Back to Journals » International Journal of Nanomedicine » Volume 21
Novel Dual Strategy Based on EPR/AT for Optimizing Therapeutic Effect by Improving Drug Delivery System Physicochemical Properties and Regulating TME
Authors Chen L ![]() , Deng X
, Deng X ![]() , Shen Q, Chen M, Li C, Wang S
, Shen Q, Chen M, Li C, Wang S
Received 21 August 2025
Accepted for publication 9 December 2025
Published 15 January 2026 Volume 2026:21 559763
DOI https://doi.org/10.2147/IJN.S559763
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. RDK Misra
Long Chen,1,2,* Xiang Deng,1– 3,* Qian Shen,1– 3 Min Chen,4 Churong Li,2,5 Shunxi Wang1,2,6,7
1The Second Affiliated Hospital of Chengdu Medical College, Nuclear Industry 416 Hospital Chengdu, Sichuan, 610000, People’s Republic of China; 2Non-Coding RNA and Drug Discovery Key Laboratory of Sichuan Province, School of Basic Medical Sciences, Chengdu Medical College, Chengdu, Sichuan, 610550, People’s Republic of China; 3Clinical Medical College, Chengdu Medical College, Chengdu, Sichuan, 610500, People’s Republic of China; 4Department of Dermatology, Nanbu People’s Hospital, Nanchong, Sichuan, 637300, People’s Republic of China; 5Radiotherapy Center, Radiation Oncology Key Laboratory of Sichuan Province, Sichuan Clinical Research Center for Cancer, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, University of Electronic Science and Technology of China, Chengdu, 610041, People’s Republic of China; 6Institute of Pathology & Southwest Cancer Center, the First Afliated Hospital (Southwest Hospital) of Army Medical University (Third Military Medical University), Chongqing, 400038, People’s Republic of China; 7Chongqing Institute of Advanced Pathology, Jinfeng Laboratory, Chongqing, 400039, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Churong Li, Email [email protected] Shunxi Wang, Email [email protected]
Abstract: The clinical advancement of cancer nanomedicine is significantly hindered by its limited accumulation in tumors, a key factor behind the frequent failure of nanodrugs in clinical trials. The effectiveness of these nanodrugs is closely tied to their route of administration, whether oral, transdermal, intravenous, or intracerebral, as each path presents unique physiological barriers that impede bioavailability and precise tumor targeting. Among the major causes of poor accumulation are rapid clearance by the mononuclear phagocyte system, opsonization accompanied by protein corona formation, renal filtration, and the abnormal, heterogeneous nature of tumor vasculature that restricts passive targeting via the enhanced permeability and retention (EPR) effect. In response, active targeting (AT) strategies have been widely explored, including surface modification with ligands, antibodies, or aptamers designed to bind specifically to overexpressed receptors on cancer cells or blood vessels. Despite these efforts, challenges such as the dense extracellular matrix, elevated interstitial fluid pressure, and the notable inconsistency of the EPR effect between animal models and human patients continue to limit therapeutic penetration. This review offers a systematic examination of nanodrug delivery pathways and the reasons behind their inadequate accumulation, highlighting the potential of both active targeting and combined passive-active strategies to enhance tumor-specific delivery. Overcoming these biological barriers through refined nano-design is crucial for developing the next generation of nanomedicines with improved tumor accumulation and treatment outcomes.
Keywords: nanomedicine, EPR effect, active targeting, tumor accumulation
Introduction
Cancer continues to be a major global health challenge, responsible for approximately 10 million deaths each year and imposing a significant burden on healthcare systems worldwide.1 In response, nanomedicine has arisen as a promising frontier in oncology, leveraging sophisticated drug delivery systems and insights into the tumor microenvironment to create intelligent nanoparticles for targeted applications.2 However, the transition from laboratory success to clinical impact has been limited, with only a handful of nanoformulations progressing to trials or achieving regulatory approval.3 Clinically, many approved nanomedicines demonstrate a greater capacity for reducing the severe side effects of chemotherapy than for enhancing treatment efficacy or prolonging patient survival.4 A central issue underlying this translational shortfall is the inefficient accumulation of nanomedicines within tumors, a process dictated by the administration route and its accompanying biological barriers5 Each delivery path presents a unique set of physiological challenges that shape the nanoparticles’ journey, ultimately affecting their distribution throughout the body and their final concentration at the tumor site.6 The oral route, while preferred for its ease and patient adherence, must contend with the harsh environment of the gastrointestinal tract—encompassing enzymatic breakdown, acidic conditions, and poor epithelial permeability—all of which contribute to low bioavailability.7 Transdermal delivery utilizes the skin’s extensive surface to bypass first-pass metabolism but is constrained by the formidable stratum corneum, which blocks the penetration of most nanoparticles unless assisted by permeation-enhancing technologies like microneedles.8 Targeting central nervous system tumors requires navigating the selective blood-brain barrier, whose tight junctions and efflux pumps typically prevent nanocarriers from entering, even with the aid of receptor-specific strategies. Intravenous injection, as the predominant systemic method, offers immediate bioavailability and wide distribution, yet it faces hurdles such as swift clearance by immune organs, protein corona formation, and renal excretion, which all reduce the fraction of dose reaching the tumor.9 These delivery-specific complications highlight the importance of improving tumor accumulation, a phenomenon historically linked to the Enhanced Permeability and Retention (EPR) effect.10 First described in 1986, the EPR effect has long supported passive targeting, relying on the leaky vasculature and poor lymphatic drainage in tumors to allow nanoparticle deposition.11 However, its general applicability is now questioned, as emerging research underscores the contribution of active, cell-driven processes to the transport and clearance of nanoparticles in tumor tissue12–14 Crucially, the EPR effect is highly variable, differing between and within cancer types. Vascular pore sizes can vary dramatically, leading to more accumulation in vascular tumors like sarcomas and far less in stroma-rich cancers such as pancreatic ductal adenocarcinoma. This heterogeneity helps explain why positive results in uniform animal models frequently fail to predict outcomes in human patients, where dense stroma and individual pathological differences can undermine EPR-based delivery. This review systematically details the drivers of nanomedicine accumulation, covering nanoparticle design, tumor microenvironment modulation, and active targeting. We also analyze the two-way relationship between nanodrug accumulation and tumor changes. Synthesizing transport mechanisms with efficacy and toxicity data, this work provides a blueprint for developing next-generation nanocarriers with improved clinical potential.
Nanomedicine Delivery Routes
The efficacy of nanomedicine is critically dependent on its ability to reach the target site in sufficient quantities. Nanoparticles must navigate a series of biological barriers that vary significantly depending on the administration route. The choice of delivery path is therefore paramount, influencing nanoparticle biodistribution, accumulation at the tumor site, and eventual therapeutic outcome. This section systematically reviews the primary administration routes for nanocarriers, highlighting the unique biological barriers encountered and the corresponding nanoparticle design strategies employed to overcome them.
Oral Administration
Oral administration is the most common and patient-preferred route of drug delivery, attributed to its convenience, pain avoidance, high patient compliance, and reduced risks of cross-infection and needle stick injuries, which also helps overcome the drawbacks of injections (eg, tissue injury, pain, adverse reactions, poor compliance).15 However, oral drug bioavailability depends on the compound’s solubility and permeability,16 and oral delivery of peptides/proteins often suffers from degradation by the gastrointestinal tract (GIT)’s acidic environment and enzymatic system—lowering therapeutic value—prompting the exploration of strategies like site-specific delivery systems, peptide chemical modification (eg, lipophilic derivatives, peptidomimetics), bioadhesive systems, and co-administration of penetration enhancers/protease inhibitors to enhance drug stability and absorption.9,17 After oral administration, nanocarriers encounter the GIT’s physicochemical environment: the human intestinal epithelium, featuring villi that expand the absorptive surface area to 300–400 m2 and acting as a physical barrier to drug absorption, consists of absorptive enterocytes, mucus-secreting goblet cells, endocrine cells, and immunocompetent cells, while biological fluids affect particle stability even before intestinal cell contact.18 Nanotechnology offers unique advantages for oral drug delivery, enabling (i) delivery of poorly water-soluble drugs, (ii) GIT site-specific targeting, (iii) transcytosis across the tight intestinal barrier, and (iv) intracellular/transcellular delivery of large macromolecules.19 Nanoparticle encapsulation, for instance, overcomes the GI barrier, protects drugs from enzymatic degradation, and enables controlled/systemic release,20 and biodegradable polymeric nanoparticles represent another promising approach for oral delivery of protein/peptide drugs to improve efficacy (Figure 1).21–23 The stability of polymeric nanocarriers in the GIT is strongly influenced by their composition: nanoparticles made of insoluble polymers resist degradation and rapid drug release, whereas water-soluble polymers forming polyelectrolyte nanoparticles are prone to destabilization by pH/ionic strength. Even with better kinetic stability than surfactant micelles, polymeric micelles must maintain concentrations above the critical micelle concentration after GIT dilution and be exposed to ionic strength below their flocculation point to avoid premature drug release.24 Other strategies for oral delivery include transport carriers (eg, transferrin for insulin delivery) and prodrug approaches, with polymeric micelles reported to cross the intestinal barrier post-oral administration for effective use.25 Examples of advancements include Zhang et al’s development of pH-responsive starch nanoparticles-grafted-poly(L-glycolic acid) (SNP-g-PGA)—using starch nanoparticles as the backbone and poly(L-glycolic acid) as the graft—as a carrier for oral insulin delivery,26 and Tyr-Gly-Leu-Phe (YF4)-loaded lipid nanoparticles (YF4-LNPs)—a novel oral peptide delivery system leveraging the advantages of polymer nanoparticles and liposomes— which showed superior performance: in vitro, free YF4 had 80% burst release within 6 h while YF4-LNPs released <40% over 24 h, and in animal models, YF4-LNPs reduced systolic blood pressure (SBP) by 43.5 mmHg ~2 h post-administration compared to a 15.6 mmHg SBP decrease with free YF4 at 4 h.27 Despite these progresses, considerable advancements in innovative materials and technologies are still required to maximize drug absorption and stability in oral drug delivery.
 |
Figure 1 Oral delivery of a pH-sensitive peptide drug. (A) Orally administered peptide drugs are particularly susceptible to proteolytic degradation in the stomach, thereby leading to their low bioavailability. (B) Nanoparticulate carriers protect the encapsulated drug from enzymatic cleavage. Reproduced with permission from Ref.28 |
Transdermal Drug Delivery (TDD)
Of the various administration routes for nanocarriers, transdermal drug delivery (TDD) presents a compelling non-invasive strategy that leverages the skin’s extensive surface area (1.8–2.0 m2) to systemically administer therapeutics while bypassing first-pass metabolism, thereby enabling efficient delivery with reduced dosage and toxicity. The primary obstacle is the stratum corneum (10–20 μm), the skin’s outermost layer, which exhibits a formidable “brick-and-mortar” structure of keratinocytes and lipids, effectively blocking the penetration of high molecular weight drugs (>500 Da).29–31 To overcome this barrier, engineered nanocarriers (NCs) utilize three principal penetration pathways: the intercellular route (through the lipid matrix), the transcellular route (through keratinocytes), and the transappendageal route (via hair follicles and sebaceous/sweat glands).31,32 Although appendages cover a mere 0.1% of the skin surface, the follicular pathway is a significant reservoir; studies demonstrate that nanoparticles (eg, 320 nm) can be stored in hair follicles for up to 10 days, exhibiting time-dependent accumulation as visualized by CLSM.33,34 A diverse array of NCs has been developed for this purpose. Liposomes, the pioneering closed colloidal systems composed of phospholipids and steroids, can encapsulate hydrophilic drugs in their aqueous core and lipophilic drugs within their lipid bilayers; their clinical translation is exemplified by Doxil®, the first FDA-approved PEGylated liposomal doxorubicin.35–37 To enhance skin permeation, second-generation lipid vesicles were engineered. Transfersomes, incorporating surfactants (eg, sodium cholate) and ethanol, possess ultradeformability, allowing them to squeeze through pores less than one-tenth their diameter (up to 500 nm).38 Niosomes, assembled from non-ionic surfactants, prolong drug residence in the stratum corneum and reduce systemic absorption, as demonstrated for Minoxidil and ellagic acid.39 Ethosomes, characterized by a high ethanol content (~30%), effectively fluidize the stratum corneum lipids, facilitating deep skin penetration for drugs like Tacrolimus and Testosteron.40,41 Furthermore, nanoemulsions (100–1000 nm), which are transparent dispersions of oil and water stabilized by emulsifiers, also serve as effective carriers for enhanced transdermal permeation.42 Consequently, the strategic design of nanocarriers provides a powerful means to circumvent the skin’s barrier properties, making TDD a viable platform for effective drug administration.
