle size <200 nm, with the drug present in the amorphous form. It improved the solubility (32.51±2.41 μg/mL), dissolution (~85% in 10 minutes), and oral bioavailability (~2.5-fold) of the drug compared with the properties of the free drug; accordingly, this formulation is a potential drug delivery system to deliver poorly water-soluble fenofibrate with greater bioavailability. Further studies on its efficacy, including measurement of serum triglycerides, will be carried out for the development of novel fenofibrate-loaded electrosprayed nanospherule.
Acknowledgment
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (No. 2015R1A2A2A05027872 & 2015R1A2A2A01004118).
Disclosure
The authors report no conflicts of interest in this work.
References
Guay D. Micronized fenofibrate: a new fibric acid hypolipidemic agent. Ann Pharmacother. 1999;33(10):1083–1103. | ||
Genest JJ, Nguyen N-H, Theroux P, et al. Effect of micronized fenofibrate on plasma lipoprotein levels and hemostatic parameters of hypertriglyceridemic patients with low levels of high-density lipoprotein cholesterol in the fed and fasted state. J Cardiovasc Pharmacol. 2000; 35(1):164–172. | ||
Munoz A, Guichard JP, Reginault P. Micronised fenofibrate. Atherosclerosis. 1994;110(suppl):S45–S48. | ||
Ming-Thau S, Ching-Min Y, Sokoloski TD. Characterization and dissolution of fenofibrate solid dispersion systems. Int J Pharm. 1994;103(2): 137–146. | ||
Dressman J, Butler J, Hempenstall J, et al. The BCS: where do we go from here? Pharm Technol. 2001;25(7):68–77. | ||
Jia Z, Lin P, Xiang Y, et al. A novel nanomatrix system consisted of colloidal silica and pH-sensitive polymethylacrylate improves the oral bioavailability of fenofibrate. Eur J Pharm Biopharm. 2011;79(1):126–134. | ||
Xing F, Cheng G, Yi K, et al. Nanoencapsulation of capsaicin by complex coacervation of gelatin, acacia, and tannins. J Appl Polym Sci. 2005;96(6):2225–2229. | ||
Pinto Reis C, Neufeld RJ, Ribeiro AJ, et al. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine. 2006;2(1):8–21. | ||
Soppimath KS, Aminabhavi TM, Kulkarni AR, et al. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70(1):1–20. | ||
Vauthier C, Bouchemal K. Methods for the preparation and manufacture of polymeric nanoparticles. Pharm Res. 2009;26(5):1025–1058. | ||
Cho JH, Kim YI, Kim DW, et al. Development of novel fast-dissolving tacrolimus solid dispersion-loaded prolonged release tablet. Eur J Pharm Sci. 2014;54:1–7. | ||
Kim DW, Kwon MS, Yousaf AM, et al. Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohydr Polym. 2014;114:365–374. | ||
Yang KY, Hwang du H, Yousaf AM, et al. Silymarin-loaded solid nanoparticles provide excellent hepatic protection: physicochemical characterization and in vivo evaluation. Int J Nanomedicine. 2013;8: 3333–3343. | ||
Luo CF, Yuan M, Chen MS, et al. Pharmacokinetics, tissue distribution and relative bioavailability of puerarin solid lipid nanoparticles following oral administration. Int J Pharm. 2011;410(1–2):138–144. | ||
Frézard F, Martins PS, Bahia AP, et al. Enhanced oral delivery of antimony from meglumine antimoniate/beta-cyclodextrin nanoassemblies. Int J Pharm. 2008;347(1–2):102–108. | ||
Mora-Huertas C, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1):113–142. | ||
Arya N, Chakraborty S, Dube N, et al. Electrospraying: a facile technique for synthesis of chitosan-based micro/nanospheres for drug delivery applications. J Biomed Mater Res B Appl Biomater. 2009;88(1): 17–31. | ||
Hong Y, Li Y, Yin Y, et al. Electrohydrodynamic atomization of quasi-monodisperse drug-loaded spherical/wrinkled microparticles. J Aerosol Sci. 2008;39(6):525–536. | ||
