Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 11
Novel approaches to treating advanced systemic mastocytosis
Authors Gilreath JA ![]() , Tchertanov L
, Tchertanov L ![]() , Deininger MW
, Deininger MW
Received 24 February 2019
Accepted for publication 26 April 2019
Published 10 July 2019 Volume 2019:11 Pages 77—92
DOI https://doi.org/10.2147/CPAA.S206615
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
JA Gilreath,1 L Tchertanov,2 MW Deininger3
1Department of Pharmacotherapy, College of Pharmacy and Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 84112, USA; 2Centre de Mathématiques et de Leurs Applications (CMLA-CNRS), ENS Paris-Saclay, Cachan 94235, France; 3Division of Hematology and Hematologic Malignancies and Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 84112, USA
Abstract: Mastocytosis is a myeloproliferative neoplasm characterized by expansion of abnormal mast cells (MCs) in various tissues, including skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes. Subtypes include indolent systemic mastocytosis, smoldering systemic mastocytosis and advanced systemic mastocytosis (AdvSM), a term collectively used for the three most aggressive forms of the disease: aggressive systemic mastocytosis, mast cell leukemia, and systemic mastocytosis with an associated clonal hematological non-mast cell disease (SM-AHNMD). MC activation and proliferation is physiologically controlled in part through stem cell factor (SCF) binding to its cognate receptor, KIT. Gain-of-function KIT mutations that lead to ligand-independent kinase activation are found in most SM subtypes, and the overwhelming majority of AdvSM patients harbor the KITD816V, mutation. Several approved tyrosine kinase inhibitors (TKIs), such as imatinib and nilotinib, have activity against wild-type KIT but lack activity against KITD816V,. Midostaurin, a broad spectrum TKI with activity against KIT,D816V has a 60% clinical response rate, and is currently the only drug specifically approved for AdvSM. While this agent improves the prognosis of AdvSM patients and provides proof of principle for targeting KITD816V, as a driver mutation, most responses are partial and/or not sustained, indicating that more potent and/or specific inhibitors are required. Avapritinib, a KIT and PDGFRα inhibitor, was specifically designed to inhibit KITD816V,. Early results from a Phase 1 trial suggest that avapritinib has potent antineoplastic activity in AdvSM, extending to patients who failed midostaurin. Patients exhibited a rapid reduction in both symptoms as well as reductions of bone marrow MCs, serum tryptase, and KITD816V, mutant allele burden. Adverse effects include expected toxicities such as myelosuppression and periorbital edema, but also cognitive impairment in some patients. Although considerable excitement about avapritinib exists, more data are needed to assess long-term responses and adverse effects of this novel TKI.
Keywords: avapritinib, BLU285, systemic mastocytosis, KIT, tyrosine kinase inhibitor
Introduction
Mastocytosis is a myeloproliferative neoplasm (MPN) characterized by expansion of abnormal mast cells (MC) in various tissues, including skin, bone marrow (BM), gastrointestinal tract, liver, spleen, or lymph nodes. Based on the sites of organ involvement and extent of organ dysfunction, several subtypes are distinguished according to the 2016 World Health Organization (WHO) classification of myeloid neoplasms (Table 1).1 Cutaneous mastocytosis is strictly limited to the skin, and may present in several patterns, most commonly as urticaria pigmentosa (maculopapular CM). All other subtypes are systemic by definition, and encompass a very broad clinical spectrum which includes indolent systemic mastocytosis (ISM), smoldering systemic mastocytosis (SSM), aggressive systemic mastocytosis (ASM), mast cell leukemia (MCL), and systemic mastocytosis with an associated clonal hematological non-mast cell disease (SM-AHNMD).1 The term advanced systemic mastocytosis (AdvSM) refers to the three most aggressive forms of the disease, namely ASM, MCL, and SM-AHNMD, which will be focus for this review. The BM is involved in nearly every case of AdvSM.2 Activating mutations of KIT, a receptor tyrosine kinase (RTK) also known as stem cell factor (SCF) receptor, are central to mastocytosis pathogenesis, enabling proliferation and survival of abnormal MCs in affected tissues.3 Although various activating mutations have been described, the D816V mutation in the activation loop of KIT (KITD816V) accounts for more than 90% of all KIT mutations in AdvSM.
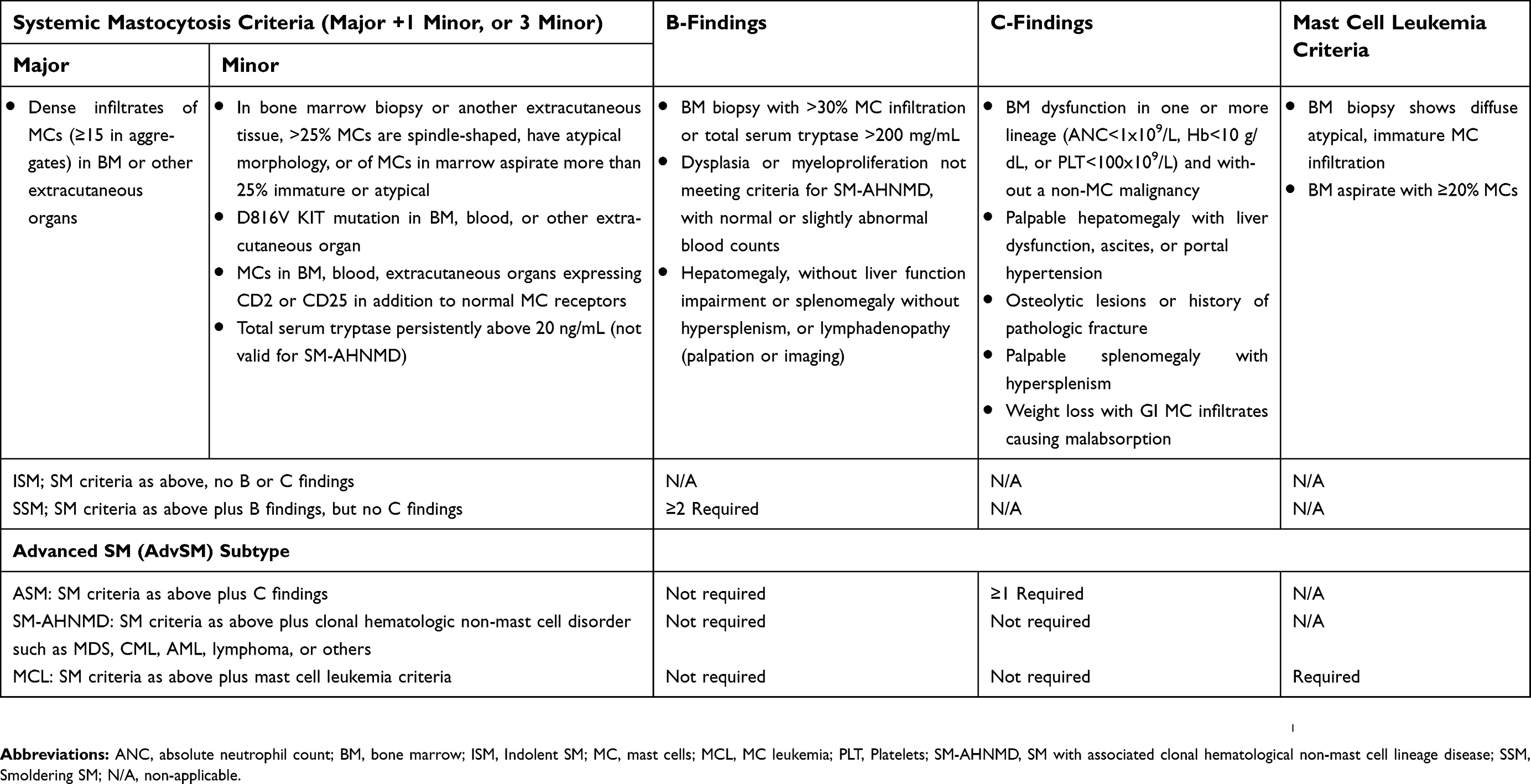 |
Table 1 World Health Organization diagnostic criteria for systemic mastocytosis |
KITD816V was recognized as an activating mutation in mastocytosis as early as 1993,4 however targeting KITD816V proved a formidable challenge for drug development and until recently, AdvSM was an inexorably progressive malignancy associated with a median overall survival of 3.5 years or less.5–8 The introduction of midostaurin, a broad spectrum tyrosine kinase inhibitor (TKI) with activity against KITD816V improved the prognosis of patients with AdvSM, providing proof of principle for targeting this mutant.5 Unfortunately, most responses to midostaurin are partial and not sustained, indicating that more potent and/or specific inhibitors are required to fully exploit the potential of targeting KITD816V in AdvSM. After reviewing classification and pathophysiology of AdvSM, including key components of KIT receptor biology, we will cover the goals of AdvSM treatment, including symptomatic management and reduction of MC burden. Lastly, we will focus on KIT inhibitors, with particular attention paid to avapritinib (BLU-285), a novel KIT inhibitor currently in early clinical trials.
