Back to Journals » International Medical Case Reports Journal » Volume 11
Novel codon 15 RHO gene mutation associated with retinitis pigmentosa
Authors Vilela MAP ![]() , Menna Barreto RK, Menna Barreto PK, Sallum JMF
, Menna Barreto RK, Menna Barreto PK, Sallum JMF ![]() , Mattevi VS
, Mattevi VS ![]()
Received 4 July 2018
Accepted for publication 10 September 2018
Published 20 November 2018 Volume 2018:11 Pages 339—344
DOI https://doi.org/10.2147/IMCRJ.S179105
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Manuel AP Vilela,1 Roberta K Menna Barreto,2 Pedro K Menna Barreto,1 Juliana MF Sallum,3 Vanessa S Mattevi1
1Department of Ophthalmology, Federal Health Sciences University of Porto Alegre, Porto Alegre, Brazil; 2Medical School, Federal University of Rio Grande do Sul, Rio Grande do Sul, Brazil; 3Department of Ophthalmology, Federal University of São Paulo, São Paulo, Brazil
Objective: To describe, in a multimodal way, a new RHO gene mutation with lysine-for-asparagine substitution in autosomal dominant retinitis pigmentosa.
Methods: Case report. Retrospective data analysis.
Results: The mutation is located within codon 15 of exon 1 of the RHO gene. A single base-pair transversion lead to a specific lysine-for-asparagine substitution (Asn15Lys). Hypoacusis, myopia, dyschromatopsis and diffuse retinitis pigmentosa were detected. Detailed multimodal images for the posterior segment are presented.
Conclusion: We present a new mutation with a specific substitution that may cause eye disease and which has not been described previously. There is no description of this variant in the genetic databases.
Keywords: tapetoretinal degeneration, rod-cone dystrophy, pigmentary retinopathy, opsins, retinal pigments
Introduction
Retinitis pigmentosa (RP; OMIM #268000) is the term used to characterize a heterogeneous group of hereditary diseases that cause variable degeneration and loss of photoreceptor (PR) cells (cones and rods) and retinal pigment epithelium (RPE). In most forms of the disease, cones and rods are the cells initially affected. As such, the first symptoms characteristic of RP are night blindness (nyctalopia) and loss of peripheral vision. Usually, these complaints progressively limit patients’ everyday activities, such as driving, going down stairs, practicing sports or moving around at night. As they progress, there is loss of central vision, although in some specific forms cones can be affected from a very early stage. Electrophysiological alterations are critical and must be included in assessment.1
The pathogenesis is still not clear and there is no approved treatment options for RP. Notwithstanding this, knowledge of the genetic mutations that cause the disease, together with studies of gene and cell therapy, are moving toward the discovery of efficient interventions.2–4
It is known that the pattern of genetic inheritance of RP can be autosomal recessive (more frequent), autosomal dominant, linked to the X chromosome (rarer), and sporadic. The majority of cases occur owing to a mutation in the RHO gene (OMIM #180380) or in the PRPH gene (OMIM #170710), both of which are present in the PR cells.5 RP does however show considerable genetic heterogeneity and mutations in various other genes have been identified in affected patients. In 1989, by means of genetic analysis, an autosomal dominant RP locus was identified on chromosome 3.6 Following this, numerous different mutations in the RHO gene have been described and considered to cause RP.7–15 In 1993, a variant that leads to the amino acid asparagine being replaced by serine in codon 15 (Asn15Ser) of the RHO gene was described and associated with sectorial RP in five generations of an Australian family.7 This mutation was described in Japan (one family) and France (ten unrelated families). For both, the phenotype were similar, with sectorial RP.8–10
In our study, we report on the occurrence of a new mutation in codon 15 of the RHO gene related to RP, a variant with the substitution of the amino acid asparagine by lysine (Asn15Lys) and present repercussions through multimodality imaging studies.