Blood Brain Barrier Drug Delivery
The blood-brain barrier (BBB), one of the tightest endothelia in the central nervous system (CNS) coined by Lewandowsky in 1900, consists of endothelial cells, astrocytes, pericytes, microglial cells and adjacent neurons and covers a total surface area of approximately 20 m2 via about 100 billion brain capillaries with endothelium spanning around 650 km.43 It acts as a bottleneck for CNS drug development as most neurotherapeutic compounds fail to reach the market due to their inability to cross the BBB together with the blood-cerebrospinal fluid barrier that regulates molecular entry into the brain.44 The BBB’s high selectivity originates from cerebral endothelial cells including physical barriers (adherens junctions and tight junctions providing >1500 Ωcm2 transelectrical resistance), immunological barriers (microglia, perivascular macrophages, mast cells), transport barriers (para- and transcellular routes such as carrier-mediated transport, receptor-mediated transcytosis, adsorptive mediated transcytosis, cell-mediated transport) and metabolic barriers (intra- and extracellular enzymes against lipophilic substances).45,46 Several nanocarriers like liposomes and solid lipid nanoparticles have enabled successful drug delivery across the BBB with hexapeptide dalargin (a leucine-enkephalin analogue) being the first drug delivered to the brain via polysorbate 80-coated nanoparticles while dalargin bound to nanoparticles without polysorbate 80 showed no analgesic effect.47 Most nanosized systems rely on adsorptive mediated transcytosis and receptor-mediated transcytosis for neurotherapeutic delivery.48 Negatively charged cerebral endothelial cells can interact with nanoparticles by introducing positive charges either through preparing nanoparticles positively charged at physiological pH 7.4 or surface-functionalizing nanoparticles with positively charged molecules and cell-penetrating peptides (eg, HIV-derived TAT-peptides) or cationic proteins (eg, albumin) are widely used for nanoparticle anchoring to facilitate drug passage across the BBB.49 The BBB expresses various receptors (eg, transferrin receptor, insulin receptor, low density lipoprotein receptor) so ligands can decorate delivery systems such as lactoferrin-covalently conjugated PEG-coated Fe3O4 nanoparticles for receptor-mediated delivery.50 Carrier-mediated transcytosis uses carriers for glucose, amino acids, purine bases, nucleosides and choline (which supply brain nutrients and deliver drugs) like liposome-incorporated mannose derivatives that crossed the BBB via glucose transporters in mouse brains.51 Approaches for BBB drug delivery include direct injection and implantation, chemical modifications (lipidization adding lipid molecules to drug polar ends to enhance permeability, prodrug approach with enzyme-catalyzed conversion to active drugs in the brain), temporary BBB opening via permeability enhancers (eg, bradykinin, histamine, serotonin, tumor necrosis factor-α) and nano-enabled platforms via intravenous or intranasal routes.52
Intravenous Delivery
Nanoparticles can be delivered to tumor sites via multiple routes, each offering distinct advantages. Intravenous injection, as a method of systemic administration, is valued for its clinical convenience and ability to reach a wide range of tissues. Furthermore, nanocarriers delivered through this route typically demonstrate significantly greater tumor accumulation compared to free, non-encapsulated drugs. The intravenous (IV) route for nanoparticle administration provides almost instantaneous response wide-ranging control over drug input rate and complete bioavailability even at low doses while overcoming first-pass metabolism and proteolytic enzyme degradation making it suitable for expensive peptide or protein drugs though it carries risks like patient pain high cost need for experienced healthcare personnel and direct systemic drug exposure.53 The first IV nanoparticulate product Abraxane® a paclitaxel reformulation was FDA-approved in 2006.54,55 Nanomedicines address key cancer therapy challenges including drug side effects from accumulation cancer recurrence and delayed disease stabilization as controlled-release nanocarriers can be IV–infused to target tumors enhancing solid tumor treatment efficacy reducing drug toxicity and achieving prolonged remission with drug-polymer conjugates and nanoemulsions mainly explored for prostate cancer targeting.56 Paclitaxel-LDE a cholesterol-rich nanoemulsion showed lower toxicity higher anticancer activity in mice concentrated in solid tumors (binding to LDL receptor-overexpressing cancer cells) and in human gynecological cancers had a longer half-life (14.51 ± 3.23 h vs 6.62 ± 2.05 h for paclitaxel-cremophor) and 3.5-fold higher targeting in tumor than normal tissues.57 IV emulsions require oil droplets <5 μm (the size of the smallest lung blood vessels) with mean sizes of 200–400 nm hence called nanoemulsions.58 Third-generation IV iron therapies (eg Cosmofer iron dextran) significantly improve efficacy without old-generation toxicity and accelerated Cosmofer administration is effective safe time-saving and reduces nursing time for chronic kidney disease (CKD) patients though long-term toxicity evaluation is needed for iron–oxyhydroxide core/carbohydrate shell formulations.59
Obstacles to Effective Tumoral Nanoparticle Delivery
Effective tumoral nanoparticle delivery faces multiple critical obstacles rooted in nano-bio interactions and physiological barriers. Upon entering physiological fluids (eg, blood plasma, mucus, ascites, interstitial fluids), nanoparticles undergo dynamic nano–bio interface changes that drive the formation of a protein corona—a multilayered adsorbed layer of endogenous proteins formed via electrostatic and hydrophobic interactions with nanoparticle cores.60,61 This corona includes a rapidly formed inner “hard corona” of tightly bound proteins and an outer “soft corona” of loosely associated, environment-exchangeable proteins, and contains opsonins (eg, immunoglobulins, fibrinogen, complement proteins) that enhance nanoparticle uptake by circulating immune cells, even though dysopsonins (eg, albumin, clusterin, apolipoproteins) may mitigate such interactions, collectively impairing targeted delivery.62–64 A second major obstacle is the extremely low accumulation of passively targeted nanoparticles in solid tumors: Wilhem et al conducted a literature review and calculated that only approximately 0.7% of injected nanoparticles accumulate in solid tumors in mice, a median value that, while not capturing the full spectrum of nanoparticle potential, highlights poor tumoral targeting efficiency.6 Physiologically, nanoparticles injected into systemic circulation must evade clearance mechanisms, including the reticuloendothelial system (RES, also called the mononuclear phagocytic system) which mediates nanoparticle phagocytosis in liver and splenic tissue 25, and rapid renal clearance of small (5–8 nm) nanoparticles.65 As summarized by Hui et al, the obstacles further encompass five key steps: opsonization of nanoparticles by serum proteins, destruction by the immune system, inefficient extravasation of nanoparticles from vasculature into tumors, limited infiltration of nanoparticles into tumor tissue, and insufficient endocytosis of nanoparticles into individual tumor cells.66 These obstacles can be categorized into three interconnected groups: barriers within the vasculature and first-pass organs (protein corona formation, RES phagocytosis, renal clearance), barriers to extravasation from the vasculature to tumor tissue, and barriers to nanoparticle internalization into tumor cells, all of which collectively hinder effective tumoral nanoparticle delivery.
Mechanisms of Nanomedicine Accumulation in Tumors
Nanomedicines have emerged as a transformative approach in targeted cancer therapy, primarily due to their “passive-active” dual targeting strategy that promotes selective drug accumulation at tumor sites.67 This targeted enrichment is governed by the synergistic interaction of nanoparticle properties with the distinctive pathophysiology of the tumor microenvironment.
Physiochemical Properties of the Nanoparticles
Size-Dependent Accumulation
The size of nanoparticles is a fundamental parameter governing their biodistribution and tumor accumulation profiles. To avoid rapid renal clearance (cutoff ~10 nm), nanocarriers are typically engineered with dimensions exceeding this threshold.68,69 Conversely, their upper size limit is constrained by vascular endothelial pore sizes in different tumors, generally capping at several hundred nanometers.70 Furthermore, the reticuloendothelial system (RES) actively clears nanoparticles, with hepatic and splenic macrophages demonstrating particularly high phagocytic activity. This RES-mediated clearance exhibits a strong size dependence: Liu et al observed biphasic hepatic sequestration for liposomes <50 nm or >300 nm, while those >400 nm were predominantly cleared by the spleen. In contrast, liposomes within the 90–200 nm range demonstrated optimal tumor targeting.71 This size-dependent behavior is corroborated by PEG-PLA nanoparticles, where diameters of 111 nm and 141 nm enhanced tumor deposition, while 166 nm particles were associated with accelerated hepatic clearance.72 A prolonged circulation half-life increases vascular exposure time, yet it complexly modulates three critical processes: the efficiency of vascular extravasation, the depth of tissue penetration post-extravasation, and the ultimate internalization by malignant cells.73 Recent investigations in 3D tumor spheroids further elucidate these dynamics, demonstrating that smaller nanoparticles (~10 nm) achieve deeper intratumoral penetration (up to ~85 µm depth) via enhanced Brownian motion and passive diffusion, albeit with lower overall accumulation, whereas larger counterparts (50–65 nm) exhibit superior peripheral accumulation but are restricted to the outer ~30–40 µm (approximately 2 cell layers) due to extracellular matrix (ECM) barriers (Figure 2).74 The definition of a single, universally optimal nanoparticle size is complicated by the interplay of multiple material characteristics. While the conventional EPR paradigm typically utilizes carriers between 10–200 nm, stimuli-responsive nanoplatforms can undergo size transformation after extravasation, thereby improving both tumor deposition and interstitial diffusion.75 Consequently, the rational design of size-tunable nanocarriers emerges as a promising frontier to simultaneously optimize circulation kinetics, vascular extravasation, and tissue penetration for superior therapeutic efficacy.
 |
Figure 2 The influence of nanoparticle size on spheroid permeation and cellular uptake. Figure panels (A–D) and (E–P) present typical confocal micrographs showing the distribution of AuNS nanoparticles of varying sizes (10, 30, 50, and 65 nm) within two-dimensional monolayers and three-dimensional spheroids of A549 cells, respectively. The two-dimensional cultures were treated with a nanoparticle concentration of approximately 8 × 1010 particles/mL for a duration of 3 hours. In contrast, the three-dimensional spheroids were exposed to a lower concentration of about 8 × 109 NPs/mL over a longer period of 24 hours. In these visualizations, the photoluminescence (PL) signal from the nanoparticles is pseudocolored in green. Cellular structures are highlighted in magenta, achieved by using CellMask™ Deep Red for staining plasma membranes in 2D models and Phalloidin CruzFluor™ 647 for labeling the cytoskeletal network in the 3D spheroids. Reproduced with permission from Ref.74 |
Surface Charge Effects
The surface charge of nanoparticles serves as a key determinant of their colloidal stability, aggregation behavior, blood compatibility, and systemic clearance kinetics.76 Negatively charged surfaces promote colloidal stability through electrostatic repulsion and are less prone to opsonization, as their anionic character generates repulsive forces against the similarly charged vasculature and cellular membranes. This confers superior stealth properties, thereby minimizing recognition by the mononuclear phagocyte system (MPS) and extending circulatory half-life.77 In contrast, cationic nanoparticles readily adsorb anionic plasma proteins, leading to aggregation and subsequent rapid clearance by the MPS.78 Strategic surface engineering is therefore critical for balancing circulation time with tumor engagement. To achieve prolonged blood circulation, nanoparticles are often shielded with hydrophilic polymers like PEG or specific polysaccharides, which enhance stability and reduce opsonin adsorption.79 Conversely, a positive surface charge generally promotes stronger electrostatic interactions with anionic cell membranes and extracellular matrix components, thereby enhancing tumor retention, tissue penetration, and cellular internalization. Thus, the rational modulation of surface potential is a vital strategy for optimizing the pharmacokinetic profile and tumor-specific accumulation of nanomedicines.80
Shape
The geometry of nanoparticles plays a critical role in their hemodynamic behavior and subsequent tumor deposition. After evading RES clearance, nanocarriers must marginate toward and adhere to the vascular endothelium in diseased tissues to enable extravasation. Although spherical morphologies are most common, anisotropic structures such as rods, disks, and stars are increasingly explored.81 Spherical particles typically follow laminar flow streams, facilitating wall positioning and tissue migration.82 In contrast, rod-shaped nanoparticles induce hydrodynamic disturbances, promoting deviation from streamlines and enhancing trans-endothelial migration.83 These non-spherical forms often provide superior biological performance. For instance, cylindrical micelles exhibit improved cellular uptake and extended circulation, leading to greater tumor accumulation. Pharmacokinetic studies in H22 tumor models showed that high-aspect-ratio micelles had an elimination half-life of ~24 hours, significantly longer than their shorter-rod (14 hours) and spherical (8 hours) counterparts. Corresponding biodistribution data confirmed a 1.4-fold and 2.3-fold increase in tumor deposition for these elongated micelles over 24 hours.84 Furthermore, disk-shaped nanoparticles demonstrate the highest propensity for vascular wall contact and adhesion under laminar flow, as evidenced by Decuzzi et al.85 Therefore, strategic manipulation of nanoparticle shape can markedly improve intravascular margination, endothelial capture, and extravasation efficiency, collectively boosting tumoral delivery.
Tumor Microenvironment
Tumor Vasculature Anomalies
The rapid proliferation of cancer cells drives the formation of new blood vessels to meet escalating nutritional and oxygen requirements. However, these nascent tumor vasculature networks are often structurally and morphologically abnormal,86,87 exhibiting features such as tortuous paths, irregular contours, disorganized endothelial cells, pronounced fenestrations, and an underdeveloped smooth muscle layer.88 These structural defects result in vascular hyperpermeability, which forms the basis of the EPR effect and facilitates the initial extravasation of nanoparticles into tumors.86 Paradoxically, this same hyperpermeability, coupled with impaired lymphatic drainage, leads to the excessive leakage of plasma components and their subsequent accumulation. This process significantly elevates interstitial fluid pressure (IFP) and increases fluid viscosity within the tumor, creating a barrier that hinders the further penetration of nanoparticles into the tumor parenchyma. Furthermore, blood flow within tumor vasculature is markedly heterogeneous compared to healthy tissues, leaving substantial regions under-perfused.86 For nanoparticles to reach these poorly accessed areas, they must navigate the challenging interstitial space, ultimately resulting in an uneven intratumoral distribution and diminished therapeutic efficacy.