Physiological mast cell function
MCs are highly differentiated hematopoietic cells of myeloid lineage origin. Their most striking morphological feature is secretory granules with strong affinity to basophilic dyes. The abundance of these granules led Paul Ehrlich to coin the name mast cell (for “well fed cell”) in his doctoral thesis.9 Although best known for their role in allergy and anaphylaxis, MCs are thought to play important roles in wound healing, angiogenesis, immune tolerance, defense against pathogens, and blood–brain barrier function.10,11 However the true physiological function of MCs remains a matter of debate, as there is no known disease state related to their absence.12
MCs express high affinity receptors for the Fc region of IgE antibodies (FcεRI) and MCs become coated with IgE molecules. Upon IgE crosslinking, degranulation ensues, which releases highly bioactive substances, commonly called mediators.13 Histamine is the best known MC mediator, but many other pre-formed molecules are released, including serotonin, heparin, neutral proteases (tryptase and chymase, carboxypeptidase, cathepsin G), peroxidase, phospholipases, acid hydrolases, lipid mediators (LTB4, LTC4, PGE2, PGD2, PAF), cytokines (TNF-α, interferons, inflammatory interleukins), chemokines (IL-8 and members of the CCL-family), and growth factors (SCF, granulocyte-macrophage colony stimulating factor (GM-CSF), amongst others).13,14 For a comprehensive review of physiological MC function the reader is referred to an excellent publication.14
Diagnosis and classification of advanced systemic mastocytosis
A diagnosis of SM is established, when one major and one minor or at least three minor criteria are met (Table 1). The one major criterion requires the demonstration of dense, multifocal MC clusters (>15 MCs in aggregate) in BM or other extracutaneous organs such as the liver, spleen, or gastrointestinal tract.1,15 Multifocal MC clusters are the most important finding to establish a SM diagnosis, while the minor criteria either confirm the clonal nature or aberrant phenotype of the MCs.2,16,17 SM is then further categorized as indolent, smoldering or aggressive, depending on the presence or absence of “B findings” or “C findings” (Table 1).1,16 A subset of patients with AdvSM has an associated myeloid neoplasm, most commonly chronic myelomonocytic leukemia. The prognosis of these patients is poor, and addressing the non-MC component of these complex myeloid neoplasms is challenging.1
Biology of the KIT receptor
Physiological roles
KIT, also known as CD117, is a transmembrane receptor encoded by the KIT gene located on chromosome 4q12 that functions as the receptor for SCF. Together with other transmembrane receptors such as platelet-derived growth factors receptors α and β (PDGFRα and PDGFRβ), colony stimulating factor 1 receptor (CSF1R), and Fms-like tyrosine kinase 3 receptor (FLT3), KIT belongs to the class III family of receptor tyrosine kinases (RTKs), which serves a range of functions.18,19 KIT is expressed on multiple and diverse tissues, including hematopoietic stem and progenitor cells (HSPC), a subset of natural killer cells, epithelial cells in the skin adnexa, melanocytes, breast tissue, interstitial cells of Cajal (pacemaker cells in the gastrointestinal tract) and brain cells.20–22 Germline loss-of-function mutations of KIT result in piebaldism, an autosomal dominant condition characterized by patches of white hair and skin23 and KIT-deficient mice have reduced fertility.24 High expression of KIT in the adult murine hippocampus suggests a possible role for KIT in learning and memory. KIT null mice (heterozygous) show a deficiency in normal synaptic transmission between dentate gyrus neurons and hippocampal CA3 pyramidal cells. Moreover KIT mutation led to poorer performance in completing configural learning tasks.25 KIT-deficient mice develop macrocytic anemia, reflecting KIT’s role in erythropoiesis.26 Importantly, SCF/KIT signaling is required not only for MC differentiation from HSPCs, but also for the growth, migration, and survival of mature MCs, which uniquely retain high-level KIT expression.27 Understanding the phenotypic consequences of KIT knockout is important for assessing potential long-term consequences of suppressing KIT, and the reader is referred to a comprehensive review of KIT function.28
KIT structure and function
Full length human KIT spans 976 amino acids that are organized in structurally and functionally distinct domains (Figure 1), including extracellular, transmembrane, and intracellular components.29 The extracellular domain comprises nearly one-half of the receptor and consists of 5 IgG-like domains (amino acids 23–520), while the transmembrane domain only spans amino acids (AAs) 521–543. Intracellular components are comprised of a juxtamembrane (JM) domain; (AAs 544–581), coupled to a complex tyrosine kinase (TK) domain (AAs 582–937), and a carboxy terminus. The intracellular TK domain is further subdivided into TK1 and TK2 domains which are separated by a linker region, also known as the kinase insert (KI, AAs 685–761). Together, the TK1, KI, and TK2 triad are referred to as the “split kinase domain.”19
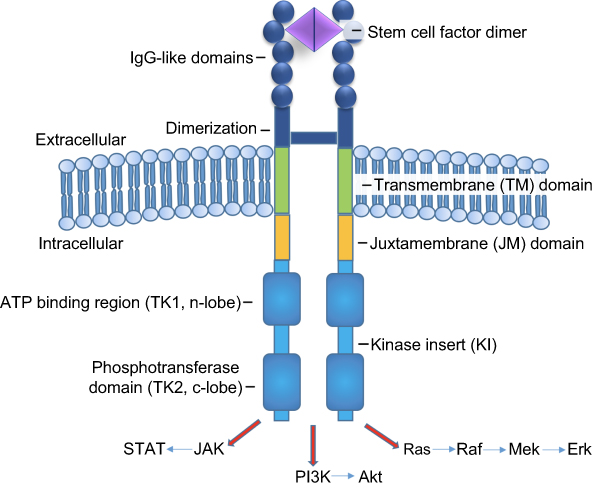 |
Figure 1 Mast cell cKIT (CD117) receptor structure.Note: KIT receptor, a class III RTK, includes extracellular (dark blue), transmembrane (green), and intracellular domains. The intracellular TK domain (light blue) is further subdivided into TK1 and TK2 domains which are separated by the KI. Together, the TK1, KI, and TK2 triad are referred to as the “split kinase domain.” The extracellular domain binds SCF to initiate kinase activation. The TK domain then becomes activated and serves to phosphorylate substrates which transduce various signals downstream via pathways such as JAK/STAT, PI3K, and Ras/Raf/Mek/Erk. |
Structure-function relationships during KIT activation
The TK domain of KIT follows the structural blueprint of class III RTKs, containing a small amino-terminal lobe (N-lobe) corresponding to TK1; AAs 582–671), consisting of a five stranded β-sheet, αC helix, and a hinge segment, and a larger carboxy-terminal lobe (C-lobe) corresponding to TK2. A cleft between these lobes defines the catalytic pocket, which harbors the activation loop (AL), and the aspartate/phenylalanine/glycine motif (DFG, corresponding to amino acids 816–818 in human KIT) that binds Mg2+ and coordinates the ATP β- and γ-phosphate groups for catalytic transfer28 (Figure 2). N- and C-lobes are linked by KI, represented by a “pseudo-KI” in all crystallographic structures.
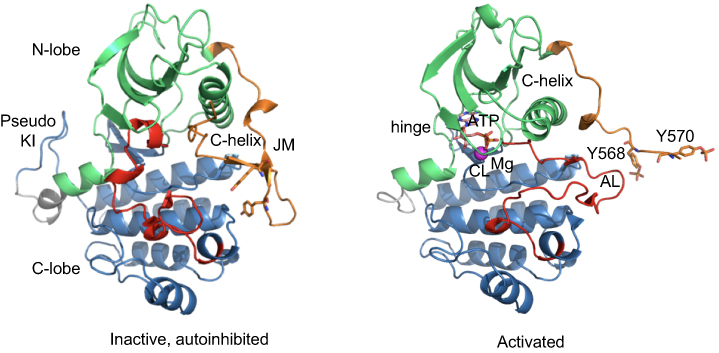 |
Figure 2 Structure of the intracellular domain of the receptor tyrosine kinase KIT in the autoinhibited and activated states. Ribbon diagrams of the cytoplasmic domain of KIT in the inactive (left) and activated (right) forms according to crystal structures (PDB: 1PKG and 1T45, respectively). The intracellular domain is composed of a juxtamembrane (JM) region (in orange), and a tyrosine kinase (TK) consisting of an ATP-binding region (N-lobe, in green) and the phosphotransferase domain (C-lobe, in blue) separated by a kinase insert (KI, shown as a pseudo-KI, in grey). In the inactive state (left), the JM is in the autoinhibited conformation, stabilized through multiple contacts with the activation loop (AL, in red), the αC helix and the catalytic loop (CL). The AL is in the folded conformation packed to TK. Both JM and AL protect the catalytic site from substrate binding. In the activated state (right), the JM is moved from the TK to the solvent exposed position. AL adopts an extended conformation and deploys out of the active site, allowing substrates to access its binding site. ATP an Mg2+ cation are shown as sticks and ball, respectively. The tyrosine residues, Y568 and Y570, (non-phosphorylated in the inactive KIT and phosphorylated in the activated protein) within JM are shown as sticks. |
Kinase activity is conformationally controlled by JM insertion into the TK domain by constraining the relative movements of the two TK lobes around a flexible hinge which maintains the receptor in the energetically favored, auto-inhibited state. JM is stabilized through contacts with both the activation loop (C-lobe), the αC helix (N-lobe) and the catalytic loop (C-lobe). JM thus exerts dual autoinhibitory action through dynamic and steric constraints, thereby preventing conformational change and subsequent substrate binding. In the inactive form, catalytic triad DFG is in the “out” conformation, which permits the unphosphorylated JM domain to dock to the catalytic cleft, shielding the ATP-binding pocket. In the DFG-out conformation, aspartate (hydrophilic) is oriented away from the ATP binding pocket and acts to stabilize this inactive closed conformation through multiple hydrogen bonds.18
Physiological KIT activation begins when SCF binds to the N-terminal IgG-like motifs 2 and 3 of the KIT extracellular domain, which in turn activates the C-terminal IgG-like motifs 4 and 5 to initiate dimerization of the extracellular, transmembrane, and intracellular components.18,30,31 Extracellular SCF binding and receptor dimerization are followed by activation of low level kinase activity causing transphosphorylation of the JM domain at four tyrosines (primarily 568 and 570), which disrupts autoinhibitory interactions leading to destabilization of JM docking to AL.18 Upon JM phosphorylation, the JM moves away from the ATP binding pocket, the interactions of aspartate 816 are disrupted and the DFG motif reorients to the inward facing position (aspartate inward and phenylalanine outward), thereby allowing rotation of the N-lobe toward the C-lobe, triggering the AL to depart from the catalytic site and allowing for ATP binding. Through this conformational change in the AL, an exposed β-strand in the catalytic site provides a functional platform for substrate binding.32 This transition from the inactive to active state switches the catalytic site to the “on” position allowing for substrate binding, and controls phosphorylation of JM at multiple tyrosine residues which serve as recognition/docking sites for phospho-tyrosine binding proteins that transmit intracellular signals downstream to their effector sites.28,33