Ding L, Lee T, Wang C-H. Fabrication of monodispersed Taxol-loaded particles using electrohydrodynamic atomization. J Control Release. 2005;102(2):395–413. | ||
Bohr A, Kristensen J, Stride E, et al. Preparation of microspheres containing low solubility drug compound by electrohydrodynamic spraying. Int J Pharm. 2011;412(1):59–67. | ||
Yu D-G, Williams GR, Yang J-H, et al. Solid lipid nanoparticles self-assembled from electrosprayed polymer-based microparticles. J Mater Chem. 2011;21(40):15957–15961. | ||
Enayati M, Ahmad Z, Stride E, et al. Size mapping of electric field-assisted production of polycaprolactone particles. J R Soc Interface. 2010;7:S393–S402. | ||
Trotta M, Cavalli R, Trotta C, et al. Electrospray technique for solid lipid-based particle production. Drug Dev Ind Pharm. 2010;36(4):431–438. | ||
Cavalli R, Bisazza A, Bussano R, et al. Poly (amidoamine)-cholesterol conjugate nanoparticles obtained by electrospraying as novel tamoxifen delivery system. J Drug Deliv. 2011;0:1–9. | ||
Chakraborty S, Liao I-C, Adler A, et al. Electrohydrodynamics: a facile technique to fabricate drug delivery systems. Adv Drug Deliv Rev. 2009; 61(12):1043–1054. | ||
Yousaf AM, Kim DW, Choi HG, et al. Validation of a highly sensitive RP-HPLC method for quantification of fenofibrate in pure and pharmaceutical dosage forms. Curr Pharm Anal. 2014;10(2): 97–104. | ||
Kim KS, Jin SG, Mustapha O, et al. Novel fenofibric acid-loaded controlled release pellet bioequivalent to choline fenofibrate-loaded commercial product in beagle dogs. Int J Pharm. 2015;490:273–280. | ||
Shah VP, Noory A, Noory C, et al. In vitro dissolution of sparingly water-soluble drug dosage forms. Int J Pharm. 1995;125(1):99–106. | ||
Sanganwar GP, Gupta RB. Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int J Pharm. 2008;360(1–2):213–218. | ||
Yousaf AM, Kim DW, Kim JO, et al. Characterization of physicochemical properties of spray-dried solid dispersions loaded with unmodified crystalline fenofibrate. Curr Pharm Anal. 2015;11(2):139–144. | ||
Windbergs M, Strachan CJ, Kleinebudde P. Understanding the solid-state behaviour of triglyceride solid lipid extrudates and its influence on dissolution. Eur J Pharm Biopharm. 2009;71(1):80–87. | ||
Hens B, Brouwers J, Corsetti M, et al. Gastrointestinal behavior of nano- and microsized fenofibrate: in vivo evaluation in man and in vitro simulation by assessment of the permeation potential. Eur J Pharm Sci. 2015;77:40–47. | ||
Oh DH, Park YJ, Kang JH, et al. Physicochemical characterization and in vivo evaluation of flurbiprofen-loaded solid dispersion without crystalline change. Drug Deliv. 2011;18(1):46–53. | ||
Li X, Gu L, Xu Y, et al. Preparation of fenofibrate nanosuspension and study of its pharmacokinetic behavior in rats. Drug Dev Ind Pharm. 2009;35(7):827–833. | ||
Yousaf AM, Kim DW, Oh YK, et al. Enhanced oral bioavailability of fenofibrate using polymeric nanoparticulated systems: physicochemical characterization and in vivo investigation. Int J Nanomedicine. 2015; 10:1819–1830. | ||
Straka RJ, Burkhardt RT, Fisher JE. Determination of fenofibric acid concentrations by HPLC after anion exchange solid-phase extraction from human serum. Ther Drug Monit. 2007;29(2):197–202. | ||
Streel B, Hubert P, Ceccato A. Determination of fenofibric acid in human plasma using automated solid-phase extraction coupled to liquid chromatography. J Chromatogr B Biomed Sci Appl. 2000;742(2):391–400. | ||
Tang N, Lai J, Chen Y, et al. Fenofibrate solid dispersion pellets prepared by fluid-bed coating: physical characterization, improved dissolution and oral bioavailability in beagle dogs. J Chin Pharm Sci. 2009;18(2):156–161. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.