Dense ECM
The extracellular matrix (ECM) primarily consists of a cross-linked network comprising collagens, elastin fibers, proteoglycans, glycosaminoglycans such as hyaluronic acid, and various glycoproteins including fibronectin and laminins.89 This intricate scaffold provides structural support, regulates cellular behavior through biochemical and biomechanical cues, and modulates tissue homeostasis. In tumor tissues, the ECM undergoes extensive remodeling driven by dysregulated stromal cells, resulting in greater compactness and stiffness compared to normal tissues. This pathological alteration, often termed desmoplasia, is primarily attributed to elevated deposition of fibrillar collagens (eg, types I, III, and V) and upregulated expression of lysyl oxidase (LOX), a key enzyme involved in ECM maturation.41 The abundant collagen in the tumor ECM establishes a substantial physical obstacle to nanoparticle translocation, limiting their diffusion through the dense interstitial spaces. Concurrently, LOX catalyzes the covalent cross-linking of collagen and elastin fibers, thereby further rigidifying the ECM, increasing its mechanical resistance, and exacerbating hurdles for effective nanoparticle transport and deep tissue penetration. Such stiffness not only impedes passive diffusion but also influences active cellular processes, potentially reducing nanoparticle uptake by tumor cells. These charge-based bindings may lead to nanoparticle sequestration at the ECM interface, promoting aggregation or premature clearance, and consequently diminishing intratumoral distribution and therapeutic efficacy.90–92
Current Methods for Enhancing the Accumulation of Nanomedicines in Tumors
Current nano-drug delivery systems primarily utilize two distinct pathways for tumor accumulation: passive and active targeting (Figure 3). Passive targeting relies on the EPR effect, which facilitates the passive extravasation and retention of nanoscale agents within tumor tissues (representative agents summarized in Table 1). In contrast, active targeting employs specific moieties—such as aptamers, ligands, or antibodies—anchored onto the nanocarrier surface to enable precise recognition of and binding to tumor cells (as detailed in Table 2).
 |
Figure 3 Active versus Passive Targeting Mechanisms in Antitumor Nanotherapy. Passive targeting exploits the Enhanced Permeability and Retention (EPR) effect: Nanocarriers circulate systemically, extravasate through hyperpermeable tumor vasculature, and accumulate within tumor tissue. Active targeting employs ligand-modified nanoplatforms that bind overexpressed tumor cell receptors, facilitating site-specific drug release or receptor-mediated endocytosis. Reproduced with permission from Ref.93 |
 |
Table 1 Passive Strategies |
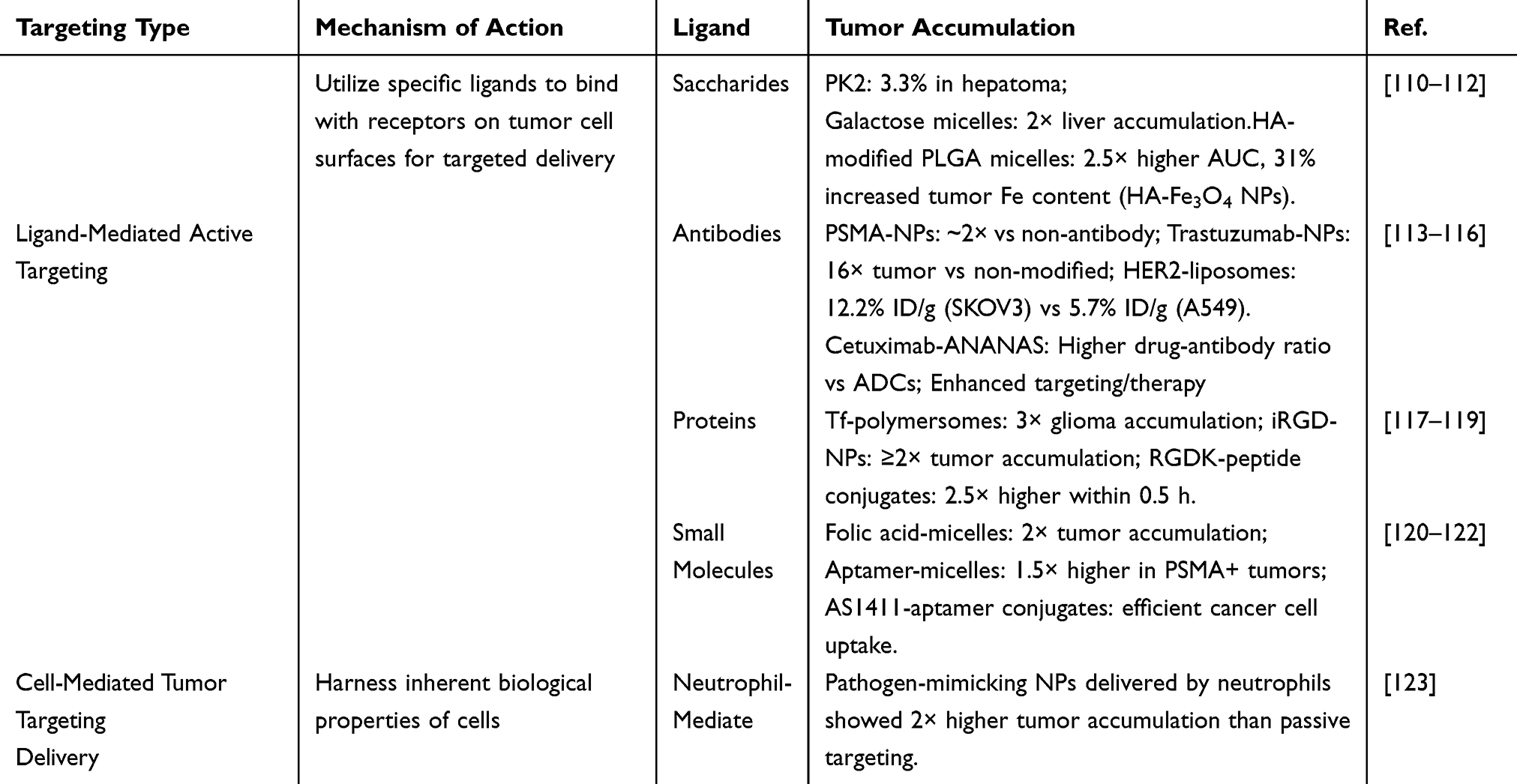 |
Table 2 Active Targeting of Nanoparticle Drug Delivery Systems |
Passive Strategy Based on EPR
Passive targeting via the EPR effect exploits the leaky vasculature and impaired lymphatic drainage in tumors to enable nanoparticle extravasation and retention. While this approach offers simplicity and broad applicability, its efficacy is highly variable due to tumor heterogeneity, with limited translation from preclinical models to humans where EPR is often less pronounced.124
Particle Size Modulation
The dimensions of nanoparticles can be adaptively modulated by various tumor-specific stimuli, such as overexpressed proteases, mild acidosis, or external photonic irradiation.94,95 Capitalizing on the elevated levels of matrix metalloproteinases (MMPs) in tumors,96,97 Hu et al engineered an Angio-DOX-DGL-GNP system responsive to MMP-2. Enzyme-triggered disassembly reduced the particle size from 185.7 nm to 55.6 nm, which significantly improved tumor penetration and yielded a 74.1% antitumor rate in 4T1 models.98 Similarly, acid-labile bonds (eg, imine and hydrazone) can be exploited for size transformation. Li et al developed PEG-b-PAEMA-PAMAM/Pt nanoparticles that rapidly shrink from 80 nm to 10 nm in the acidic tumor microenvironment. This system leverages the prolonged circulation of larger particles for initial accumulation and the superior penetrability of smaller ones for deep tissue infiltration, thereby enhancing treatment in poorly permeable BxPC-3 pancreatic tumors.99 Moving beyond single-stimulus responses, multi-stage systems have been designed for sequential activation. For instance, Nagel et al created MMP-7-responsive nanogels (pNG-DOX) that shrink from 100 nm to 50 nm upon enzyme exposure, with subsequent acid-enhanced drug release promoting tumor accumulation.100 In a parallel approach, Ruan et al developed gelatin-coated, contractile gold nanoparticles (G-AuNPs–DOX-PEG). These particles underwent MMP-2-induced size reduction from 186.5 nm to 59.3 nm for improved penetration, followed by acid-triggered hydrolysis to release DOX, eliciting a potent therapeutic response.100 Size-responsive strategies offer advantages in balancing circulation stability with deep tumor penetration, reducing off-target effects and enhancing therapeutic indices. However, challenges include potential premature activation in non-tumor acidic environments and manufacturing complexity, which may hinder scalability. Their potential lies in combination with imaging modalities for real-time monitoring, making them promising for heterogeneous tumors where uniform size optimization is insufficient.125
Surface Charge
Charge-reversal nanocarriers are engineered to maintain neutral or negative charges in the bloodstream, thereby extending their circulation time. Once they reach the tumor tissue, stimuli such as pH, enzymes, light, or temperature trigger a reversal, which significantly enhances their ability to penetrate deep into the tumor and promotes cellular internalization. Among strategies for modulating surface charge, pH-responsive systems represent the predominant category for achieving charge reversal. The mechanism of charge conversion in these systems generally operates via one of two pathways: the cleavage of specific chemical bonds or the protonation/deprotonation of ionizable functional groups. A schematic of the charge-reversal mechanism is provided in Figure 4. For instance, cationic lipid - polymer nanohybrids may be enveloped by the anionic PEI - DMMA through electrostatic forces, this coating allows the nanoparticles to circulate longer in the bloodstream. TME exhibits an acidic pH, a condition primarily driven by lactic acid generation from aerobic glycolysis and the release of protons during ATP hydrolysis.126 Consequently, while the physiological pH of blood is maintained at approximately 7.4, the interstitial pH in solid tumors is typically measured between 6.5 and 6.8. Upon entering the tumor microenvironment, the nanoparticles undergo a charge transition, reverting to a positive charge. This positive charge facilitates more efficient cellular uptake Biodistribution experiments revealed that charge-reversal nanoparticles accumulated at tumor sites at approximately twice the level of non-charge-reversal nanoparticles. Notably, there was little difference in their blood circulation profiles.105
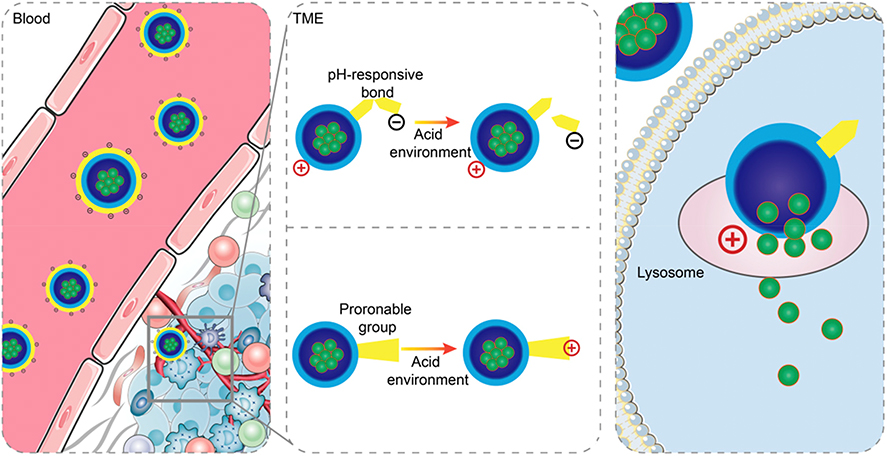 |
Figure 4 Two approaches for realizing positive charge reversal in acid-responsive charge-reversal systems: breakage of pH-responsive bonds or protonation. Reproduced with permission from Ref.127 |
Shaping Tumor Microenvironment
Before systemically administered nanoparticles can reach cancer cells, they must navigate two principal obstacles: the vascular endothelium and the dense tumor extracellular matrix (ECM). The aberrant tumor microenvironment (TME), marked by leaky vasculature, impaired lymphatic drainage, and a pathologically compacted ECM, exacerbates these delivery challenges. Therefore, strategies that modulate the TME—such as normalizing tumor vasculature or reprogramming the ECM—hold promise for enhancing nanoparticle infiltration and amplifying the Enhanced Permeability and Retention (EPR) effect. Supporting this approach, Ghandehari et al demonstrated that pre-conditioning tumors with gold nanoparticle-mediated photothermal therapy boosted the subsequent accumulation of polymeric nanocarriers in prostate tumors by approximately 1.5-fold.106 Radiation therapy has been demonstrated to induce endothelial cell apoptosis, which enhances tumor vascular permeability while reducing interstitial fluid pressure (IFP).107 Similarly, the sonoporation effect generated by high-intensity focused ultrasound can disrupt the integrity of the endothelial barrier, thereby facilitating nanoparticle extravasation. Theek et al validated this by showing that ultrasound treatment enhanced the EPR effect and consistently improved liposome accumulation in tumor models, regardless of their initial EPR status.108 In a related approach, the reactive oxygen species generated during photodynamic therapy also trigger vascular endothelial apoptosis, resulting in increased vascular leakage and improved nanoparticle delivery.109 Microenvironment modulation enhances EPR by transiently increasing vascular permeability, offering synergistic benefits with other therapies. Advantages include applicability to low-EPR tumors, but limitations encompass off-target damage to healthy tissues and dependency on external stimuli like ultrasound, which may require specialized equipment. This approach shows high promise for clinical translation, especially in combination therapies where it can amplify nanoparticle efficacy in resistant tumors.