Activation of signal transduction downstream of KIT
Upon phosphorylation, three tyrosines in the KI (Tyr703, Tyr721, and Tyr730) attract the adaptor protein Grb2, phosphatidylinositol 3-kinase (PI3K), and phospholipase Cγ, respectively.28 Additionally, phosphorylated Tyr900 in the distal kinase domain (C-terminal) also binds PI3K, and plays an essential role in MC migration.28,34 Activated PI3K activates AKT to promote MC survival.35 Grb2 activation also plays a role in activating both the Rat Sarcoma (RAS)/Rapidly Accelerated Fibrosarcoma (RAF)/MAPK ERK Kinase (MEK)/Extracellular Signal-Regulated Kinase (ERK), and Janus Kinase (JAK)/Signal Transducer and Activator of Transcription (STAT) pathways to increase cell proliferation, differentiation, survival, cytokine production, chemotaxis, and adhesion (Figure 1).36
Negative regulation of KIT
Wild-type KIT receptors activated by SCF binding are quickly downregulated to limit duration of signaling. Negative feedback loops such as KIT ubiquitinization and internalization are activated which dephosphorylate KIT to terminate kinase activation.18 Furthermore, activation of protein kinase C (PKC) may deplete phosphate from KIT and induce shedding of its extracellular domain thereby decreasing SCF binding ability. Phosphatase SHP1 has also been shown to negatively regulate activated KIT.34
Constitutive KIT activation in malignancy
Mutation types and tissue tropism
Constitutive activation of KIT occurs as the result of missense mutations or in-frame deletions/insertions that disrupt auto-inhibition. Activating somatic mutations occur in different KIT domains, and exhibit considerable tissue specificity.28 Acquisition of secondary KIT mutations (compound mutations) is common in patients with gastrointestinal stromal tumors (GISTs) who develop clinical resistance to TKIs. For instance, GIST patients with exon 11 mutations (JM domain) initially respond to imatinib.37 Relapse is usually associated with mutations in the ATP binding pocket (exons 13 and 14) or the activation loop (exons 17 and 18).38–41 Thus far experience with potent KIT inhibitors in AdvSM is limited, and no compound mutations have been described, including patients with acquired resistance to midostaurin.42 For the purpose of this review we will concentrate on single mutations observed in patients with mastocytosis.
Mutations in the extracellular domain (exons 8 and 9)
Mutations of residues encoding the extracellular domain impart a conformational change that increases affinity to SCF, promoting KIT activation in lower concentrations of SCF than physiologically observed.28,31,39 However, ligand is still required for kinase activation. Extracellular domain mutations primarily occur in pediatric (cutaneous) mastocytosis and in core binding factor (CBF)-mutated acute myeloid leukemia (AML).31,43
Mutations in the transmembrane domain (exon 11)
Mutations in the transmembrane domain (TM) are rare. The first report described a Phe522Cys point mutation in a patient with mastocytosis. While the bone marrow biopsy revealed excessive numbers of mature-appearing MCs, CD2 and CD25 markers were lacking. Oral Imatinib therapy resulted in dramatic improvement in symptoms and MC burden.44
Mutations in the juxtamembrane domain (exon 11)
The majority of mutations (67%) seen in GISTs occur in the JM domain (AA 544–581).45,46 Most mutations are deletions of a short or extended sequence (from 2 to 16 amino acids), most commonly involving AAs 557–559. In ASM, one single JM-mutant case has been described with an isoleucine for valine exchange at codon 559, resulting in gain-of-function and imatinib resistance.47 Although JM mutations are typically imatinib-sensitive, KITV559I may stabilize an active KIT conformation, thereby reducing imatinib affinity.
Mutations in the activation loop (exon 17 and 18)
Somatic mutations in the activation loop (AL) of the kinase are most frequently seen in adults with AdvSM (Figure 2).18,31 AL mutations of class III RTKs most often involve the aspartate residue of the DFG motif (816 in KIT, 835 in FLT3, and 842 in PDGFRα). The most common variant in AdvSM is a valine for aspartate substitution at codon 816 in exon 17 (KITD816V), which occurs in more than 90% of cases. One study reported KITD816V in 100% of patients with ASM and SM-AHNMD, 97% of patients with ISM, and 67% of MCL cases.48 Rarely, AdvSM patients have other amino acid substitutions in this position, such as KITD816Y or KITD816H or more distally within the activation loop such as KITD820G. In KITD816V, because the hydrophilic aspartic acid residue is involved in stabilizing the DFG-out conformation, when DFG becomes VFG, the hydrophobic valine forces this new triad into the “in” conformation which destabilizes the AL and allows it to move away from the catalytic site, releasing the open conformation of active KIT.18
Mutations in the ATP binding pocket (exons 13 and 14)
ATP binding pocket mutations do not exist independently but occur exclusively in GIST patients with exon 11 mutations who develop imatinib resistance. Whether or no such mutations will be seen in mastocytosis as the experience with KIT inhibitors grows remains to be seen.
Consequences of constitutive KIT activation
All activating KIT mutations ultimately lead to enhanced downstream signaling involving canonical pathways such as PI3K/AKT/mammalian target of rapamycin (mTOR), or JAK/STAT, or RAS/RAF/MEK/ERK that confer resistance to apoptosis and increase proliferation. Of these, AKT and STAT5 appear to be most critical.49 Constitutive activation of AKT is seen in patients with KITD816V and AKT inhibition is associated with growth inhibition of KITD816V-expressing neoplastic MCs.49,50 GAB2, a scaffold protein, enhances cytoplasmic retention of STAT5 which is excessively activated to pSTAT5 by way of KITD816V to enhance PI3K/AKT activity.49 It is likely that differences in the utilization of signaling pathways used by the various KIT mutants contribute to their predilection for certain tissues over others. In mastocytosis, MCs accumulate slowly over time due to constitutive KIT activity and are easily triggered to degranulate leading to the typical symptoms of mastocytosis.16,29,31,51
Clinical presentation
The clinical spectrum of SM is highly variable. Patients with ISM may be highly symptomatic with low quality of life, but their survival is close to that of matched control populations. In contrast patients with AdvSM may have fewer mediator-release symptoms, but much poorer survival outcomes.52
Mediator release-related symptoms
Activated MCs release potent mediators that induce pathologic responses in various organs, causing a host of symptoms. Skin reactions include pruritus, extreme flushing under emotional or physical stress, urticaria, and Darier’s sign (dermatographism). Gastrointestinal symptoms include diarrhea, abdominal pain or cramping, nausea or vomiting, or heartburn. Cardiovascular symptoms include dizziness, palpitations, anaphylaxis with hypotension and syncopal events, and angioedema. Many patients have bone and muscle pain, as well as neuropsychiatric disturbances such as memory impairment, anxiety, and depression.53 In some patients MC mediator release is elicited by minimal triggers such as temperature changes, exercise, or emotional stress.17,54 Certain foods may cause severe vasomotor responses and lead to anaphylaxis.51 Ironically, some of the same medications used to treat symptoms (eg NSAIDS and opiate analgesics) may increase histamine release from mutated MCs.55 For these reasons substantial morbidity, diminished quality of life, and inability to perform activities of daily living are common. Symptom assessment in mastocytosis is challenging, particularly quantifying severity, and standardized symptom assessment tools specifically for mastocytosis are under development.56
End organ damage
Malignant MCs may infiltrate every tissue, but typical target organs in SM are the hematopoietic system (BM, spleen, lymph nodes), liver, and GI tract. The correlation between the density of MC infiltration and the degree of functional impairment is poor. C-findings describe typical consequences of MC-induced end organ damage (Table 1). Importantly, for organ damage to count as a C-finding, MC infiltrates must be demonstrated histologically and other causes excluded.2
Therapy
Goals of therapy
ISM therapy is focused on symptom relief, while prevention of further organ damage by reducing MC burden is the goal in AdvSM. MC reducing agents such as cladribine, IFN-alfa, and midostaurin are associated with significant and potentially life-threatening side effects and are therefore typically avoided in ISM patients who otherwise have an excellent prognosis with less aggressive therapies. For instance, cladribine effectively reduces MC burden in 60–70% of AdvSM patients, but is highly immunosuppressive, which limits its use in ISM to patients with symptoms refractory to maximal mediator-directed management.57 This paradigm may change with the arrival of better tolerated MC reducing agents. Current recommendations for managing ISM and SSM still focus exclusively on symptomatic measures, while MC reducing therapies are reserved for AdvSM.58
Compared to ISM, AdvSM is associated with greatly shortened overall survival, estimated at 3.5 years for those with ASM, 2 years for SM-AHNMD, and less than 6 months for patients with MCL, implying that the goal of AdvSM therapy must be fundamentally different.5,7,8 While controlling symptoms remains important and relies on the same armamentarium of drugs, MC reducing therapy is indicated to prevent further organ damage, and extend survival, often at the expense of significant side effects. Reversal of C-findings is the basis for improving survival, but current definitions pose significant challenges in measuring therapeutic efficacy. For instance, “large lytic bone lesions” qualify as a C-finding, but they rarely improve on therapy, which led to the exclusion of this C-finding from clinical trial response assessments.5 Attributing cytopenias to hypersplenism vs marrow replacement by MCs vs associated hematologic neoplasm vs therapy-induced myelosuppression can be impossible.