Case report
For this case report, written informed consent was obtained from the patient before performing the study procedures and examinations. Our subject is a 53-year-old woman who was born and still lives at Porto Alegre, city capital of the state of Rio Grande do Sul, Brazil. She is of Italian descent. Her parents, both deceased, were consanguineous (first cousins). She has three siblings and none of them (their offspring included) were diagnosed with any eye diseases. She was currently treated with a beta-blocker for systemic arterial hypertension. Besides the use of a hearing aid, no other comorbidities or daily-use medications were reported. Her first symptoms were hypoacusis, progressive myopia (detected at age 10), and nyctalopia. For the first 20 years after the first symptoms appeared, she was treated at another facility with pulse therapy (due to the appearance of bilateral pericentral scotomas mistakenly attributed to neuritis). There was no improvement. The pericentral and paracentral scotomas and nyctalopia slowly progressed up until approximately 2 years ago.
Her ophthalmological examination showed:
- Visual acuity (Snellen): right eye (RE) 20/20 (–2,50 spherical) and left eye (LE) 20/30 (–3,0 spherical).
- Complete dyschromatopsy in LE, blue-green in RE.
- Normal ocular motricity, pupils reactions and anterior segment in both eyes (BE).
- Applanation tonometry: 12 mmHg in BE.
- Binocular indirect fundoscopy: diffuse pallor of the optic discs, generalized vascular thinning, disperse pigment spicules on the four quadrants also coming close to the posterior pole, macular alterations on pigment epithelium (RPE) plane and internal surface reflexes. Superficial micro-hemorrhage cluster near to left optic disc. Visual fields with sensitivity alterations on all quadrants and with better perceptions in the lower hemifields of BE.