Active Targeting of Nanomedicines: Ligand-Driven Recognition and Cellular Internalization
Nanoparticles offer a promising approach to circumvent biological barriers within the tumor microenvironment (TME) and improve the delivery of therapeutic agents to malignant tissues through passive or active targeting mechanisms (Figure 3). Passive accumulation relies on the enhanced permeability and retention (EPR) effect, whereby nanocarriers extravasate through the disorganized, leaky vasculature characteristic of tumors, resulting in non-selective deposition within the tumor interstitium. In contrast, active targeting involves functionalizing nanoparticle surfaces with specific ligands or molecular recognition elements that bind to biomarkers overexpressed on cancer cells, enabling more precise tissue- and cell-specific delivery (Figure 5). The selection of appropriate targeting ligands is guided by the differential expression and internalization capacity of corresponding receptors at the target site. Ideally, these molecular targets should be abundantly expressed on malignant cells while minimally present on healthy tissues to maximize specific homing of nanocarriers. Furthermore, stimuli-responsive nanoplatforms have emerged as an advanced active targeting strategy, as they maintain stability during circulation while triggering controlled drug release upon exposure to tumor-specific physiological signals. Various receptor types are frequently overexpressed on malignant cell membranes, providing a molecular basis for distinguishing them from normal counterparts. Advances in tumor proteomics and bioinformatics have significantly accelerated the identification of such tumor-associated receptors.128 By decorating nanocarrier surfaces with corresponding targeting ligands, researchers can achieve selective binding to cancer cells. Following ligand-receptor interaction, the nanoplatforms are typically internalized via receptor-mediated endocytosis, facilitating efficient intracellular delivery of encapsulated therapeutics. Consequently, receptor-targeted drug delivery approaches have garnered substantial research interest in recent years for their potential to enhance tumor-specific accumulation. Table 2 provides a representative list of receptors overexpressed across different cancer types along with their cognate ligands. The implementation of cell surface-specific targeting methodologies has significantly refined oncological therapeutic strategies, with several prominent examples detailed in subsequent sections.
 |
Figure 5 Design of nanoparticles for active targeting. (A) Monoclonal Antibodies, (B) Fabs, (C) Small Peptides, (D) Natural Proteins, (E) Aptamers, (F) Carbohydrates, (G) Small molecules. Reproduced with permission from Ref.129 |
Saccharide-Mediated Targeting
Saccharide-directed targeting strategies have emerged as a powerful approach for receptor-specific tumor accumulation. These utilize specific ligands, including N-Acetylgalactosamine (GalNAc) and hyaluronic acid (HA), to bind to receptors that are abundantly expressed on malignant cells. A prominent example is the GalNAc-decorated PolyHPMA-doxorubicin conjugate (PK2), which exhibited preferential uptake in hepatocellular carcinoma. Studies confirmed that approximately 3.3% of the injected PK2 dose localized to tumor sites within one day post-IV administration, a marked improvement over non-targeted polymers.110 In a similar approach, Paliwal et al engineered TPGS micelles functionalized with galactose, observing a doubling of hepatic drug deposition relative to their plain counterparts.111 Regarding HA-mediated strategies, research by Zhong et al indicated that HA-modified PLGA micelles possessed an area under the curve (AUC) 2.5 times greater and demonstrated superior tumor fluorescence intensity compared to uncoated particles. This was ascribed to synergistic effects involving CD44 receptor-driven cellular uptake and prolonged systemic circulation. Corroborating this, Dai et al utilized HA-coated Fe3O4 nanoparticles and documented a 31% rise in intra-tumoral iron levels versus uncoated controls, providing direct evidence that HA-CD44 binding facilitates improved tumor retention and accumulation.112 Despite the marked potential of saccharide ligands to boost the specificity and efficiency of drug delivery, their clinical translation necessitates a concerted effort to maximize targeting precision and minimize off-target accumulation.
Antibody-Mediated Targeting
Through active targeting mechanisms, ligands conjugated to antibodies significantly improve the specificity and extent of nanoparticle accumulation in tumors. For instance, HER2 antibody-functionalized poly(lactide-co-glycolide) (PLG) nanoparticles loaded with DM1 exhibited approximately sixfold greater tumoral deposition versus non-targeted IgG counterparts at 4 h post-intravenous administration.130 The modification of nanoparticles with targeting ligands significantly enhances their tumor-specific delivery. Trastuzumab-functionalized, rod-like bismuth-based nanoparticles achieved a 16-fold increase in bismuth concentration at tumor sites relative to their non-targeted counterparts, concurrently reducing off-target deposition in peritumoral and pulmonary tissues.113 The critical importance of receptor specificity was further evidenced by HER2-targeted liposomes, which accumulated at 12.2% ID/g in HER2-positive SKOV3 tumors, a level more than double the 5.7% ID/g observed in HER2-negative A549 models.114 This targeting precision, pivotal for therapeutic efficacy, can be engineered through specific surface modifications.57 Roncato et al developed an anticancer nanoplatform by integrating cetuximab, an anti-EGFR antibody, into avidin-nucleic-acid nanoassemblies (ANANAS).115 Their methodology centered on a poly-avidin core that was surface-modified with biotinylated targeting ligands, which was then used to assess the efficacy of antibody-drug conjugates (ADCs)—cornerstone agents in personalized oncology. The study demonstrated that the cetuximab-directed ANANAS system attained a superior drug-to-antibody ratio relative to traditional ADCs. This improvement is attributed to the high-affinity avidin-biotin interaction, which substantially augments the drug-carrying capability of the nanocarrier. These findings demonstrate that antibody-directed nanoengineering effectively circumvents the constraints of passive targeting, thereby establishing a framework for designing more potent and tumor-selective nanomedicines.
Protein- and Peptide-Directed Targeting
Active targeting strategies employing proteins and peptides enhance tumor accumulation via receptor-specific recognition and subsequent cellular internalization. For instance, biodegradable polymersomes conjugated with transferrin (Tf) exhibit a threefold increase in glioma accumulation relative to non-targeted counterparts, facilitated by Tf receptor-mediated endocytosis—a process upregulated in malignant glioma cells—and Tf’s capacity to traverse the blood–brain barrier.117 Similarly, polycaprolactone–polyvinylpyrrolidone (PCL-PVP) nanoparticles modified with the iRGD peptide achieve at least a twofold enhancement in tumor accumulation through dual actions: integrin receptor engagement for primary targeting and tumor-penetrating motifs that promote extravascular dissemination. Although iRGD modification marginally shortens plasma half-life, it fosters deeper stromal infiltration by perturbing integrin-dependent cell adhesion.118 In another example, micelles comprising SN-38 prodrug conjugated to the neuropilin-1-targeting peptide (RGDK) display a 2.5-fold elevation in tumor accumulation (5% injected dose per gram tissue versus 2% injected dose per gram tissue) at 0.5 hours post-injection compared with unmodified oligo(ethylene glycol) micelles. This accelerated uptake stems from selective neuropilin-1 receptor affinity on tumor endothelial and neoplastic cells, culminating in swift clathrin-mediated endocytosis.119 A class of fluorescent peptide nanoparticles (f-PNPs), self-assembled from cyclo[-(d-Ala-L-Glu-D-Ala-L-Trp)2-] cyclopeptides, has recently been engineered and assessed for its potential in both drug delivery and bioimaging. These f-PNPs have undergone comprehensive characterization of their nanoscale architecture, photonic traits, therapeutic efficacy, and biocompatibility. To confer specific targeting toward endometrial cancer (EC) cells, the nanoplatform was functionalized with RGD peptides, which recognize overexpressed αvβ3 integrins (Figure 6a). Subsequently, the chemotherapeutic agent epirubicin (EPI) was loaded into these RGD-conjugated f-PNPs (RGD-f-PNPs) through π-π stacking and electrostatic interactions. As depicted in Figure 6b, the resulting RGD-f-PNPs/EPI complexes preferentially accumulate in tumor tissue, leveraging both the enhanced permeability and retention (EPR) effect and active targeting. Upon binding to αvβ3 integrins, the complexes are internalized by EC cells, leading to intracellular EPI release and its eventual nuclear accumulation to induce cytotoxicity. The dual targeting and enhanced uptake capabilities were confirmed in EC cell cultures, while targeted delivery and drug tracking were validated in EC xenograft models. Collectively, in vitro and in vivo studies demonstrate that the RGD-f-PNPs/EPI system significantly augments anticancer efficacy while minimizing systemic toxicity by localizing drug action.131 The peptide-ligand interaction boosts initial targeting and promotes subsequent drug release within the tumor microenvironment, thereby synergistically improving both accumulation kinetics and the resulting therapeutic outcome.
 |
Figure 6 (A) The RGD-f-PNPs were initially fabricated through the co-assembly of Zn2⁺ ions and cyclo[-(d-Ala-L-Glu-D-Ala-L-Trp)2-] peptides, followed by surface conjugation with RGD ligands. EPI was subsequently loaded into the resulting nanoparticles via π-π stacking and electrostatic attractions. (B) The obtained EPI-loaded RGD-f-PNPs enable targeted imaging and eradication of EC cells by leveraging active targeting and improved tissue penetration. These nanoconjugates preferentially localize in tumor tissue over healthy tissue, a phenomenon driven by the EPR effect. Furthermore, RGD ligands on the nanoconjugates specifically recognize and bind to overexpressed αvβ3 integrin receptors on EC cells, promoting cellular internalization. Reproduced with permission from Ref.131 |
Small Molecule- and Aptamer-Mediated Targeting
Active targeting strategies employing small molecules or aptamers enhance tumor-specific delivery by exploiting their high affinity for receptors uniquely expressed on cancer cells. A prime example is folic acid-functionalized polymeric micelles, which achieve a two-fold increase in tumor accumulation compared to non-targeted versions. This enhancement stems from folate receptor-mediated endocytosis in tumors that overexpress this receptor. Supporting in vivo evidence shows that the presence of folic acid ligands promotes specific binding and significantly prolongs the retention of drugs like doxorubicin within tumor tissue.120 Similarly, aptamer-guided systems demonstrate superior targeting. For instance, micelles conjugated with the A10 aptamer accumulate 1.5-fold more in PSMA-positive tumors than their non-targeted counterparts, as the aptamer facilitates receptor-mediated internalization and bypasses the inefficiency of passive diffusion.121 The AS1411 aptamer operates on a similar principle, enabling efficient cellular uptake of drug conjugates by binding to nucleolin receptors on the cancer cell surface, thus ensuring highly specific intracellular drug delivery.122 Collectively, these approaches underscore the distinct strengths of both small molecules and aptamers: folic acid capitalizes on inherent metabolic pathways, while aptamers provide programmable recognition with low immunogenic risk. By honing in on unique molecular fingerprints of tumors, both strategies counter the problem of tumor heterogeneity and mark a significant step toward precision nanomedicine. Small molecules and aptamers are advantageous for their stability, low cost, and ease of conjugation, but aptamers may degrade enzymatically, and small molecules risk non-specific uptake. Their potential is significant in combinatorial therapies, with aptamers particularly suited for RNA-based diagnostics integration.132
Ligand-directed approaches collectively outperform passive targeting in specificity but require biomarker validation to avoid inefficacy in low-expression tumors. Challenges like manufacturing scalability and ligand desorption persist, yet their application potential is vast in precision oncology, especially when hybridized with passive elements.
Cell-Mediated Tumor Targeting Delivery Systems
In recent years, cell-based drug delivery systems have continued to attract substantial interest owing to their unique biological attributes and intrinsic functionalities. A broad spectrum of cellular carriers—including red blood cells (RBCs), T lymphocytes, stem cells, macrophages, neutrophils, natural killer (NK) cells, and adipocytes—has been rigorously explored for drug delivery purposes.133 Tumor-specific delivery is achieved either through direct nanoparticle loading into cells or via biomimetic engineering, such as membrane coatings that confer immune evasion and tropism. Neutrophils, comprising the majority of circulating leukocytes, exemplify this strategy due to their upregulated integrin and chemokine receptors (eg, CXCR2), which drive rapid infiltration into inflammatory tumor niches. In a seminal 2017 study, Li et al employed subcutaneous nano-CpG adjuvants to immunomodulate neutrophils, priming them for enhanced tumor homing; isolated neutrophils from treated mice exhibited significantly greater infiltration than controls, laying the foundation for neutrophil-mediated nanodrug transport.134 Extending this, Wang et al developed pathogen-mimetic nanoparticles internalized by circulating neutrophils, enabling “hitchhiking” to inflamed tumors and subsequent payload release; this yielded approximately twofold higher nanoparticle retention compared to passive EPR alone.123 In a recent innovative study, Wang et al established a “cell-hitchhiking” strategy for tumor targeting. They designed gold nanoparticles that mimic bacteria, enabling their uptake by phagocytic immune cells. These cells then naturally migrated to tumor tissues, guided by their intrinsic inflammatory tropism, thereby delivering the nanoparticles. The accumulation of these cell-based carriers was further enhanced when combined with a preconditioning photothermal treatment, highlighting a promising new direction for targeted therapy.135 Contemporary innovations have refined these neutrophil-centric systems for superior tumor penetration and therapeutic indices. For instance, neutrophil membrane-coated poly(lactic-co-glycolic acid) nanoparticles exploit ICAM-1 mimicry to promote endothelial adhesion and endocytosis, achieving 2- to 3-fold greater accumulation in breast cancer xenografts versus uncoated particles, coupled with augmented T-cell infiltration and 40% survival prolongation.129 Similarly, neutrophils internalizing paclitaxel-loaded liposomes via CXCR2 homing have traversed the blood-brain barrier in glioma models, delivering over 1162-fold higher drug levels relative to free paclitaxel, with 80% enhanced accumulation and 50% extended median survival.136 Macrophage-based “cellular backpacks” loaded with immunomodulatory nanoparticles similarly promote sustained intratumoral retention,137 while MSC-derived carriers exhibit innate tropism to cancer stem cell niches, enabling low-dose delivery with 8-fold dose reductions in lung metastases.138 These cell-mediated nanotherapeutics not only surmount physicochemical barriers but also reprogram the tumor microenvironment to foster immunogenicity, underscoring their translational promise for recalcitrant solid tumors despite challenges in scalability and biodistribution consistency.