Symptom-directed therapy
The management of MC release-related symptoms in AdvSM is similar for patients with ISM. Anti-mediator medications include sedating H1-antihistamines (eg diphenhydramine), non-sedating H1-antihistamines (eg cetirizine), H2-antihistamines (eg ranitidine), MC stabilizers such as cromoglicic acid or ketotifen, leukotriene inhibitors (eg montelukast), proton pump inhibitors (eg omeprazole), corticosteroids, non-steroidal anti-inflammatory drugs (in select cases), and anti-IgE monoclonal antibodies (omalizumab). In our practice, we typically start with H1 and H2 receptor blockers, followed by montelukast and MC stabilizers if symptoms persist. Proton pump inhibitors are useful for patients with predominant gastrointestinal symptoms. As symptoms are rarely controlled by one class of medication, and polypharmacy is frequent, maximizing drug doses before adding additional agents is recommended. We minimize long-term use of systemic steroids and sedatives, and avoid narcotics whenever possible. In our experience it is extremely difficult to wean mastocytosis patients from chronic opiates, preventing them from resuming a functional life, even after responding to MC reducing therapy.
Many of the symptom-directed medications, such as antihistamines, have a short pharmacodynamic effect, necessitating frequent dosing and require a high daily pill burden that reduces adherence. While mediator-directed therapy is often effective during the initial phases of the disease, symptoms often increase with disease progression. However, symptom severity correlates poorly with MC burden, and separating MC-related symptoms from those caused by unrelated conditions is challenging. Hopefully novel, well-tolerated MC-reducing therapies will reduce the need for symptom-directed medications.59
Mast cell-directed therapy
AdvSM is defined by organ damage, and as such the diagnosis of AdvSM is typically an indication to initiate therapies to reduce MC burden. Prior to the approval of midostaurin, this relied on chemotherapeutic agents or interferon-α. After briefly reviewing these conventional approaches, we will focus on TKIs targeting mutant KITD816V.
Conventional chemotherapeutic agents
Non TKI-based cytoreductive options include cladribine and interferon-α. Based on small case series these agents have partial response rates of 35–70%,60,61 but responses are often not durable and complete remission is rare. BM MC burden often remains unchanged, and elevations in serum tryptase may persist.61–64 Importantly, cytoreductive therapies often come at a cost of additional side effects. In the case of interferon-α, one-third of patients experience depression and the adverse effects of therapy can be similar to the symptoms of mastocytosis.61,65 Cladribine causes DNA strand breaks without requiring active cell division, making it an ideal agent for AdvSM with its typically low mitotic rate.66 Unfortunately, nearly half of patients experience grade III/IV neutropenia, and 80% prolonged lymphopenia, increasing the risk of life-threatening opportunistic infections.66 With the advent of KIT-targeted TKIs, the use of cytoreductive therapy has declined and may soon be of historical interest only, except during pregnancy, when interferon-α is one of few agents that may be used safely.
KIT inhibitors
Overview
Numerous orally-administered TKIs (imatinib, nilotinib, regorafenib, sunitinib, crenolanib, dasatinib, masitinib, midostaurin, quizartinib, and avapritinib) have been tested in patients with GIST and AdvSM. Imatinib has been in use for nearly two decades to treat GIST, while sunitinib and regorafenib have been approved as second and third-line agents. In contrast, the use of TKIs to treat AdvSM is a much more recent development, and will be the focus of our review.
Structure/activity relationships
TKIs are broadly classified into type I and type II inhibitors. Type I inhibitors bind both active and inactive kinase conformations, while type II inhibitors recognize only inactive conformations. As TKs exhibit a great deal of structural similarity in their active conformations, but are relatively unique in their inactive conformations, type I inhibitors are typically less selective than type II inhibitors.67 Thus, the specific binding mode of TKIs is particularly important with respect to targeting mutant KIT, where the kinase can be activated by several different mechanisms. Type II TKIs bind KIT through an indirect competition with substrates and maintain KIT in the inactive non-autoinhibited conformation. The binding site of these inhibitors is not limited to the ATP pocket and the adjacent hydrophobic pockets but also includes the “allosteric site” (Figure 3). AL mutations, such as KITD816, stabilize a constitutively active conformation, with the DFG motif in an inward position.68 The resulting conformational change renders KITD816V inaccessible to type II inhibitors. In contrast, type II inhibitors can access KIT mutants with preserved KD conformation, such as transmembrane or juxtamembrane domain mutants (Figure 1).32 Based on these considerations it was predictable that imatinib would be largely ineffective in unselected SM patients, given the high frequency of KITD816V in this population.58,69,70
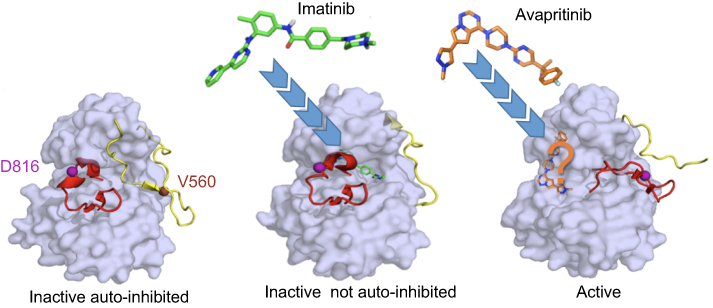 |
Figure 3 KIT inhibition by tyrosine kinase inhibitors (TKIs). Imatinib, a type II TKI, targets inactive auto-inhibited KIT (left) and stabilizes the inactive not auto-inhibited form (middle). Imatinib occupies the ATP binding pocket, the adjacent hydrophobic cavity and the “allosteric” cleft (PDB: 1T46). Avapritinib, a type I TKI, targets the active state of KIT (right) and not only occupies the space where the ATP adenine group binds to KIT, but likely extends into proximal front-pocket regions of the receptor as well. As no published data characterize avapritinib binding to these targets, prediction of its position is based on the binding modes of other type I TKIs (eg, ceritinib binding to ALK RTK, PDB: 4MKC). Protein surface of KIT is in gray. For clearer presentation, only the basic activation fragments, the activation loop (AL, red) and juxtamembrane (JM) region (yellow), with secondary structures observed in the inactive and active states are shown. Inhibitors are shown as sticks. Residues D816 and V560, mutations of which are associated with mastocytosis, are indicated as balls. |
Clinical activity of type II KIT TKIs
Three type II inhibitors (imatinib, nilotinib, and masitinib) have been tested in patients with SM. Predicted based on preclinical data, the activity of these TKIs was limited on a population basis. However, as type II inhibitors are important for the rare patients with KIT variants other than activation loop mutations, they are briefly discussed here.
Imatinib
Imatinib, a potent inhibitor of wild-type KIT kinase activity, was studied in patients with a variety of KIT mutations.71 Early studies established a clear link between mutation type and response. Of ten patients with symptomatic MC disease who prospectively received imatinib at doses of 100–400 mg orally daily, eight patients without KITD816V responded to treatment, and three had complete clinical and hematologic remissions, whereas the two patients with KITD816V did not respond.72 A Phase II clinical study of 20 SM patients tested imatinib 400 mg daily in patients with and without KITD816V. After a median 9 months of therapy, only one KITD816V-negative patient achieved complete remission. While several patients reported symptomatic improvement, including two with KITD816V-positive SM (decreased diarrhea and fatigue), others had no benefit.70 In another study 11 of 14 KITD816V SM patients (five with ASM or SSM and nine with ISM) responded to imatinib 400 mg daily. Notably, serum tryptase levels decreased in 71% and BM MCs in 61% of evaluable patients. Improvements in skin involvement and reduction of hepatomegaly were noted in approximately half of the patients with abnormal findings at entry.73 The reason for the stark discrepancy between these results and other studies remains unclear. In our experience imatinib is not effective in patients with KITD816V-positive SM, and we believe its use should be limited to patients with mutations outside of the kinase domain.
Nilotinib
The second generation TKI nilotinib was evaluated in a Phase II, multicenter study of SM patients with or without KITD816V. Patients received 400 mg orally twice daily until disease progression or unnaceptable toxicity. A total of 61 patients were included (37 ASM, 19 ISM, 5 other). Patients with KITD816V comprised 78% (48/61) of the study population. In the ASM subgroup (all KITD816V-positive), only 21.6% had a minor response. Improvements in BM MCs and laboratory parameters were seen in some patients, including a 29.8% decrease of serum tryptase declined in responders with ASM. The authors concluded that while response rates were low, nilotinb may have modest clinical benefit in select patients.69 It is reasonable to assume that only KITD816V-negative patients may benefit from nilotinib.