- The mean deviation, pattern SD, and visual fields indices were found to be altered in BE (Humphrey Field Analyzer 3; Carl Zeiss Meditec AG, Jena, Germany).
- Fundus autofluorescence (FAF) showed inferior and temporal perimacular islands of hypo-autofluorescence in the central zone of BE.
- Fluorescein angiography (FA) showed preserved filling times, diffuse hypofluorescence of both discs, extensive defects in window (RPE atrophy either isolated or combined with the choriocapillaris), reaching the edges of the superior and inferior temporal vascular arcades, hypofluorescence caused by spicules in BE.
- Optical coherence tomography (OCT) and OCT angiography (OCT-A) showed foveal thickness as being RE 255 μm and LE 321 μm; presence of bilateral epiretinal membrane, more significant in LE, with disappearance of clivus and formation of folds; bilateral sectoral discontinuities in the ellipsoid zones. OCT-A identified points of non-perfusion in the outer retina and central choriocapillaris and peri-discs, without corresponding to pigments (DRI Swept Source OCT Triton Plus; Topcon, Tokyo, Japan) (Figures 1 and 2).
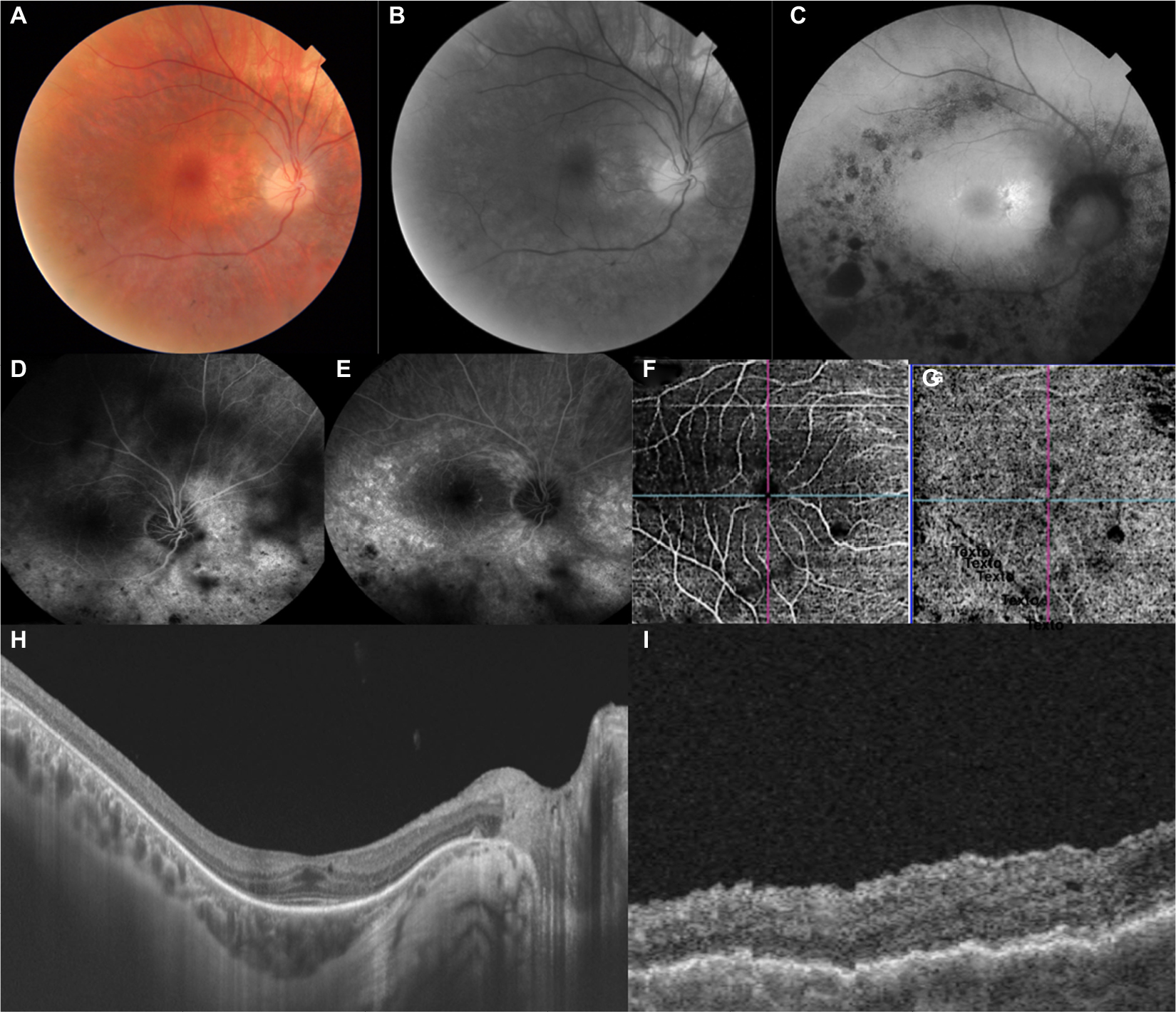 | Figure 1 Multimodal images of the right eye. Notes: Color photo (A) shows peri-macular discoloration caused by RPE alterations; Red-free image (B) with consonant changes in RPE; (C) FAF with clumps of hypo-autofluorescence around the posterior pole and tiny halo of hyper-autofluorescence around the fovea. IVFA (D, E) demarcates the window defects, as well as the spiculated pigments, with impaired edges of the fovea. (F, G) OCTA locate zones of non-perfusion in both outer retina and choriocapillaris. OCT sections (H, I) show flat clivus, folds on the outer limiting membrane, discontinuity of the Henle and ellipsoidal layers, with unequal choroidal thickness. Abbreviations: FAF, fundus autofluorescence; IVFA, intravenous fluorescein angiography; OCT-A, OCT angiography; RPE, retinal pigment epithelium. |