Translational Challenges: Discrepancies Between Preclinical Animal Models and Human Clinical Trials in Nanomedicine Tumor Accumulation Strategies
Despite promising results in preclinical animal models, the translation of nanomedicine accumulation strategies to human clinical trials has been fraught with challenges, often resulting in suboptimal efficacy. Passive and active targeting approaches, particularly those relying on the EPR effect, demonstrate robust tumor accumulation in rodents, with accumulation rates frequently exceeding 5–10% of the injected dose.139 However, in humans, these strategies typically achieve only 0.5–1% dose accumulation, leading to therapeutic failures in numerous Phase II/III trials.140 This section critically examines the underlying reasons for these discrepancies, drawing on tumor biology, physiological differences, and methodological limitations to inform future design improvements.
Tumor Heterogeneity and EPR Variability
One primary reason for translational failure is the pronounced heterogeneity of human tumors compared to animal models. In preclinical studies, xenograft or syngeneic mouse models often feature rapidly growing tumors with highly permeable, immature vasculature, amplifying the EPR effect and facilitating passive accumulation. These models exhibit uniform leaky vessels due to induced angiogenesis, enabling nanoparticles to extravasate efficiently. In contrast, human tumors display significant inter- and intra-patient variability in EPR, influenced by tumor type, stage, location, and microenvironmental factors such as fibrosis and necrosis.141 For instance, pancreatic and desmoplastic tumors in humans often have dense stroma and compressed vessels, reducing permeability and retention, whereas rodent models rarely recapitulate this complexity.142 Clinical imaging studies confirm that only a subset of patients (eg, 20–30% in some cohorts) exhibit detectable EPR, underscoring the need for patient stratification biomarkers absent in animal testing.143
Physiological and Immunological Differences
Physiological scaling issues further exacerbate discrepancies. Animal models, particularly mice, have faster metabolic rates, shorter circulation times, and smaller body sizes, which alter nanoparticle pharmacokinetics. Doses normalized by body weight in rodents often overestimate human-equivalent exposure, leading to inflated efficacy signals.144 Moreover, many preclinical tumors are subcutaneously implanted for ease of monitoring, differing from orthotopic human sites where interstitial fluid pressure (IFP) is elevated, impeding nanoparticle penetration.145 Immunological factors also play a critical role. Immunodeficient mice (eg, nude or SCID strains) commonly used in xenograft studies lack robust immune responses, allowing unhindered nanoparticle circulation and tumor homing. In humans, the mononuclear phagocyte system (MPS) rapidly clears nanoparticles, reducing bioavailability.146 Active targeting ligands, effective in immune-compromised models, may trigger immunogenicity or opsonization in patients, further diminishing accumulation.
Methodological and Trial Design Limitations
Preclinical studies often employ homogeneous tumor cohorts with early-stage lesions, contrasting with heterogeneous, advanced-stage human cancers in trials. This mismatch contributes to overestimation of benefits; for example, active targeting via antibodies shows 2–5 fold enhancement in mice but marginal improvements in humans due to receptor downregulation in late-stage tumors.147 Additionally, animal experiments rarely account for comorbidities or prior treatments common in patients, which can alter vascular integrity and EPR. Cell-mediated strategies, while promising in rodents for exploiting inflammation, face scalability issues in humans, including cell sourcing, viability, and regulatory hurdles for autologous therapies. Clinical failures highlight the need for better models, such as patient-derived xenografts (PDXs) or organoids, to bridge this gap.148
In summary, the success of nanomedicine strategies in animal models stems from simplified tumor biology and exaggerated EPR, while human failures arise from biological complexity and inadequate translation. Future prospects lie in personalized approaches, such as EPR imaging for patient selection and hybrid strategies combining microenvironment modulation with active targeting. Addressing these challenges through advanced preclinical models and biomarker-driven trials is essential for clinical success.149,150
Future Perspectives
Rational design of cancer nanomedicines for advancing clinical translation necessitates a synergistic optimization of physicochemical properties, exploitation of biological transport mechanisms, and rigorous mitigation of nanotoxicity—all of which collectively govern tumor drug delivery efficacy and clinical safety. For physicochemical tuning, nanoparticle size should be optimized within the 12–60 nm range to maximize the EPR effect for solid tumor targeting, while mitigating size-dependent toxicity: ultrasmall nanoparticles (<10 nm) evade renal clearance but trigger reactive oxygen species (ROS) bursts through mitochondrial respiratory chain disruption, leading to oxidative stress and cellular apoptosis.151 Conversely, larger particles (>400 nm) provoke splenic clearance and heightened phagocytic activation, exacerbating systemic inflammation.152 Furthermore, as evidenced in lip osomal formulations, sizes around 100 nm, akin to Doxil/Caelyx, facilitate accumulation via leaky tumor vasculature but require careful assessment to avoid off-target uptake in liver and spleen, potentially inducing complement activation and hypersensitivity.153 Non-spherical nanoparticles (eg, rods, discs) improve tumor margination and penetration via enhanced vascular interactions but elevate toxicity risks, including amplified macrophage-mediated inflammation relative to spherical counterparts, driven by increased cellular uptake and activation of NF-κB/MAPK pathways that promote pro-inflammatory cytokine release (eg, TNF-α, IL-6).154 Moreover, these shapes can induce genotoxicity by direct DNA interaction or ROS-mediated damage, potentially causing chromosomal aberrations and epigenetic alterations.155 Surface engineering necessitates careful trade-offs in nanomedicine design. Neutral or PEGylated surfaces can reduce clearance by the mononuclear phagocyte system (MPS) and limit opsonization.156 However, repeated administration may induce anti-PEG antibodies, leading to accelerated blood clearance and potential hypersensitivity reactions.157 In contrast, cationic surfaces promote transvascular flux and cellular uptake but often cause plasma protein aggregation, oxidative stress through electrostatic membrane disruptions, and immunotoxicity via complement activation and cytokine release.158 Beyond physicochemical modifications, leveraging biological transport mechanisms offers promising delivery strategies. For instance, validating the role of nanoparticle-transcytosing endothelial cells (N-TECs) in soft nanomedicine transport—and their prevalence in human tumors—could enable targeted delivery using ligand-modified nanoparticles or through induced transcytosis via endothelial gene transfection. Similarly, repolarizing tumor-associated macrophages (TAMs) or other immune cells toward anti-tumor phenotypes may facilitate active nanoparticle transport into tumors.159 Addressing the heterogeneity of nanoparticle tumor targeting is critical for improving clinical efficacy. Biomarker-guided strategies are indispensable in this regard. Non-invasive imaging combined with companion nanodiagnostics or nanotheranostics allows real-time visualization and quantification of targeting efficiency, aiding in patient stratification. Histopathological biomarkers—such as blood vessel density or TAM presence in biopsies—can also predict nanoformulation accumulation.160 For example, Figure 7 outlines an experimental protocol for identifying tissue biomarkers correlated with nanomedicine accumulation. It includes a schematic of tumor models and biomarker assessment (panel a), fluorescence reflectance imaging of DY750-labeled PHPMA showing variable accumulation across tumor types (panel b), longitudinal CT–FMT visualization in organs (panel c), and quantification of the percentage of injected dose normalized to tumor volume over time (panel d), revealing significant inter-tumor differences.161 Throughout the design process, comprehensive nanotoxicity assessment remains essential. An integrated design framework—balancing physicochemical customization, biological transport mechanisms, biomarker-guided patient selection, and systematic nanotoxicity control—is vital to minimizing clinical trial failures and realizing the full potential of cancer nanomedicines. Table 3 summarizes key candidates in Phase I–III trials, highlighting nanocarrier designs, targeting strategies, and preliminary clinical outcomes that reflect the crucial balance between efficacy and safety required for regulatory approval.
 |
Figure 7 (A) Schematic overview of the experimental workflow for identifying tumor tissue biomarkers associated with nanocarrier accumulation. The tumor-targeting efficiency of a prototypical polymer nanocarrier (PHPMA) was quantified via CT-FMT across three different mouse models. (B) Serial in vivo fluorescence reflectance imaging (FRI) of DY750-labeled PHPMA accumulation in A431 tumor-bearing mice. (C and D) Corresponding longitudinal CT-FMT reconstructions. *p-value≤0.05,**p-value≤0.01. Reproduced with permission from Ref.161 |
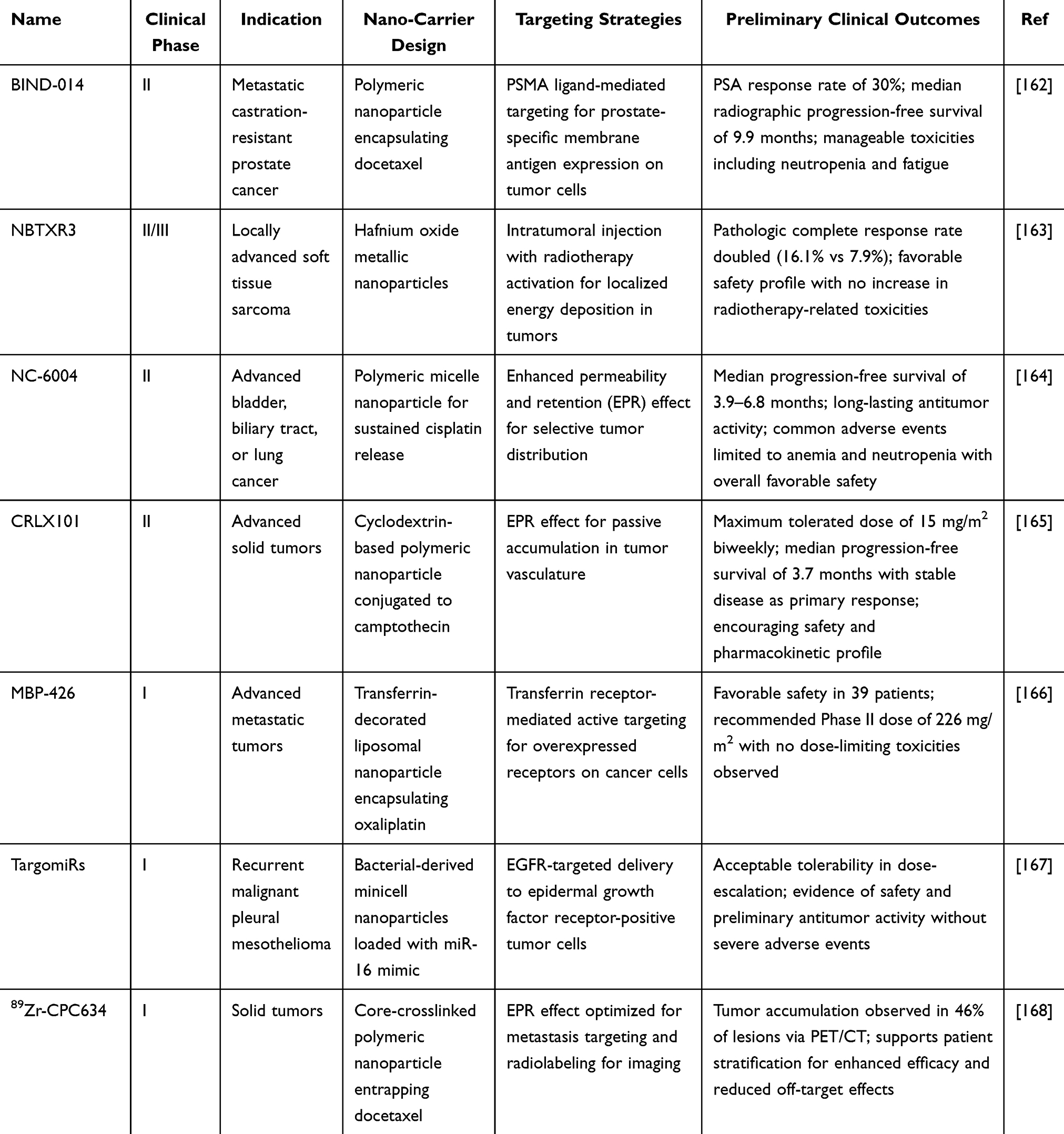 |
Table 3 Key Candidate Agents in Phase I–III Clinical Trials |
Conclusion
The clinical translation of nanomedicine is fundamentally constrained by its inefficient accumulation in solid tumors, a central challenge stemming from physiological barriers, a complex tumor microenvironment, and profound intertumoral EPR heterogeneity. This review has provided a mechanistic framework for understanding this issue, evaluating both passive strategies for optimized biodistribution and active targeting for enhanced specificity. The bidirectional relationship between nanodrug accrual and TME modulation necessitates next-generation designs that harmonize therapeutic impact with biosafety. Looking forward, the field must embrace a new paradigm centered on smart, multi-stage nanocarriers and personalized approaches based on biomarker profiling. Overcoming the longstanding challenges of EPR variability and delivery route limitations is the definitive path toward nanotherapeutics that fulfill their clinical promise in oncology.