Masitinib
Masitinib is an oral TKI developed to target wild-type KIT with greater potency in vitro than KITD816V (IC50 150 vs 500 nmol/L) with greater activity and selectivity than imatinib.74 At submicromolar concentrations it additionally inhibits LYN and FYN, kinases thought to play a role in MC function and survival. While molecular modeling suggested that masitinib would more strongly inhibit MC degranulation, cytokine production, and cell migration than imatinib, response rates of only 18.7% were seen in a severely symptomatic ISM and SSM population (n=67, KITD816V=94%).75 Masitinib has yet to be tested clinically in AdvSM.
Clinical activity of type I TKIs
Type I TKIs tested in patients with SM include dasatinib, midostaurin, and avapritinib.
Dasatinib
Activity profile
Dasatinib is a multikinase inhibitor that was initially conceived as a SRC kinase inhibitor. Clinical development focused on its activity against ABL1 and led to its approval for the treatment of CML and Ph+ acute lymphoblastic leukemia. Dasatinib is structurally unrelated to imatinib, and less susceptible to AL mutations.76 In cellular assays, dasatinib has activity against both KITWT and KITD816V at low nanomolar concentrations. For instance, the IC50 in BaF/3 cells expressing KITD816V is 100–250 nmol/L,77 giving rise to expectations of considerable in vivo activity. Dasatinib was also shown to re-localize KITD816V from the cytoplasm to the cell surface, suggesting reversal of key features of pathogenesis.78
Pharmacology
Oral dosing of dasatinib exhibits dose proportional increases in AUC. High fat meals increase the mean AUC by 14%. Like other TKIs, it is primarily metabolized by the cytochrome P450 system, and is a major substrate for the 3A4 enzyme. UGT enzymes are also involved in formation of metabolites. Dasatinib differs from other TKIs in that its mean terminal half-life is 3–5 hours, precluding prolonged or even continuous suppression of the target kinase.
Clinical efficacy
In a Phase II study of chronic or acute myeloid neoplasms, including 33 patients with SM (18 ISM, 9 ASM, 6 SM-AHNMD), the overall response rate in mastocytosis was 30%. Only two patients, one with SM-myelofibrosis and one with SM-chronic eosinophilic leukemia, achieved a complete response lasting for 5 and 16 months, respectively. Both patients were KITD816V-negative. Additionally, nine SM patients had symptomatic response, lasting 318+ months. Dasatinib’s poor in vivo effectiveness despite the substantial in vitro activity may reflect its short half-life.79 Based on these disappointing results, dasatinib was not developed further for the treatment of mastocytosis. The striking difference between the high level of clinical activity seen with dasatinib in CML and its poor performance in SM suggest that, while constitutively active tyrosine kinase signaling is essential to both, there is a fundamental difference how cells respond to intermittent vs continuous blockade of signaling.80
Midostaurin
Activity profile
Midostaurin (formerly PKC412), a type I inhibitor initially developed to target protein kinase C, is currently the only FDA-approved KIT inhibitor for patients with AdvSM.81 Midostaurin has activity against a very broad spectrum of kinases and is frequently referred to as a multikinase inhibitor. Its activity against FLT3 led to its approval for the treatment of FLT3 ITD mutant acute myeloid leukemia (AML), in combination with chemotherapy.82 In cell-based assays, midostaurin inhibits KITD816V with an IC50 of 44 nmol/L, and KITWT with an IC50 of 138–345 nmol/L.83
Pharmacology
With oral dosing, midostaurin exhibits time-dependent pharmacokinetics, where Cmin is greatest after 1 week of dosing and declines thereafter to reach steady-state after roughly 28 days. Cmax is decreased by 20–27% when the drug is taken with food. Midostaurin is a major substrate for the CYP3A4 enzyme and like most other TKIs, is subject to drug-drug interactions that inhibit (eg azole antifungals, diltiazem, grapefruit juice) or induce (eg carbamazepine, rifampin, St. John’s wort) this enzyme. In the absence of drug-drug interactions, the geometric mean terminal half-life is 19 hours, but more than 25-fold longer for one of its active metabolites (CGP52421; T½ 482 hours) which continues to increase in plasma concentration after 1 month of treatment.84
Clinical efficacy
In an open-label, multicenter study, patients with AdvSM (ASM, n=16; SM-AHNMD, n=57; MCL, n=16), and at least one measurable C-finding were treated with midostaurin 100 mg orally twice daily. Dose reductions to 50 mg twice daily or interruption for up to 21 days were allowed for drug-related AEs. Midostaurin demonstrated a 60% overall response rate in evaluable patients. Nearly half (45%) of patients had a major response, defined as complete resolution of at least one type of C-finding, while 15% had a partial response (>20% improvement in ≥1 C-finding). Overall response rate (ORR) by subtype was 75% for ASM, 58% for SM-AHNMD, and 50% for MCL. Response rates were independent of KIT mutation status, and number of previous therapies. The median duration of response was not reached for patients with ASM (95% CI, 24.1 months to not estimated) or in patients with MCL (95% CI, 3.6 months to not estimated), and was 12.7 months (95% CI, 7.431.4) for SM-AHNMD patients. Of the eight responses in MCL, seven were major, and four were ongoing at 8, 19, 33, and 49 months (time of data cutoff). Additional benefits included independence from red blood cell transfusions in 8/20 (40%) patients and independence from platelet transfusions in 4/4 (100%). The median greatest percentage decrement for MC burden was −59% (BM MCs) and −58% (serum tryptase). Median overall survival was not reached (95% CI, 28.7 months to not reached) for ASM, 20.7 months (95% CI, 16–44.4) for SM-AHNMD and 9.4 months (95% CI, 7.5 to not reached) for MCL. In a post-hoc analysis, non-responders had a significantly lower median overall survival compared to those that did respond (15.4 months, [95% CI, 7.5 to not reached] vs 44.4 months (95% CI 7.5 to not reached), respectively; HR for death 0.42; p=0.005).5
Adverse effects
Midostaurin therapy is associated with a high incidence of AEs. Table 2. Likely owing to the fact that HSPCs express KIT, new or worsening cytopenias (neutropenia, anemia, thrombocytopenia) were observed. The midostaurin dose was reduced in 56% of patients, mostly due to AEs.5 Although re-escalation to the initial dose was feasible in about one-third of patients, maintaining dose intensity may be a barrier to achieving and/or maintaining response.5
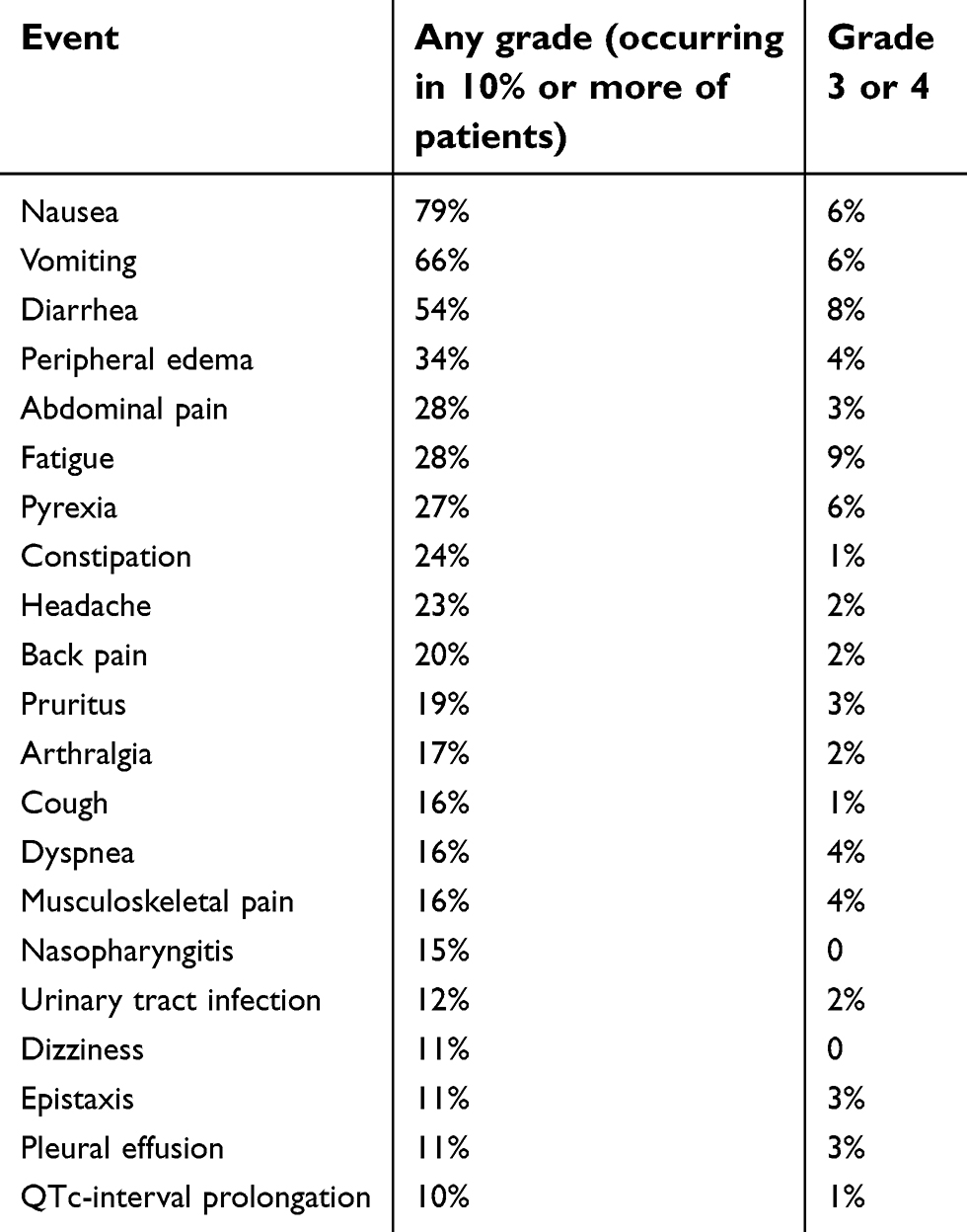 |
Table 2 Adverse effects of midostaurin (non-hematologic) |
Avapritinib
Activity profile
Avapritinib is a rationally designed type I TKI with selective activity against KITD816V vs KITWT31 (Figure 3). Avapritinib binds an active “DFG-in” kinase conformation, as it exclusively occupies the ATP binding pocket without using the adjacent hydrophobic pocket.18,31 Avapritinib is a potent inhibitor of KITD816V (IC50 0.27 nmol/L), roughly 10-fold more potent than midostaurin (IC50 30–40 nmol/L).30,85 Avapritinib also inhibits PDGFRα, a class III tyrosine kinase receptor with similar architecture to KIT. Compared to midostaurin, avapritinib is more selective.30
Pharmacology
After oral administration, avapritinib is rapidly absorbed with a time to maximum plasma concentration (Tmax) of 2–4 hours and exposure increases linearly with increasing dosages. Doses for AdvSM have been studied in a range of 30–400 mg. In a KITD816V mutant xenograft model, 300 mg daily lead to a mean steady state AUC and Cmax above the exposure requirement for maximum effectiveness.86 As a major CYP3A4 substrate, avapritinib drug-drug interactions are likely to be similar to those with midostaurin. The half-life of avapritinib is estimated at 25 hours, supporting once daily dosing. Data on avapritinib metabolism are not yet available.