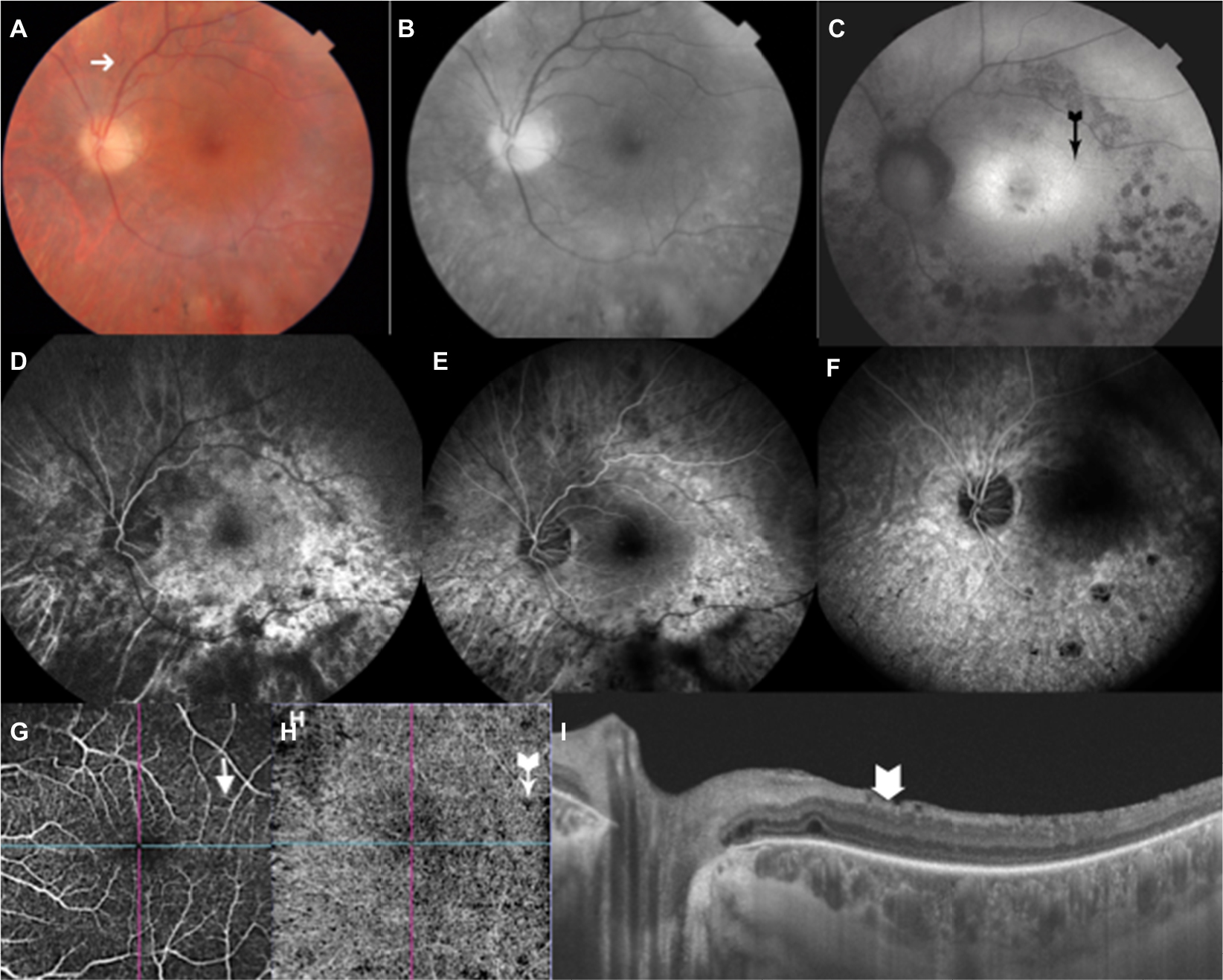 | Figure 2 Multimodal images of the left eye. Notes: Color photo (A) and red-free image (B) show peri-macular discoloration and greater visibility of the pigment alterations below the macula. Superficial micro- hemorrhage (A, arrow). FAF (C) shows the numerous islands of hypo-autofluorescence around the macula, with the fovea being limited by a hyper-autofluorescent halo (C, black arrow). IVFA (D–F) highlights the diffuse hypofluorescence of the disc and bordering the zones of pigment epithelium and/or choriocapillaris atrophy. OCTA shows small zones of nonperfusion (G, H, arrows) especially in outer retina and choriocapillaris. OCT section (I) shows the surface folds (arrow). Abbreviations: FAF, fundus autofluorescence; IVFA, intravenous fluorescein angiography; OCT-A, OCT angiography; OCT, optical coherence tomography. |
The electrophysiological studies (Retiport; Roland Consult, Brandenburg an der Havel, Germany) included visual evoked potential (VEP), full field electroretinogram (ERG), and electrooculogram (EOG). VEP recorded alterations in the bilateral occipital cortical response, with reduction in wave amplitude and morphology, with increased retino-cortical conduction time in BE. ERG showed overall and significant reduction in cell responses at all structural levels and at all stages of the examination in BE. We detected accentuated impairment of rods and with greater intensity in LE. EOG showed subnormal RPE functional responses, with an Arden index of 1.20 in BE.