Abbreviations
5-hmC, 5-hydroxymethylcytosine; 5-mC, 5-methylcytosine; ADCs, Antibody-Drug Conjugates; AECs, Airway Epithelial Cells; ANANAS, Avidin-Nucleic-Acid Nanoassemblies; AP-1, Activator Protein-1; AT, Active targeting; AUC, Area Under the Curve; BBB, Blood-Brain Barrier; CAFs, Cancer-Associated Fibroblasts; CD44, Cluster of Differentiation 44; CLSM, Confocal Laser Scanning Microscopy; CNS, Central Nervous System; CNTs, Carbon Nanotubes; CpG, Cytosine-phosphate-Guanine; CT, Computed Tomography; CXCR2, C-X-C Chemokine Receptor Type 2; Dnmt, DNA methyltransferase; DOX, Doxorubicin; ECM, Extracellular Matrix; EGF, Epidermal Growth Factor; EGFR, Epidermal Growth Factor Receptor; EPI, Epirubicin; EPR, Enhanced Permeability and Retention; ERK, Extracellular Signal-Regulated Kinase; FMT, Fluorescence Molecular Tomography; FRI, Fluorescence Reflectance Imaging; GalNAc, N-Acetylgalactosamine; GIT, Gastrointestinal Tract; HA, Hyaluronic Acid; HDAC, Histone Deacetylase; HER2, Human Epidermal Growth Factor Receptor 2; ICAM-1, Intercellular Adhesion Molecule 1; IFP, Interstitial Fluid Pressure; IL, Interleukin; iRGD, Tumor-penetrating peptide; IV, Intravenous; LOX, Lysyl Oxidase; MAPK, Mitogen-Activated Protein Kinase; MBD2, Methyl-CpG-Binding Domain Protein 2; MMPs, Matrix Metalloproteinases; MPS, Mononuclear Phagocyte System; NADPH, Nicotinamide Adenine Dinucleotide Phosphate; NF-κB, Nuclear Factor kappa B; NK cells, Natural Killer cells; NPs, Nanoparticles; N-TECs, Nanoparticle-Transcytosing Endothelial Cells; PAMAM, Polyamidoamine; PCL, Poly(ε-caprolactone); PDXs, Patient-Derived Xenografts; PDT, Photodynamic Therapy; PEG, Polyethylene Glycol; PEI, Polyethylenimine; PHPMA, Poly(N-(2-hydroxypropyl)methacrylamide); PK2, PolyHPMA-Doxorubicin conjugate; PLA, Polylactic Acid; PLGA, Poly(lactic-co-glycolic acid); PSA, Prostate-Specific Antigen; PSMA, Prostate-Specific Membrane Antigen; PVP, Polyvinylpyrrolidone; RBCs, Red Blood Cells; RES, Reticuloendothelial System; RGDK, Neuropilin-1 receptor-targeting peptide; RNS, Reactive Nitrogen Species; ROS, Reactive Oxygen Species; SCID, Severe Combined Immunodeficiency; SINE, Short Interspersed Nuclear Element; STAT, Signal Transducer and Activator of Transcription; SWCNTs/MWCNTs, Single-/Multi-Walled Carbon Nanotubes; TAMs, Tumor-Associated Macrophages; TAT, Trans-Activator of Transcription; TDD, Transdermal Drug Delivery; Tf, Transferrin; TLR, Toll-Like Receptor; TME, Tumor Microenvironment; TNF-α, Tumor Necrosis Factor-alpha; TPGS, Tocopheryl Polyethylene Glycol Succinate; VEGFA, Vascular Endothelial Growth Factor A; WNT, Wingless-type signaling pathway protein.
Author Contributions
Long Chen and Xiang Deng are co-first authors who contributed equally to this study. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Sponsored by National Natural Science Foundation of China (No. 82504308), University-Level Natural Science Foundation General Project of Chengdu Medical College (2024CDYXY-01), Clinical Science Research Foundation of Chengdu Medical College & the First Affiliated Hospital of Chengdu Medical College (24LHLNYX1-08), Clinical Science Research Foundation of Chengdu Medical College & Nanbu People’s Hospital (2024LHFBM1-04) and Clinical Science Research Foundation of Chengdu Medical College & Chengdu Pidu People’s Hospital (2024LHFYSZ1-41).
Disclosure
The authors declare that there are no competing interests associated with the paper.
References
1. Li T, Li J, Chen Z, et al. Glioma diagnosis and therapy: current challenges and nanomaterial-based solutions. J Control Release. 2022;352:338–26. doi:10.1016/j.jconrel.2022.09.065
2. Mitchell MJ, Billingsley MM, Haley RM, et al. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
3. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med. 2019;4(3):e10143. doi:10.1002/btm2.10143
4. Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
5. Rosenblum D, Joshi N, Tao W, et al. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi:10.1038/s41467-018-03705-y
6. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nature Rev Mater. 2016;1(5):16014. doi:10.1038/natrevmats.2016.14
7. Thanki K, Gangwal RP, Sangamwar AT, et al. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. doi:10.1016/j.jconrel.2013.04.020
8. Prausnitz MR, Langer R. Transdermal drug delivery. Nature Biotechnol. 2008;26(11):1261–1268. doi:10.1038/nbt.1504
9. Tan ML, Choong PF, Dass CR. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides. 2010;31(1):184–193. doi:10.1016/j.peptides.2009.10.002
10. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392.
11. Wu J. The enhanced permeability and retention (EPR) Effect: the significance of the concept and methods to enhance its application. J Pers Med. 2021;11(8):771. doi:10.3390/jpm11080771
12. Kwak B, Ozcelikkale A, Shin CS, et al. Simulation of complex transport of nanoparticles around a tumor using tumor-microenvironment-on-chip. J Control Release. 2014;194:157–167. doi:10.1016/j.jconrel.2014.08.027
13. Chan WCW. Principles of nanoparticle delivery to solid tumors. BME Front. 2023;4:0016. doi:10.34133/bmef.0016
14. Minh Hoang CN, Nguyen SH, Tran MT. Nanoparticles in cancer therapy: strategies to penetrate and modulate the tumor microenvironment – a review. Smart Mater Med. 2025;6(2):270–284. doi:10.1016/j.smaim.2025.07.004
15. Quadir SS, Saharan V, Choudhary D, et al. Nano-strategies as oral drug delivery platforms for treatment of cancer: challenges and future perspectives. AAPS Pharm Sci Tech. 2022;23(5):152. doi:10.1208/s12249-022-02301-0
16. Plapied L, Duhem N, Des Rieux A, et al. Fate of polymeric nanocarriers for oral drug delivery. Curr Opin Colloid Interface Sci. 2011;16(3):228–237. doi:10.1016/j.cocis.2010.12.005
17. Allémann E, Leroux J, Gurny R. Polymeric nano- and microparticles for the oral delivery of peptides and peptidomimetics. Adv Drug Deliv Rev. 1998;34(2–3):171–189. doi:10.1016/S0169-409X(98)00039-8
18. Wilczewska AZ, Niemirowicz K, Markiewicz KH, et al. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64(5):1020–1037. doi:10.1016/S1734-1140(12)70901-5
19. Agrawal U, Sharma R, Gupta M, et al. Is nanotechnology a boon for oral drug delivery? Drug Discov Today. 2014;19(10):1530–1546. doi:10.1016/j.drudis.2014.04.011
20. Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64(6):557–570. doi:10.1016/j.addr.2011.12.009
21. Makhlof A, Tozuka Y, Takeuchi H. Design and evaluation of novel pH-sensitive chitosan nanoparticles for oral insulin delivery. Eur J Pharm Sci. 2011;42(5):445–451. doi:10.1016/j.ejps.2010.12.007
22. Pridgen EM, Alexis F, Farokhzad OC. Polymeric nanoparticle technologies for oral drug delivery. Clin Gastroenterol Hepatol. 2014;12(10):1605–1610. doi:10.1016/j.cgh.2014.06.018
23. Kim JH, Kim Y-S, Park K, et al. Self-assembled glycol chitosan nanoparticles for the sustained and prolonged delivery of antiangiogenic small peptide drugs in cancer therapy. Biomaterials. 2008;29(12):1920–1930. doi:10.1016/j.biomaterials.2007.12.038
24. Lee H, Lee K, Park TG. Hyaluronic acid-paclitaxel conjugate micelles: synthesis, characterization, and antitumor activity. Bioconjug Chem. 2008;19(6):1319–1325. doi:10.1021/bc8000485
25. Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11(19–20):905–910. doi:10.1016/j.drudis.2006.08.005
26. Zhang Z, Shan H, Chen L, et al. Synthesis of pH-responsive starch nanoparticles grafted poly (l-glutamic acid) for insulin controlled release. Eur Polym J. 2013;49(8):2082–2091. doi:10.1016/j.eurpolymj.2013.04.032
27. Zhao S, Li J, Zhou Y, et al. Lipid nanoparticles-encapsulated YF4: a potential therapeutic oral peptide delivery system for hypertension treatment. Front Pharmacol. 2019;10:102. doi:10.3389/fphar.2019.00102
28. Chenthamara D, Subramaniam S, Ramakrishnan SG, et al. Therapeutic efficacy of nanoparticles and routes of administration. Biomater Res. 2019;23(1):20. doi:10.1186/s40824-019-0166-x
29. Prow TW, Grice JE, Lin LL, et al. Nanoparticles and microparticles for skin drug delivery. Adv Drug Deliv Rev. 2011;63(6):470–491. doi:10.1016/j.addr.2011.01.012
30. Silva GA. Introduction to nanotechnology and its applications to medicine. Surg Neurol. 2004;61(3):216–220. doi:10.1016/j.surneu.2003.09.036
31. Lane ME. In vitro permeation testing for the evaluation of drug delivery to the skin. Eur J Pharm Sci. 2024;201:106873. doi:10.1016/j.ejps.2024.106873
32. Desai P, Patlolla RR, Singh M. Interaction of nanoparticles and cell-penetrating peptides with skin for transdermal drug delivery. Mol Member Biol. 2010;27(7):247–259. doi:10.3109/09687688.2010.522203
33. Attama A, Ogbonna JDN, Uchechi O. Nanoparticles for dermal and transdermal drug delivery. In: Sezer AD, editor. Application of Nanotechnology in Drug Delivery. London: IntechOpen; 2014.
34. Lademann J, Richter H, Teichmann A, et al. Nanoparticles--an efficient carrier for drug delivery into the hair follicles. Eur J Pharm Biopharm. 2007;66(2):159–164. doi:10.1016/j.ejpb.2006.10.019
35. Kraft JC, Freeling JP, Wang Z, et al. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci. 2014;103(1):29–52. doi:10.1002/jps.23773
36. Bladé J, Sonneveld P, Miguel JFS, et al. Efficacy and safety of pegylated liposomal Doxorubicin in combination with bortezomib for multiple myeloma: effects of adverse prognostic factors on outcome. Clin Lymphoma Myeloma Leuk. 2011;11(1):44–49. doi:10.3816/CLML.2011.n.005
37. Barenholz Y. Doxil®--the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi:10.1016/j.jconrel.2012.03.020
38. Mathur V, Satrawala Y, Rajput M. Physical and chemical penetration enhancers in transdermal drug delivery system. Asian J Pharm. 2010;4.