Clinical efficacy
Clinical trials of Avapritinib in GIST were initiated in 2015, and in AdvSM in 2016. Preliminary results from a Phase I study in AdvSM (EXPLORER trial) were reported in abstract form. A total of 52 adult patients with AdvSM (ASM, n=25; SM-AHNMD, n=15; MCL, n=5; smoldering systemic mastocytosis, n=2; pending central pathology diagnosis, n=5) were treated in either Part 1: a 3+3 dose escalation design (n=32) in one of seven cohorts at doses ranging between 30 mg and 400 mg orally once daily or in Part 2: a dose expansion arm with avapritinib 300 mg orally once daily (n=20). Inclusion criteria included a diagnosis of AdvSM, at least 1 C-finding, age ≥18 years, and ECOG performance status ≤3. Antineoplastic activity was assessed by BM MC burden, serum tryptase, and KITD816V mutant allele burden. ORR was determined by modified International Working Group Myeloproliferative Neoplasms Research and Treatment and European Competence Network on Mastocytosis criteria (m-IWG-MRT-ECNM). Forty-six patients (88%) harbored a KIT mutation (D816V, n=45; D816Y, n=1), and six patients (12%) were wild type KIT.87 Of the 32 patients treated in the dose escalation Phase, the majority of patients had co-occurring mutations (the same patient can have more than one) in TET2 (n=17), DNMT3A (n=9), ASXL1 (n=7), SRSF2 (n=6), and GATA2 (n=6).86
During the dose escalation Phase, most patients experienced first evidence of response at the start of Cycle 3 (after 8 weeks), with median time to first resolution of ≥1 evaluable C-finding of 35 days. At all dose levels, patients exhibited a rapid reduction in MC and KITD816V mutant allele burden. Of 12 patients with urticaria pigmentosa, 10 (83%) noted a reduction in lesions. Across the study population there was a median weight gain of 5 kg, and serum albumin increase of 0.5 mg/dL. Reduction in MCs and KITD816V allele burden was durable, with sustained disease control in seven patients taking avapritinib for more than 1 year. Two of the three MCL patients refractory to midostaurin at study entry showed a marked reduction of MC burden, and two ASM patients with intolerance to midostaurin remained on avapritinib for 5 months or longer.86 Overall, avapritinib had antineoplastic activity across all AdvSM subtypes, and an ORR of 83%, with mostly durable responses. Avapritinib induced BM MC reductions of more than 50% in 33% (n=12/36), reduction of serum tryptase to <20 μg/L in 66% (n=24/36), and undetectable BM MCs in 58% (n=21/26) of evaluable patients (Table 3). Moreover, reductions in splenomegaly, and KITD816V mutant allele were seen, regardless of prior therapy.
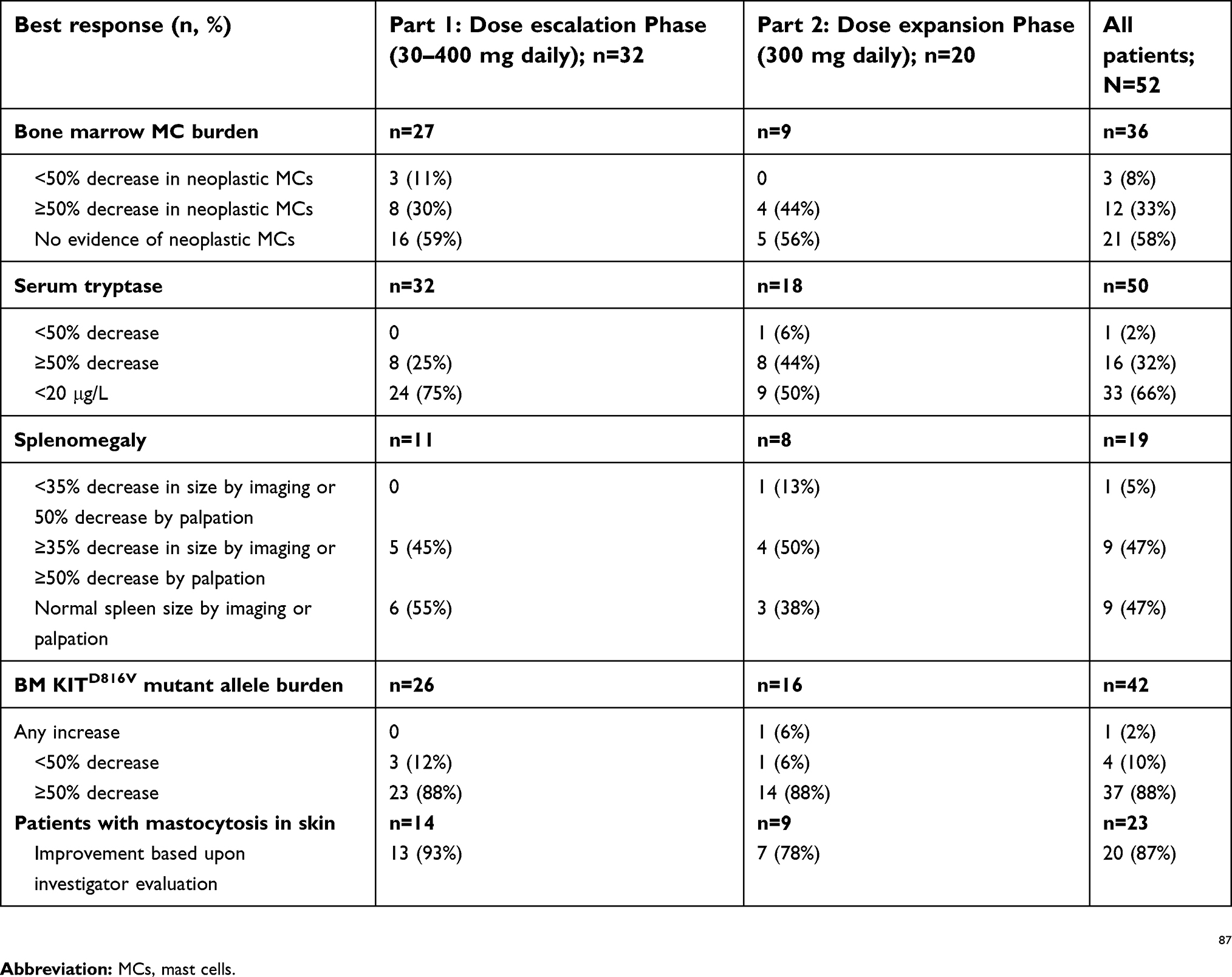 |
Table 3 Antineoplastic activity of avapritinib in a Phase I study (EXPLORER) of patients with AdvSM (median duration of treatment 14 months (Part 1) and 5 months (Part 2) |
Adverse effects
In the early Phase trial of BLU-285 in AdvSM (n=30), avapritinib was noted to be well-tolerated with most adverse events (AE) scored as either grade 1 or 2 by CTCAE using doses between 30–400 mg daily. The most common AEs were periorbital edema (n=13; 43%), anemia, diarrhea, fatigue, peripheral edema (n=8; 27% each), headache (n=7; 23%), thrombocytopenia and nausea (n=6; 20% each). Grade 3 or higher neutropenia occurred in four patients (13%) and grade 3 or higher anemia and periorbital edema occurred in two patients (7%) each. An update to these initial findings included the dose expansion Phase using avapritinib at a dose of 300 mg daily. A total of 52 patients were included. At higher doses AEs were slightly more common. 50/52 (96%) of patients reported an AE at any grade. Treatment-related Grade ≥3 AEs occurred in 28 (54%) of patients. Of more significant concern, specific AEs included ascites (10%, including 4% grade 3/4], pleural effusion (10%), and cognitive impairment unexplained by other potential causes, including thiamine deficiency. Ten patients discontinued therapy, with 6/10 discontinuing due to a drug-related AE (ascites, n=4; encephalopathy, n=1; confusional state, n=1). Avapritinib caused reversible poliosis (whitening of the hair due to lack of melanin in hair follicles), reflecting KIT inhibition in melanocytes, with an onset approximately 1 month into therapy.88 AEs seen in ≥20% of patients are summarized in Table 4. Based upon these findings, a 300 mg daily dose was recommended for Phase II studies. Regarding tolerability and common reasons for drug discontinuation, avapritinib had a lower rate of nausea than midostaurin (37% vs 79%), but at higher doses it can adversely affect cognitive function, particularly short-term memory. The mechanism of this has not been elucidated.