DNA analysis
Exome analysis was performed by next-generation sequencing using the Nextera Exome Capture kit in the Illumina HiSeq platform. The gene panel analyzed comprised the following genes: ABCA4, ABCC6, ABCD1, ABHD12, ACO2, ADAM9, ADGRV1, AHI1, AIPL1, ALMS1, AMACR, ARL13B, ARL6, ATF6B9D1, B9D2, BBS1, BBS10, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, BEST1, C12ORF65, C1QTNF5, C2ORF71C5ORF42, C8ORF37, CA4, CABP4, CACNA1F, CACNA2D4, CASK, CC2D2A, CDH23, CDH3, CDHR1, CEP290, CEP41, CERKL, CFH, CHM, CIB2, CISD2, CLN3, CLN5, CLN6, CLN8, CLRN1, CNGA1, CNGB1, CNGB3, CNNM4, CRB1, CRXCTSD, CYP4V2, DFNB31, DHDDS, DNAJC5, EFEMP1, ELOVL4, EYSFAM1614, FLVCR1, FRMD7, FSCN2, FZD4, GDF6, GJB2, GJB6, GNAT1, GNAT2, GNPTG, GPR143, GPR179, GRK1, GRM6, GRN, GUCA1A, GUCA1B, GUCY2D, GUCY2DHARS, HGSNAT, HK1, HMCN1, HMX1, IDH3B, IFT140, IMPDH1, IMPG2, IQCB1, ITM2B, KCNJ13, KCNV2, KCTD7, KIF7KLHL7, LAMA1, LCA5, LRAT, LRP5, LZTFL1, MAK, MERTK, MFN2, MFRP, MFSD8, MKKS, MKS1, MMACHC, MVKMYO7A, NDP, NEUROD1, NMNAT1, NHPHP1, NPHP3, NPHP4, NR2F1, NRL, NYX, NYX, OAT, OFD1, OPA1, OPA3OPN1LW, OPN1MW, OTX2, PAX6, PCDH15, PDE6A, PDE6B, PDE6C, PDE6G, PDE6H, PDZD7, PEX1, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6, PEX7, PHYH, PITPNM3, PNMPLA6, PPT1PRCD, PROM1, PRPF3, PRPF31, PRPF6, PRPF8, PRPH2, PRPS1, RAB28, RAX2, RBP3, RBP4, RD3, RDH12, RDH5RGR, RGS9, RGS9BP, RHO, RIMS1, RLBP1, ROM1, RP1, RP1L1, RP2, RP9, RPE65, RPGR, RPGRIP1, RPGRIP1L, RS1, SAG, SDCCAG8, SEMA4A, SLC24A1, SNRNP200, SPATA7, TCTN1, TCTN2, TEAD1, TIMM8A, TIMP3, TMEM126, ATMEM138, TMEM216, TMEM237, TMEM67, TOPORS, TPP1, TRIM32, TRPM1, TSPAN12, TTC21B, TTCB, TUBGCP4, TUBGCP6, TULP1, TYR, UNC19, USH1C, USH1G, USH2A, VPS13B, WDPCP, WDR19, WFS1, ZNF513. Average coverage was 121× and 96.7% of bases were read at least 10×.
In the RHO gene, variant Chr3 was identified: 129.247.621 T>G in heterozygosis, promoting the substitution of the amino acid asparagine in codon 15 with lysine (p.Asn15Lys), being predicted as deleterious by the SIFT predictor and as probably damaging by PolyPhen.16 No other variation in the remaining genes was found. The reference sequence assembly used was GRCh37 - hg19. A previous description of this variant was not found in searches performed in the OMIM,14 dbSNP,18 EnSembl,19 ClinVar,20 ExAC,21 and UniProt22 databases (Table 1).
 | Table 1 Genetic findings |
Discussion
RP affects approximately 1–3 in every 5,000 people. It can be caused by mutations in several genes and different transmission routes.1,5 In the gene panel analyzed we found a mutation in the RHO gene, which would characterize type 4 RP (OMIM, # 613731). The forms of RP caused by mutations in this gene are usually of autosomal dominant inheritance, which is compatible with this case, in which the patient is heterozygous for Asn15Lys (N15K) mutation. This form of inheritance accounts for 20%–40% of all RP cases.1,23 Asn-15 is important for activity in signal transduction and its substitution is related to an opsin which generates a defective chromophore that is only 10% as efficient as RHO.24
Tapetoretinal degeneration is associated with syndromes in approximately 25% of cases.1,4 However, the only simultaneous finding in our case report was hypoacusis. Usher, Bardet-Biedl, and Alström syndromes have many other signs apart from hearing difficulty.