39. Moammeri A, Chegeni MM, Sahrayi H, et al. Current advances in niosomes applications for drug delivery and cancer treatment. Mater Today Bio. 2023;23:100837. doi:10.1016/j.mtbio.2023.100837
40. Zhan B, Wang J, Li H, et al. Ethosomes: a promising drug delivery platform for transdermal application. Chemistry. 2024;6(5):993–1019. doi:10.3390/chemistry6050058
41. Barua S, Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: a review of current status and future prospects. Nano Today. 2014;9(2):223–243. doi:10.1016/j.nantod.2014.04.008
42. Wilson RJ, Li Y, Yang G, et al. Nanoemulsions for drug delivery. Particuology. 2022;64:85–97. doi:10.1016/j.partic.2021.05.009
43. Chen Y, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 2012;64(7):640–665. doi:10.1016/j.addr.2011.11.010
44. Bicker J, Alves G, Fortuna A, et al. Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: a review. Eur J Pharm Biopharm. 2014;87(3):409–432. doi:10.1016/j.ejpb.2014.03.012
45. Katz JS, Slika H, Sattari SA, et al. Overcoming the blood-brain barrier for drug delivery to the brain. ACS Omega. 2025;10(30):32544–32563. doi:10.1021/acsomega.5c00364
46. Jiao Y, Yang L, Wang R, et al. Drug delivery across the blood-brain barrier: a new strategy for the treatment of neurological diseases. Pharmaceutics. 2024;16(12):1611. doi:10.3390/pharmaceutics16121611
47. Wei M, Qian N, Gao X, et al. Single-particle imaging of nanomedicine entering the brain. Proc Natl Acad Sci. 2024;121(5):e2309811121. doi:10.1073/pnas.2309811121
48. Posadas I, Monteagudo S, Ceña V. Nanoparticles for brain-specific drug and genetic material delivery, imaging and diagnosis. Nanomedicine. 2016;11(7):833–849. doi:10.2217/nnm.16.15
49. Masserini M. Nanoparticles for brain drug delivery. ISRN Biochem. 2013;2013:238428. doi:10.1155/2013/238428
50. Shadab A, Farokhi S, Fakouri A, et al. Hydrogel-based nanoparticles: revolutionizing brain tumor treatment and paving the way for future innovations. Eur J Med Res. 2025;30(1):71. doi:10.1186/s40001-025-02310-2
51. Ding L, Kshirsagar P, Agrawal P, et al. Crossing the blood–brain barrier: innovations in receptor- and transporter-mediated transcytosis strategies. Pharmaceutics. 2025;17(6):706. doi:10.3390/pharmaceutics17060706
52. Bogadi S, Bhaskaran M, Ravichandran V, et al. Functionalized nanoparticles: a promising approach for effective management of alzheimer’s disease. Mol Neurobiol. 2025;62(8):10915–10934. doi:10.1007/s12035-025-04917-2
53. Parker SE, Davey PG. Pharmacoeconomics of intravenous drug administration. Pharmacoeconomics. 1992;1(2):103–115. doi:10.2165/00019053-199201020-00007
54. Kalepu S, Nekkanti V. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm Sin B. 2015;5(5):442–453. doi:10.1016/j.apsb.2015.07.003
55. Wong J, Brugger A, Khare A, et al. Suspensions for intravenous (IV) injection: a review of development, preclinical and clinical aspects. Adv Drug Deliv Rev. 2008;60(8):939–954. doi:10.1016/j.addr.2007.11.008
56. Sasikumar A, Kamalasanan K. Nanomedicine for prostate cancer using nanoemulsion: a review. J Control Release. 2017;260:111–123. doi:10.1016/j.jconrel.2017.06.001
57. Dias ML, Carvalho JP, Rodrigues DG, et al. Pharmacokinetics and tumor uptake of a derivatized form of paclitaxel associated to a cholesterol-rich nanoemulsion (LDE) in patients with gynecologic cancers. Cancer Chemother Pharmacol. 2007;59(1):105–111. doi:10.1007/s00280-006-0252-3
58. Hörmann K, Zimmer A. Drug delivery and drug targeting with parenteral lipid nanoemulsions - A review. J Control Release. 2016;223:85–98. doi:10.1016/j.jconrel.2015.12.016
59. Cooke M, Lamplugh A, Naudeer S, et al. Efficacy and tolerability of accelerated-dose low-molecular-weight iron dextran (Cosmofer) in patients with chronic kidney disease. Am J Nephrol. 2012;35(1):69–74. doi:10.1159/000334877
60. Zhang Y, Wu JLY, Lazarovits J, et al. An analysis of the binding function and structural organization of the protein corona. J Am Chem Soc. 2020;142(19):8827–8836. doi:10.1021/jacs.0c01853
61. Corbo C, Molinaro R, Taraballi F, et al. Unveiling the in vivo protein corona of circulating leukocyte-like carriers. ACS Nano. 2017;11(3):3262–3273. doi:10.1021/acsnano.7b00376
62. Mosquera J, García I, Henriksen-Lacey M, et al. Reducing protein corona formation and enhancing colloidal stability of gold nanoparticles by capping with silica monolayers. Chem Mater. 2019;31(1):57–61. doi:10.1021/acs.chemmater.8b04647
63. Hadjidemetriou M, Al-Ahmady Z, Kostarelos K. Time-evolution of in vivo protein Corona onto blood-circulating PEGylated liposomal doxorubicin (DOXIL) nanoparticles. Nanoscale. 2016;8(13):6948–6957. doi:10.1039/C5NR09158F
64. Xiao W, Gao H. The impact of protein Corona on the behavior and targeting capability of nanoparticle-based delivery system. Int J Pharm. 2018;552(1–2):328–339. doi:10.1016/j.ijpharm.2018.10.011
65. Dawidczyk CM, Kim C, Park JH, et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–144. doi:10.1016/j.jconrel.2014.05.036
66. Hui Y, Yi X, Hou F, et al. Role of nanoparticle mechanical properties in cancer drug delivery. ACS Nano. 2019;13(7):7410–7424. doi:10.1021/acsnano.9b03924
67. Chen Z, Kankala RK, Long L, et al. Current understanding of passive and active targeting nanomedicines to enhance tumor accumulation. Coord Chem Rev. 2023;481:215051. doi:10.1016/j.ccr.2023.215051
68. Choi HS, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–1170. doi:10.1038/nbt1340
69. Recordati C, De Maglie M, Bianchessi S, et al. Tissue distribution and acute toxicity of silver after single intravenous administration in mice: nano-specific and size-dependent effects. Part Fibre Toxicol. 2016;13:12. doi:10.1186/s12989-016-0124-x
70. Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756.
71. Xie S, Chen M, Song X, et al. Bacterial microbots for acid-labile release of hybrid micelles to promote the synergistic antitumor efficacy. Acta Biomater. 2018;78:198–210. doi:10.1016/j.actbio.2018.07.041
72. Schädlich A, Caysa H, Mueller T, et al. Tumor accumulation of NIR fluorescent PEG-PLA nanoparticles: impact of particle size and human xenograft tumor model. ACS Nano. 2011;5(11):8710–8720. doi:10.1021/nn2026353
73. Li X, Pan Z, Xiang C, et al. Structure transformable nanoparticles for photoacoustic imaging-guided photothermal ablation of tumors via enzyme-induced multistage delivery. Chem Eng J. 2021;421:127747. doi:10.1016/j.cej.2020.127747
74. Cybulski P, Bravo M, Chen JJ-K, et al. Nanoparticle accumulation and penetration in 3D tumor models: the effect of size, shape, and surface charge. Front Cell Dev Biol. 2024;12:1520078. doi:10.3389/fcell.2024.1520078
75. Kankala RK, Liu C-G, Yang D-Y, et al. Ultrasmall platinum nanoparticles enable deep tumor penetration and synergistic therapeutic abilities through free radical species-assisted catalysis to combat cancer multidrug resistance. Chem Eng J. 2020;383:123138. doi:10.1016/j.cej.2019.123138
76. Kankala RK, Han Y-H, Na J, et al. Nanoarchitectured structure and surface biofunctionality of mesoporous silica nanoparticles. Adv Mater. 2020;32(23):e1907035. doi:10.1002/adma.201907035
77. Zahr AS, Davis CA, Pishko MV. Macrophage uptake of core−shell nanoparticles surface modified with Poly(ethylene glycol). Langmuir. 2006;22(19):8178–8185. doi:10.1021/la060951b
78. Campbell RB, Fukumura D, Brown EB, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62(23):6831–6836.
79. Sun T, Zhang YS, Pang B, et al. Engineered nanoparticles for drug delivery in cancer therapy. Angew Chem Int Ed Engl. 2014;53(46):12320–12364. doi:10.1002/anie.201403036
80. Zhang P, Chen D, Li L, et al. Charge reversal nano-systems for tumor therapy. J Nanobiotechnology. 2022;20(1):31. doi:10.1186/s12951-021-01221-8
81. Izci M, Maksoudian C, Manshian BB, et al. The use of alternative strategies for enhanced nanoparticle delivery to solid tumors. Chem Rev. 2021;121(3):1746–1803. doi:10.1021/acs.chemrev.0c00779
82. Decuzzi P, Lee S, Bhushan B, et al. A theoretical model for the margination of particles within blood vessels. Ann Biomed Eng. 2005;33(2):179–190. doi:10.1007/s10439-005-8976-5
83. Geng Y, Dalhaimer P, Cai S, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nature Nanotechnol. 2007;2(4):249–255. doi:10.1038/nnano.2007.70
84. Kinnear C, Moore TL, Rodriguez-Lorenzo L, et al. Form follows function: nanoparticle shape and its implications for nanomedicine. Chem Rev. 2017;117(17):11476–11521. doi:10.1021/acs.chemrev.7b00194
85. Lee SY, Ferrari M, Decuzzi P. Shaping nano-/micro-particles for enhanced vascular interaction in laminar flows. Nanotechnology. 2009;20(49):495101. doi:10.1088/0957-4484/20/49/495101
86. Gao H. Shaping tumor microenvironment for improving nanoparticle delivery. Curr Drug Metab. 2016;17(8):731–736. doi:10.2174/1389200217666160630203600
87. Ruan S, Cao X, Cun X, et al. Matrix metalloproteinase-sensitive size-shrinkable nanoparticles for deep tumor penetration and pH triggered doxorubicin release. Biomaterials. 2015;60:100–110. doi:10.1016/j.biomaterials.2015.05.006
88. Miao L, Lin CM, Huang L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J Control Release. 2015;219:192–204. doi:10.1016/j.jconrel.2015.08.017
89. Khawar IA, Kim JH, Kuh HJ. Improving drug delivery to solid tumors: priming the tumor microenvironment. J Control Release. 2015;201:78–89. doi:10.1016/j.jconrel.2014.12.018
90. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi:10.1038/nrclinonc.2010.139
91. Lieleg O, Baumgärtel RM, Bausch AR. Selective filtering of particles by the extracellular matrix: an electrostatic bandpass. Biophys J. 2009;97(6):1569–1577. doi:10.1016/j.bpj.2009.07.009
92. Suzuki S, Itakura S, Matsui R, et al. Tumor microenvironment-sensitive liposomes penetrate tumor tissue via attenuated interaction of the extracellular matrix and tumor cells and accompanying actin depolymerization. Biomacromolecules. 2017;18(2):535–543. doi:10.1021/acs.biomac.6b01688
93. Shi P, Cheng Z, Zhao K, et al. Active targeting schemes for nano-drug delivery systems in osteosarcoma therapeutics. J Nanobiotechnology. 2023;21(1):103. doi:10.1186/s12951-023-01826-1
94. Wang JX. Photoswitchable ultrafast Transactivator of Transcription (TAT) targeting effect for nanocarrier-based on-demand drug delivery. Adv Funct Mater. 2018;28(3):1704806.
95. Wang T, Wang D, Liu J, et al. Acidity-triggered ligand-presenting nanoparticles to overcome sequential drug delivery barriers to tumors. Nano Lett. 2017;17(9):5429–5436. doi:10.1021/acs.nanolett.7b02031
96. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi:10.1016/j.cell.2010.03.015
97. Marecko I, Cvejić D, Šelemetjev S, et al. Enhanced activation of matrix metalloproteinase-9 correlates with the degree of papillary thyroid carcinoma infiltration. Croat Med J. 2014;55(2):128–137. doi:10.3325/cmj.2014.55.128
98. Hu G, Chun X, Wang Y, et al. Peptide mediated active targeting and intelligent particle size reduction-mediated enhanced penetrating of fabricated nanoparticles for triple-negative breast cancer treatment. Oncotarget. 2015;6(38):41258–41274. doi:10.18632/oncotarget.5692
99. Li HJ, Du J-Z, Liu J, et al. Smart superstructures with ultrahigh ph-sensitivity for targeting acidic tumor microenvironment: instantaneous size switching and improved tumor penetration. ACS Nano. 2016;10(7):6753–6761. doi:10.1021/acsnano.6b02326
100. Nagel G, Sousa-Herves A, Wedepohl S, et al. Matrix metalloproteinase-sensitive multistage nanogels promote drug transport in 3D tumor model. Theranostics. 2020;10(1):91–108. doi:10.7150/thno.34851
101. Lei Q, Wang S-B, Hu -J-J, et al. Stimuli-responsive “cluster bomb” for programmed tumor therapy. ACS Nano. 2017;11(7):7201–7214. doi:10.1021/acsnano.7b03088
102. Li HJ, Du J-Z, Du X-J, et al. Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy. Proc Natl Acad Sci U S A. 2016;113(15):4164–4169. doi:10.1073/pnas.1522080113
103. Sun Q, Sun X, Ma X, et al. Integration of nanoassembly functions for an effective delivery cascade for cancer drugs. Adv Mater. 2014;26(45):7615–7621. doi:10.1002/adma.201401554
104. Wang Y, Zhou K, Huang G, et al. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat Mater. 2014;13(2):204–212. doi:10.1038/nmat3819
105. Yu H, Li J-M, Deng K, et al. Tumor acidity activated triphenylphosphonium-based mitochondrial targeting nanocarriers for overcoming drug resistance of cancer therapy. Theranostics. 2019;9(23):7033–7050. doi:10.7150/thno.35748
106. Gormley AJ, Larson N, Banisadr A, et al. Plasmonic photothermal therapy increases the tumor mass penetration of HPMA copolymers. J Control Release. 2013;166(2):130–138. doi:10.1016/j.jconrel.2012.12.007
107. Ojha T, Pathak V, Shi Y, et al. Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv Drug Delivery Rev. 2017;119:44–60. doi:10.1016/j.addr.2017.07.007