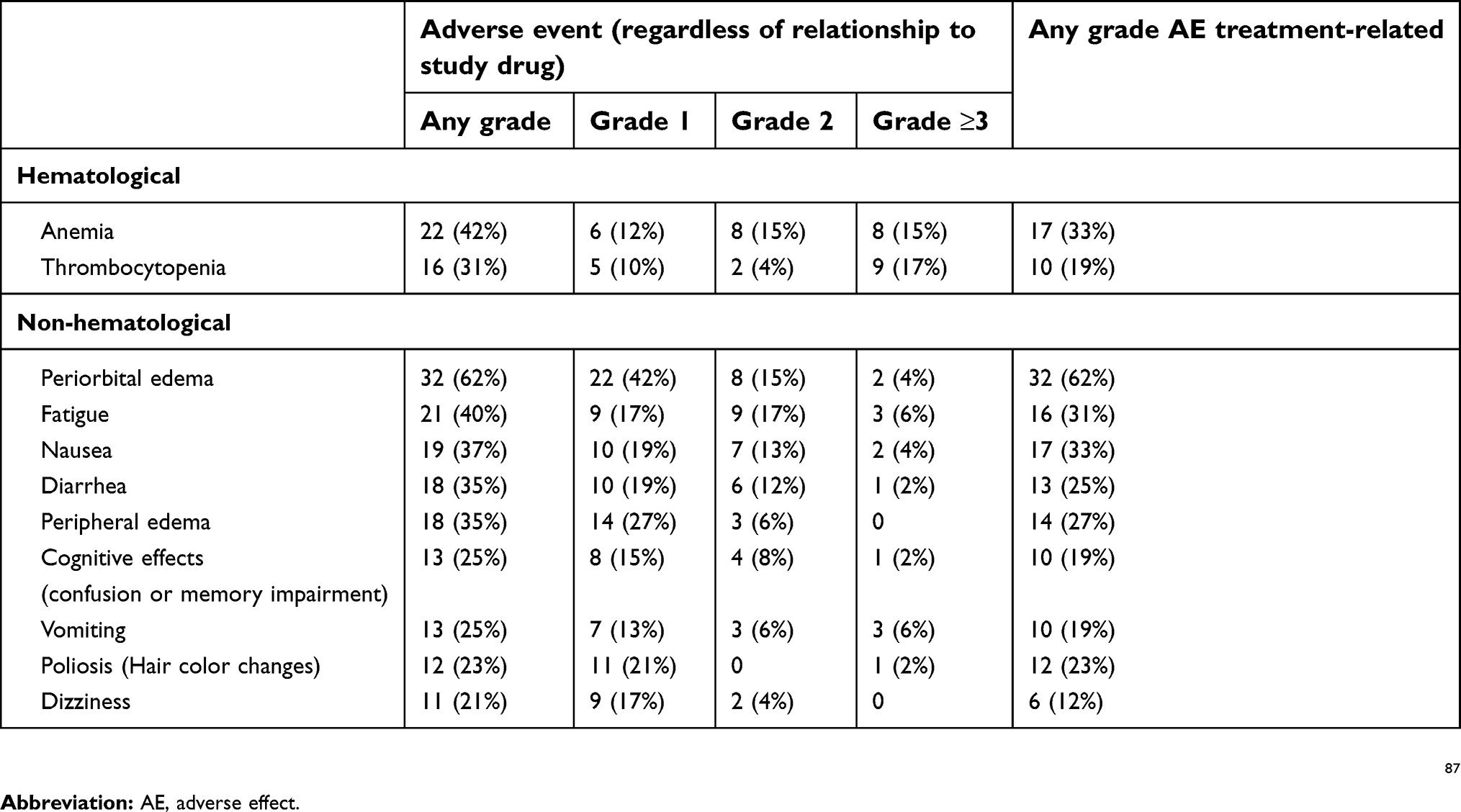 |
Table 4 Avapritinib adverse events occurring in >20% of patients from dose escalation and dose expansion Phase 1 study (n=52) |
Other type I KIT inhibitors in clinical development
DCC-2618 (ripretinib) is a potent KIT and PDGFR inhibitor with activity against KIT mutated in the extracellular domain (exon 9), transmembrane and JM domains (exon 11), ATP binding pocket (exon 13 &14), and AL (exon 17 & 18) of KIT. Ripretinib binds to the kinase “spine” and acts as a surrogate for JM to stabilize the inactive state of KIT. Clinical studies in AdvSM are underway however data are not yet available.
Conclusion
AdvSM is a heterogeneous myeloid neoplasm characterized by accumulation of abnormal MCs in extracutaneous tissues. Clinical symptoms reflect mediator release and/or organ dysfunction. The dominant driver mutation is KITD816V. Imatinib and other type II TKIs are effective in the rare AdvSM patients with KIT mutations other than KITD816V. Midostaurin has significant activity in patients in KITD816V mutant patients and became the first effective therapy for AdvSM. However, enthusiasm has been tempered based on relatively low response rates and limited durability. Moreover, gastrointestinal AEs render long-term adherence challenging. Early results from a Phase 1 trial suggest that avapritinib has potent antineoplastic activity in all subtypes of AdvSM, including patients who failed midostaurin. Patients exhibited a rapid reduction in both symptoms as well as reversal of disease markers such as BM MC infiltration and KITD816V mutant allele burden. Tolerability was generally favorable, although impairments of cognitive function are of potential concern. As a reduction in KITD816V allele burden is predictive of increased overall survival, there is considerable excitement about avapritinib, but more follow-up is clearly required.
Disclosure
Dr Jeffrey Gilreath reports personal fees from Novartis, outside of the submitted work. Dr Michael Deininger reports grants and personal fees from Blueprint; personal fees from Novartis, BMS, Pfizer, and Incyte, during the conduct of the study. The authors report no other conflicts in this work.
References
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
2. Valent P, Sperr WR, Akin C. How I treat patients with advanced systemic mastocytosis. Blood. 2010;116(26):5812–5817. doi:10.1182/blood-2010-08-292144
3. Dahlin JS, Ekoff M, Grootens J, et al. KIT signaling is dispensable for human mast cell progenitor development. Blood. 2017;130(16):1785–1794. doi:10.1182/blood-2017-03-773374
4. Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92(4):1736–1744. doi:10.1172/JCI116761
5. Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530–2541. doi:10.1056/NEJMoa1513098
6. Cohen SS, Skovbo S, Vestergaard H, et al. Epidemiology of systemic mastocytosis in Denmark. Br J Haematol. 2014;166(4):521–528. doi:10.1111/bjh.12916
7. Georgin-Lavialle S, Lhermitte L, Dubreuil P, Chandesris MO, Hermine O, Damaj G. Mast cell leukemia. Blood. 2013;121(8):1285–1295. doi:10.1182/blood-2012-07-442400
8. Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113(23):5727–5736. doi:10.1182/blood-2009-02-205237
9. Crivellato E, Beltrami C, Mallardi F, Ribatti D. Paul Ehrlich‘s doctoral thesis: a milestone in the study of mast cells. Br J Haematol. 2003;123(1):19–21. doi:10.1046/j.1365-2141.2003.04573.x
10. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10(6):440–452. doi:10.1038/nri2782
11. Strbian D, Kovanen PT, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ. An emerging role of mast cells in cerebral ischemia and hemorrhage. Ann Med. 2009;41(6):438–450. doi:10.1080/07853890902887303
12. Rodewald HR, Feyerabend TB. Widespread immunological functions of mast cells: fact or fiction? Immunity. 2012;37(1):13–24. doi:10.1016/j.immuni.2012.07.007
13. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(2):163–172. doi:10.1056/NEJMra1409760
14. Olivera A, Beaven MA, Metcalfe DD. Mast cells signal their importance in health and disease. J Allergy Clin Immunol. 2018;142(2):381–393. doi:10.1016/j.jaci.2018.01.034
15. Valent P, Akin C, Hartmann K, et al. Advances in the classification and treatment of mastocytosis: current status and outlook toward the future. Cancer Res. 2017;77(6):1261–1270. doi:10.1158/0008-5472.CAN-16-2234
16. Onnes MC, Tanno LK, Elberink JN. Mast cell clonal disorders: classification, diagnosis and management. Current Treat Options Allergy. 2016;3(4):453–464. doi:10.1007/s40521-016-0103-3
17. Arock M, Akin C, Hermine O, Valent P. Current treatment options in patients with mastocytosis: status in 2015 and future perspectives. Eur J Haematol. 2015;94(6):474–490. doi:10.1111/ejh.12544
18. Klug LR, Kent JD, Heinrich MC. Structural and clinical consequences of activation loop mutations in class III receptor tyrosine kinases. Pharmacol Ther. 2018. doi:10.1016/j.pharmthera.2018.06.016
19. Rosnet O, Birnbaum D. Hematopoietic receptors of class III receptor-type tyrosine kinases. Crit Rev Oncog. 1993;4(6):595–613.