Mendelian forms of transmission are the most common, although digenic, hereditary maternal (mitochondrial), triallelic, and disomy forms of transmission have also been described. Genetic assessment has identified dozens of genes involved in RP. These include genes that code for proteins involved in visual transduction, visual cycle, phagocytosis of the outer segments of the PRs, transcription, PR outer segment proteins, cell transport, and cell matrix, among others.5,25 Genetic characterization is important for identifying the different clinical subtypes and characterizing clinical presentation, recognizing mutation, enabling genetic counseling, and being an essential source for future options of specific case management.4,25
In this study, we found a variant with a specific substitution that may cause disease and which, as far as we know, had not been described previously. There is no description of this specific variant in the OMIM,17 dbSNP,18 Ensembl,19 and ClinVar20 databases. This mutation was also not found among the 353 RHO gene mutations already described in the ExAC database,21 which holds data on exomes of approximately 61,000 individuals from several regions of the world, nor was it found in the UniProt protein database.22 However, previous studies did show a correlation between the clinical presentation of RP in Australian, Japanese, and French families and a variant that leads to the substitution of the amino acid asparagine by serine in the same codon.11–13,16 Hemispheric pigmentary degeneration with reduced scotopic ERG and low magnitude of response density in multifocal ERG were the main characteristics. Our case presented a diffuse retinal degeneration, with severely compromised ERG.
It is important to highlight that crystallographic evidence suggests that asparagine at position 15 of the protein RHO may be glycated,7 which reinforces the possibility that amino acid substitution in this position of the protein may alter protein function. Asparagine is a neutral and apolar amino acid with two carbon atoms in its side chain. Otherwise lysine has a positive charge and has four carbons in its side chain, which also suggests that this amino acid substitution may have a functional effect.
Since other cases of this disease have not been described in the patient’s family, we suspect that the mutation may have taken place in the patient herself (de novo mutation). In order to confirm this hypothesis, the ideal situation would be to analyze her parents to exclude the presence of mutation in one of them. This, however, was not feasible as both of them are deceased.
The ophthalmoscopy, visual fields, IVFA, FAF, OCT, OCT-A, and ERG findings correspond with typical RP patterns. The multimodal analysis of the case shows that the correlations between some points of hypoperfusion compromising the deepest capillaries layers of the central area and discs (OCT-A) and the hyper-autofluorescence findings indicate the possibility of more individualized follow-up. The perfusion deficits may indicate the places where the cells have probably been lost or are severely dysfunctional, secondary to primary cell damage.
In conclusion, a new RP-associated genetic mutation has been presented with diffuse retinal pigmentary degeneration by means of color fundus photography, visual fields,IVFA, FAF, OCT, OCT-A and electrophysiological studies.
Author contributions
Manuel AP Vilela: substantial contributions to acquisition of data of examination, conception and design, collection and interpretation of data, drafting the article critically for important intellectual content, and final approval of the version to be published. Roberta K Menna Barreto: substantial contributions to design, collection of data, drafting the article critically for important intellectual content. Pedro K Menna Barreto: substantial contributions to design, collection of data, drafting the article critically for important intellectual content. Juliana MF Sallum: substantial contribution to acquisition of data of examination, interpretation of data, and final approval of the version to be published. Vanessa S Mattevi: substantial contribution to acquisition of data of examination, interpretation of data, and final approval of the version to be published. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Manuel AP Vilela, Roberta K Menna Barreto, Pedro K Menna Barreto, Vanessa S Mattevi, and Juliana MF Sallum certify that they have no affiliations with or involvement in any organization or entity with any kind of financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