108. Theek B, Baues M, Ojha T, et al. Sonoporation enhances liposome accumulation and penetration in tumors with low EPR. J Control Release. 2016;231:77–85. doi:10.1016/j.jconrel.2016.02.021
109. Chen B, Pogue BW, Luna JM, et al. Tumor vascular permeabilization by vascular-targeting photosensitization: effects, mechanism, and therapeutic implications. Clin Cancer Res. 2006;12(3 Pt 1):917–923. doi:10.1158/1078-0432.CCR-05-1673
110. Seymour LW, Ferry DR, Anderson D, et al. Hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20(6):1668–1676. doi:10.1200/JCO.2002.20.6.1668
111. Tekchandani P, Kurmi BD, Paliwal R, et al. Galactosylated TPGS micelles for docetaxel targeting to hepatic carcinoma: development, characterization, and biodistribution study. AAPS Pharm Sci Tech. 2020;21(5):174. doi:10.1208/s12249-020-01690-4
112. Yang Y, Jing L, Li X, et al. Hyaluronic acid conjugated magnetic prussian blue@quantum dot nanoparticles for cancer theranostics. Theranostics. 2017;7(2):466–481. doi:10.7150/thno.17411
113. Li L, Lu Y, Jiang C, et al. Actively targeted deep tissue imaging and photothermal-chemo therapy of breast cancer by antibody-functionalized drug-loaded X-ray-responsive bismuth sulfide@mesoporous silica core-shell nanoparticles. Adv Funct Mater. 2018;28(5). doi:10.1002/adfm.201704623
114. Li Q, Tang Q, Zhang P, et al. Human epidermal growth factor receptor-2 antibodies enhance the specificity and anticancer activity of light-sensitive doxorubicin-labeled liposomes. Biomaterials. 2015;57:1–11. doi:10.1016/j.biomaterials.2015.04.009
115. Roncato F, Rruga F, Porcù E, et al. Improvement and extension of anti-EGFR targeting in breast cancer therapy by integration with the avidin-nucleic-acid-nano-assemblies. Nat Commun. 2018;9(1):4070. doi:10.1038/s41467-018-06602-6
116. Dash A, Blasiak B, Tomanek B, et al. Target-specific magnetic resonance imaging of human prostate adenocarcinoma using NaDyF 4 –NaGdF 4 core–shell nanoparticles. ACS Appl Mater Interfaces. 2021;13(21):24345–24355. doi:10.1021/acsami.0c19273
117. Pang Z, Gao H, Yu Y, et al. Enhanced intracellular delivery and chemotherapy for glioma rats by transferrin-conjugated biodegradable polymersomes loaded with doxorubicin. Bioconjug Chem. 2011;22(6):1171–1180. doi:10.1021/bc200062q
118. Zhu Z, Xie C, Liu Q, et al. The effect of hydrophilic chain length and iRGD on drug delivery from poly(ε-caprolactone)-poly(N-vinylpyrrolidone) nanoparticles. Biomaterials. 2011;32(35):9525–9535. doi:10.1016/j.biomaterials.2011.08.072
119. Wang J, Wang J, Yang J, et al. Assemblies of peptide‐cytotoxin conjugates for tumor‐homing chemotherapy. Adv Funct Mater. 2019;29. doi:10.1002/adfm.201900893
120. Guo X, Shi C, Wang J, et al. pH-triggered intracellular release from actively targeting polymer micelles. Biomaterials. 2013;34(18):4544–4554. doi:10.1016/j.biomaterials.2013.02.071
121. Xu W, Siddiqui IA, Nihal M, et al. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted therapy of prostate cancer. Biomaterials. 2013;34(21):5244–5253. doi:10.1016/j.biomaterials.2013.03.006
122. Tran BT, Kim J, Ahn DR. Systemic delivery of aptamer-drug conjugates for cancer therapy using enzymatically generated self-assembled DNA nanoparticles. Nanoscale. 2020;12(45):22945–22951. doi:10.1039/D0NR05652A
123. Li M, Li S, Zhou H, et al. Chemotaxis-driven delivery of nano-pathogenoids for complete eradication of tumors post-phototherapy. Nat Commun. 2020;11(1):1126. doi:10.1038/s41467-020-14963-0
124. Gawali P, Saraswat A, Bhide S, et al. Human solid tumors and clinical relevance of the enhanced permeation and retention effect: a ‘golden gate’ for nanomedicine in preclinical studies? Nanomedicine. 2023;18(2):169–190. doi:10.2217/nnm-2022-0257
125. Ma T, Tran TB, Lin E, et al. Size-transformable nanotherapeutics for cancer therapy. Acta Pharmaceutica Sinica B. 2025;15(2):834–851. doi:10.1016/j.apsb.2024.11.012
126. Stubbs M, McSheehy PMJ, Griffiths JR, et al. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today. 2000;6(1):15–19. doi:10.1016/S1357-4310(99)01615-9
127. Liang Y, Wu J, Yan Y, et al. Charge-reversal nano-drug delivery systems in the tumor microenvironment: mechanisms, challenges, and therapeutic applications. Int J Mol Sci. 2024;25(18):9779. doi:10.3390/ijms25189779
128. Kwon YW, Jo H-S, Bae S, et al. Application of proteomics in cancer: recent trends and approaches for biomarkers discovery. Front Med. 2021;8:747333. doi:10.3389/fmed.2021.747333
129. Sanità G, Carrese B, Lamberti A. Nanoparticle surface functionalization: how to improve biocompatibility and cellular internalization. Front Mol Biosci. 2020;7. doi:10.3389/fmolb.2020.587012
130. Zhang H, Guo L, Li X, et al. A Modular Approach to Obtain HER2-Targeting DM1-Loaded Nanoparticles for Gastric Cancer Therapy. ACS Omega. 2024;9(49):48598–48606. doi:10.1021/acsomega.4c07442
131. Fan Z, Chang Y, Cui C, et al. Near infrared fluorescent peptide nanoparticles for enhancing esophageal cancer therapeutic efficacy. Nat Commun. 2018;9(1):2605. doi:10.1038/s41467-018-04763-y
132. Egbe Vydaline AO, Rozhkov S, Sosa G, et al. Aptamers as target-specific recognition elements in drug delivery. Adv Drug Deliv Rev. 2025;226:115685. doi:10.1016/j.addr.2025.115685
133. Yang L, Yang Y, Chen Y, et al. Cell-based drug delivery systems and their in vivo fate. Adv Drug Deliv Rev. 2022;187:114394. doi:10.1016/j.addr.2022.114394
134. Dong H, Li Y, Liu Y, et al. A nano-immunotraining strategy to enhance the tumor targeting of neutrophils via in vivo pathogen-mimicking stimulation. Biomater Sci. 2019;7(12):5238–5246. doi:10.1039/C9BM01278H
135. Gao C, Wang Q, Li J, et al. In vivo hitchhiking of immune cells by intracellular self-assembly of bacteria-mimetic nanomedicine for targeted therapy of melanoma. Sci Adv. 2022;8(19):eabn1805. doi:10.1126/sciadv.abn1805
136. Xue J, Zhao Z, Zhang L, et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat Nanotechnol. 2017;12(7):692–700. doi:10.1038/nnano.2017.54
137. Shields CW, Evans MA, Wang LL-W, et al. Cellular backpacks for macrophage immunotherapy. Sci Adv. 2020;6(18):eaaz6579. doi:10.1126/sciadv.aaz6579
138. Layek B, Sadhukha T, Panyam J, et al. Nano-engineered mesenchymal stem cells increase therapeutic efficacy of anticancer drug through true active tumor targeting. Mol Cancer Ther. 2018;17(6):1196–1206. doi:10.1158/1535-7163.MCT-17-0682
139. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. doi:10.1016/j.addr.2010.04.009
140. Chen Q, Yuan L, Chou W-C, et al. Meta-analysis of nanoparticle distribution in tumors and major organs in tumor-bearing mice. ACS Nano. 2023;17(20):19810–19831. doi:10.1021/acsnano.3c04037
141. Golombek SK, May J-N, Theek B, et al. Tumor targeting via EPR: strategies to enhance patient responses. Adv Drug Deliv Rev. 2018;130:17–38. doi:10.1016/j.addr.2018.07.007
142. Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi:10.1126/science.1171362
143. Lambris JD, Rosenthal A, Pelegrín P, et al. The future of neuroimmunology research for CNS disease therapeutics. Trends Pharmacol Sci. 2022;43(8):611–614. doi:10.1016/j.tips.2022.06.003
144. Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB j. 2008;22(3):659–661. doi:10.1096/fj.07-9574LSF
145. Heldin CH, Rubin K, Pietras K, et al. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. doi:10.1038/nrc1456
146. Kim J. Perspectives for improving the tumor targeting of nanomedicine via the EPR effect in clinical tumors. Int J Mol Sci. 2023;24(12):10082.
147. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–287. doi:10.1038/nrc3236
148. Liu Y, Wu W, Cai C, et al. Patient-derived xenograft models in cancer therapy: technologies and applications. Signal Transduct Target Ther. 2023;8(1):160. doi:10.1038/s41392-023-01419-2
149. Wu Q, Yao X, Duan X, et al. Nanomedicine reimagined: translational strategies for precision tumor theranostics. Adv Mater. 2025: e10293. doi:10.1002/adma.202510293
150. Muntimadugu E, Kommineni N, Khan W. Exploring the potential of nanotherapeutics in targeting tumor microenvironment for cancer therapy. Pharmacol Res. 2017;126:109–122. doi:10.1016/j.phrs.2017.05.010
151. Wu L, Wen W, Wang X, et al. Ultrasmall iron oxide nanoparticles cause significant toxicity by specifically inducing acute oxidative stress to multiple organs. Part Fibre Toxicol. 2022;19(1):24. doi:10.1186/s12989-022-00465-y
152. Cataldi M, Vigliotti C, Mosca T, et al. Emerging role of the spleen in the pharmacokinetics of monoclonal antibodies, nanoparticles and exosomes. Int J Mol Sci. 2017;18(6):1249. doi:10.3390/ijms18061249
153. La-Beck NM, Islam MR, Markiewski MM. Nanoparticle-induced complement activation: implications for cancer nanomedicine. Front Immunol. 2020;11:603039. doi:10.3389/fimmu.2020.603039
154. Decuzzi P, Pasqualini R, Arap W, et al. Intravascular delivery of particulate systems: does geometry really matter? Pharm Res. 2009;26(1):235–243. doi:10.1007/s11095-008-9697-x
155. Manke A, Wang L, Rojanasakul Y. Mechanisms of nanoparticle-induced oxidative stress and toxicity. Biomed Res Int. 2013;2013:942916. doi:10.1155/2013/942916
156. Suk JS, Xu Q, Kim N, et al. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. doi:10.1016/j.addr.2015.09.012
157. Fu S, Zhu X, Huang F, et al. Anti-PEG antibodies and their biological impact on pegylated drugs: challenges and strategies for optimization. Pharmaceutics. 2025;17(8):1074. doi:10.3390/pharmaceutics17081074
158. Thomas J, Kumar V, Sharma N, et al. Recent approaches in nanotoxicity assessment for drug delivery applications: challenges and prospects. Med Drug Discov. 2025;25:100204. doi:10.1016/j.medidd.2025.100204
159. Dasgupta A, Sofias AM, Kiessling F, et al. Nanoparticle delivery to tumours: from EPR and ATR mechanisms to clinical impact. Nat Rev Bioeng. 2024;2(9):714–716. doi:10.1038/s44222-024-00203-3
160. Lammers T. Nanomedicine tumor targeting. Adv Mater. 2024;36(26):2312169. doi:10.1002/adma.202312169
161. May J-N, Moss JI, Mueller F, et al. Histopathological biomarkers for predicting the tumour accumulation of nanomedicines. Nat Biomed Eng. 2024;8(11):1366–1378. doi:10.1038/s41551-024-01197-4
162. Autio KA, Dreicer R, Anderson J, et al. Safety and efficacy of BIND-014, a docetaxel nanoparticle targeting prostate-specific membrane antigen for patients with metastatic castration-resistant prostate cancer: a phase 2 clinical trial. JAMA Oncol. 2018;4(10):1344–1351. doi:10.1001/jamaoncol.2018.2168
163. Bonvalot S, Rutkowski PL, Thariat J, et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): a multicentre, phase 2–3, randomised, controlled trial. Lancet Oncol. 2019;20(8):1148–1159. doi:10.1016/S1470-2045(19)30326-2
164. Volovat SR, Ciuleanu T-E, Koralewski P, et al. A multicenter, single-arm, basket design, phase II study of NC-6004 plus gemcitabine in patients with advanced unresectable lung, biliary tract, or bladder cancer. Oncotarget. 2020;11(33):3105–3117. doi:10.18632/oncotarget.27684
165. Krasner CN, Campos SM, Young CL, et al. Sequential phase II clinical trials evaluating CRLX101 as monotherapy and in combination with bevacizumab in recurrent ovarian cancer. Gynecol Oncol. 2021;162(3):661–666. doi:10.1016/j.ygyno.2021.07.002
166. Sankhala KK, Mita AC, Adinin R, et al. A phase I pharmacokinetic (PK) study of MBP-426, a novel liposome encapsulated oxaliplatin. J clin oncol. 2009;27(15_suppl):2535. doi:10.1200/jco.2009.27.15_suppl.2535
167. I.C.L. EnGene. MesomiR 1: A Phase I Study of Intravenously Administered Epidermal Growth Factor Receptor -Targeted, EnGeneIC Delivery Vehicle (EDV)-Packaged, Mir-16 Mimic (Targomirs) for Patients with Malignant Pleural Mesothelioma (MPM) and Advanced Non-Small Cell Lung Cancer (NSCLC) Failing on Std Therapy. 2015.
168. Miedema IHC, Zwezerijnen GJC, Oprea-Lager DE, et al. First-in-human imaging of nanoparticle entrapped docetaxel (CPC634) in patients with advanced solid tumors using 89 Zr-Df-CPC634 PET/CT. J clin oncol. 2019;37(15_suppl):3093. doi:10.1200/JCO.2019.37.15_suppl.3093
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Red Blood Cell Membrane Vesicles for siRNA Delivery: A Biocompatible Carrier With Passive Tumor Targeting and Prolonged Plasma Residency

Della Pelle G, Markelc B, Bozic T, Šribar J, Krizaj I, Zagar Soderznik K, Hudoklin S, Kreft ME, Urbančič I, Kisovec M, Podobnik M, Kostevšek N
International Journal of Nanomedicine 2025, 20:3269-3301
Published Date: 15 March 2025
Targeted-Immunomodulatory Nanomedicines for the Treatment of Autoimmune Diseases via Multiple Administration Routes
Lee B, Yoo J, Lee HS, Lee Y
International Journal of Nanomedicine 2025, 20:15493-15514
Published Date: 27 December 2025