References
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. | ||
Côco M, Han SW, Sallum JMF. Terapia gênica em distrofias hereditárias de retina [Gene therapy in hereditary retinal dystrophies]. Arq Bras Oftalmol. 2009;72(4):560–566. Portuguese. | ||
He Y, Zhang Y, Su G. Recent advances in treatment of retinitis pigmentosa. Curr Stem Cell Res Ther. 2015;10(3):258–265. | ||
Sacchetti M, Mantelli F, Merlo D, Lambiase A. Systematic review of randomized clinical trials on safety and efficacy of pharmacological and nonpharmacological treatments for retinitis pigmentosa. J Ophthalmol. 2015;2015:737053. | ||
Khorana HG. Rhodopsin, Photoreceptor of the Rod Cell. J Biol Chem. 1992;267(1):1–4. | ||
McWilliam P, Farrar GJ, Kenna P, et al. Autosomal dominant retinitis pigmentosa (ADRP): localization of an ADRP gene to the long arm of chromosome 3. Genomics. 1989;5(3):619–622. | ||
Sullivan LJ, Makris GS, Dickinson P, et al. A new codon 15 rhodopsin gene mutation in autosomal dominant retinitis pigmentosa is associated with sectorial disease. Arch Ophthalmol. 1993;111(11):1512–1517. | ||
Fujiki K, Hotta Y, Murakami A, et al. Missense mutation of rhodopsin gene codon 15 found in Japanese autosomal dominant retinitis pigmentosa. Jpn J Hum Genet. 1995;40(3):271–277. | ||
Audo I, Manes G, Mohand-Saïd S, et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Invest Ophthalmol Vis Sci. 2010;51(7):3687–3700. | ||
Yoshii M, Murakami A, Akeo K, et al. Visual function in retinitis pigmentosa related to a codon 15 rhodopsin gene mutation. Ophthalmic Res. 1998;30(1):1–10. | ||
Fishman GA, Stone EM, Gilbert LD, Sheffield VC. Ocular findings associated with a rhodopsin gene codon 106 mutation. Glycine-to-arginine change in autosomal dominant retinitis pigmentosa. Arch Ophthalmol. 1992;110(5):646–653. | ||
Stone EM, Kimura AE, Nichols BE, Khadivi P, Fishman GA, Sheffield VC. Regional distribution of retinal degeneration in patients with the proline to histidine mutation in codon 23 of the rhodopsin gene. Ophthalmology. 1991;98(12):1806–1813. | ||
Fishman GA, Stone EM, Sheffield VC, Gilbert LD, Kimura AE. Ocular findings associated with rhodopsin gene codon 17 and codon 182 transition mutations in dominant retinitis pigmentosa. Arch Ophthalmol. 1992;110(1):54–62. | ||
Fishman GA, Stone EM, Gilbert LD, Kenna P, Sheffield VC. Ocular findings associated with a rhodopsin gene codon 58 transversion mutation in autosomal dominant retinitis pigmentosa. Arch Ophthalmol. 1991;109(10):1387–1393. | ||
Fishman GA, Vandenburgh K, Stone EM, Gilbert LD, Alexander KR, Sheffield VC. Ocular findings associated with rhodopsin gene codon 267 and codon 190 mutations in dominant retinitis pigmentosa. Arch Ophthalmol. 1992;110(11):1582–1588. | ||
Polymorphism Phenotyping v2 (PolyPhen-2). Available from: http://genetics.bwh.harvard.edu/pph2/. Accessed 3 April 2018. | ||
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: MIM 180380. Date last edited 02/27/2018}. Available from: https://omim.org/. Accessed September 18, 2018. | ||
Short Genetics Variations (dbSNP). Available from: https://www.ncbi.nlm.nih.gov/projects/SNP/. Accessed April 3, 2018. | ||
Ensembl GRCh37. Available from: http://grch37.ensembl.org/Homo_sapi ens/Gene/Variation_Gene/Table?g=ENSG00000163914;r=3:129247483-129254012;t=ENST00000296271. Accessed April 3, 2018. | ||
National Center for Biotechnology Information. Available from: https://www.ncbi.nlm.nih.gov/clinvar?term=180380%5bMIM. Accessed April 3, 2018. | ||
ExAC Browser Beta. Available from: http://exac.broadinstitute.org/gene/ENSG00000163914. Accessed April 3, 2018. | ||
UniProt. 2018. Available from: http://www.uniprot.org/uniprot/P08100. Accessed September 18, 2018. | ||
Zafar S, Ahmad K, Ali A, Baig R. Retinitis pigmentosa genes implicated in South Asian populations: a systematic review. J Pak Med Assoc. 2017;67(11):1734–1739. | ||
Kaushal S, Ridge KD, Khorana HG. Structure and function in rhodopsin: the role of asparagine-linked glycosylation. Proc Natl Acad Sci USA. 1994;91(9):4024–4028. | ||
Singh M, Tyagi SC. Genes and genetics in eye diseases: a genomic medicine approach for investigating hereditary and inflammatory ocular disorders. Int J Ophthalmol. 2018;11(1):117–134. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.