20. Min KW, Leabu M. Interstitial cells of Cajal (ICC) and gastrointestinal stromal tumor (GIST): facts, speculations, and myths. J Cell Mol Med. 2006;10(4):995–1013.
21. Miettinen M, Lasota J. KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol. 2005;13(3):205–220.
22. Matos ME, Schnier GS, Beecher MS, Ashman LK, William DE, Caligiuri MA. Expression of a functional c-kit receptor on a subset of natural killer cells. J Exp Med. 1993;178(3):1079–1084.
23. Oiso N, Fukai K, Kawada A, Suzuki T. Piebaldism. J Dermatol. 2013;40(5):330–335. doi:10.1111/j.1346-8138.2012.01583.x
24. Kissel H, Timokhina I, Hardy MP, et al. Point mutation in kit receptor tyrosine kinase reveals essential roles for kit signaling in spermatogenesis and oogenesis without affecting other kit responses. Embo J. 2000;19(6):1312–1326. doi:10.1093/emboj/19.6.1312
25. Motro B, Wojtowicz JM, Bernstein A, van der Kooy D. Steel mutant mice are deficient in hippocampal learning but not long-term potentiation. Proc Natl Acad Sci USA. 1996;93(5):1808–1813.
26. Metcalfe DD. Mast cells and mastocytosis. Blood. 2008;112(4):946–956. doi:10.1182/blood-2007-11-078097
27. Valent P, Spanblochl E, Sperr WR, et al. Induction of differentiation of human mast cells from bone marrow and peripheral blood mononuclear cells by recombinant human stem cell factor/kit-ligand in long-term culture. Blood. 1992;80(9):2237–2245.
28. Roskoski R
29. Arock M, Sotlar K, Akin C, et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European competence network on mastocytosis. Leukemia. 2015;29(6):1223–1232. doi:10.1038/leu.2015.24
30. Evans EK, Gardino AK, Kim JL, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017;9:414. doi:10.1126/scitranslmed.aao1690
31. Ustun C, DeRemer DL, Akin C. Tyrosine kinase inhibitors in the treatment of systemic mastocytosis. Leuk Res. 2011;35(9):1143–1152. doi:10.1016/j.leukres.2011.05.006
32. Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–1942.
33. Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109(3):275–282.
34. Heldin CH, Lennartsson J. Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb Perspect Biol. 2013;5(8):a009100. doi:10.1101/cshperspect.a009100
35. Blume-Jensen P, Janknecht R, Hunter T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of bad on Ser136. Curr Biol. 1998;8(13):779–782.
36. Cruse G, Metcalfe DD, Olivera A. Functional deregulation of KIT: link to mast cell proliferative diseases and other neoplasms. Immunol Allergy Clin North Am. 2014;34(2):219–237. doi:10.1016/j.iac.2014.01.002
37. Boichuk S, Lee DJ, Mehalek KR, et al. Unbiased compound screening identifies unexpected drug sensitivities and novel treatment options for gastrointestinal stromal tumors. Cancer Res. 2014;74(4):1200–1213. doi:10.1158/0008-5472.CAN-13-1955
38. Garner AP, Gozgit JM, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20(22):5745–5755. doi:10.1158/1078-0432.CCR-14-1397
39. Rose S. Rapid responses to avapritinib (BLU-285) in mastocytosis. Cancer Discov. 2018;8(2):133.
40. Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128(2):270–279.
41. Tamborini E, Bonadiman L, Greco A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127(1):294–299.
42. Jawhar M, Schwaab J, Naumann N, et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood. 2017;130(2):137–145. doi:10.1182/blood-2017-01-764423
43. Cammenga J, Horn S, Bergholz U, et al. Extracellular KIT receptor mutants, commonly found in core binding factor AML, are constitutively active and respond to imatinib mesylate. Blood. 2005;106(12):3958–3961. doi:10.1182/blood-2005-02-0583
44. Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222–3225. doi:10.1182/blood-2003-11-3816
45. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580.
46. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22(18):3813–3825. doi:10.1200/JCO.2004.05.140
47. Nakagomi N, Hirota S. Juxtamembrane-type c-kit gene mutation found in aggressive systemic mastocytosis induces imatinib-resistant constitutive KIT activation. Lab Invest. 2007;87(4):365–371. doi:10.1038/labinvest.3700524
48. Teodosio C, Garcia-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol. 2010;125(3):
49. Bibi S, Arslanhan MD, Langenfeld F, et al. Co-operating STAT5 and AKT signaling pathways in chronic myeloid leukemia and mastocytosis: possible new targets of therapy. Haematologica. 2014;99(3):417–429. doi:10.3324/haematol.2013.098442
50. Harir N, Boudot C, Friedbichler K, et al. Oncogenic kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood. 2008;112(6):2463–2473. doi:10.1182/blood-2007-09-115477
51. Schuch A, Brockow K. Mastocytosis and anaphylaxis. Immunol Allergy Clin North Am. 2017;37(1):153–164. doi:10.1016/j.iac.2016.08.017
52. Munoz-Gonzalez JI, Jara-Acevedo M, Alvarez-Twose I, et al. Impact of somatic and germline mutations on the outcome of systemic mastocytosis. Blood Adv. 2018;2(21):2814–2828. doi:10.1182/bloodadvances.2018020628
53. Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250–262. doi:10.1002/ajh.23931
54. Koide T, Nakajima T, Makifuchi T, Fukuhara N. Systemic mastocytosis and recurrent anaphylactic shock. Lancet. 2002;359(9323):2084. doi:10.1016/S0140-6736(02)07571-2
55. Marone G, Spadaro G, Granata F, Triggiani M. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res. 2001;25(7):583–594.
56. van Anrooij B, Kluin-Nelemans JC, Safy M, Flokstra-de Blok BM, Oude Elberink JN. Patient-reported disease-specific quality-of-life and symptom severity in systemic mastocytosis. Allergy. 2016;71(11):1585–1593. doi:10.1111/all.12920
57. Tefferi A, Li CY, Butterfield JH, Hoagland HC. Treatment of systemic mast-cell disease with cladribine. N Engl J Med. 2001;344(4):307–309. doi:10.1056/NEJM200101253440415
58. Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage). Blood. 2013;121(16):3085–3094. doi:10.1182/blood-2013-01-453183
59. Austen KF. Systemic mastocytosis. N Engl J Med. 1992;326(9):639–640. doi:10.1056/NEJM199202273260912
60. Barete S, Lortholary O, Damaj G, et al. Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Blood. 2015;126(8) quiz 1050:1009–1016. doi:10.1182/blood-2014-12-614743
61. Casassus P, Caillat-Vigneron N, Martin A, et al. Treatment of adult systemic mastocytosis with interferon-alpha: results of a multicentre phase II trial on 20 patients. Br J Haematol. 2002;119(4):1090–1097.
62. Kluin-Nelemans HC, Oldhoff JM, Van Doormaal JJ, et al. Cladribine therapy for systemic mastocytosis. Blood. 2003;102(13):4270–4276. doi:10.1182/blood-2003-05-1699
63. Kluin-Nelemans HC, Jansen JH, Breukelman H, et al. Response to interferon alfa-2b in a patient with systemic mastocytosis. N Engl J Med. 1992;326(9):619–623. doi:10.1056/NEJM199202273260907
64. Butterfield JH. Response of severe systemic mastocytosis to interferon alpha. Br J Dermatol. 1998;138(3):489–495.
65. Lim KH, Pardanani A, Butterfield JH, Li CY, Tefferi A. Cytoreductive therapy in 108 adults with systemic mastocytosis: outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am J Hematol. 2009;84(12):790–794. doi:10.1002/ajh.21561
66. Akin C. Cladribine for mastocytosis: benefits and risks. Blood. 2015;126(8):931–932. doi:10.1182/blood-2015-06-649525
67. Roskoski R
68. Treiber DK, Shah NP. Ins and outs of kinase DFG motifs. Chem Biol. 2013;20(6):745–746. doi:10.1016/j.chembiol.2013.06.001
69. Hochhaus A, Baccarani M, Giles FJ, et al. Nilotinib in patients with systemic mastocytosis: analysis of the phase 2, open-label, single-arm nilotinib registration study. J Cancer Res Clin Oncol. 2015;141(11):2047–2060. doi:10.1007/s00432-015-1988-0
70. Vega-Ruiz A, Cortes JE, Sever M, et al. hase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res. 2009;33(11):1481–1484. doi:10.1016/j.leukres.2008.12.020
71. Ma Y, Zeng S, Metcalfe DD, et al. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99(5):1741–1744.
72. Pardanani A, Elliott M, Reeder T, et al. Imatinib for systemic mast-cell disease. Lancet. 2003;362(9383):535–536.
73. Droogendijk HJ, Kluin-Nelemans HJ, van Doormaal JJ, Oranje AP, van de Loosdrecht AA, van Daele PL. Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial. Cancer. 2006;107(2):345–351. doi:10.1002/cncr.21996
74. Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4(9):e7258. doi:10.1371/journal.pone.0007258
75. Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389(10069):612–620. doi:10.1016/S0140-6736(16)31403-9
76. Shah NP, Lee FY, Luo R, Jiang Y, Donker M, Akin C. Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood. 2006;108(1):286–291. doi:10.1182/blood-2005-10-3969
77. Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66(1):473–481. doi:10.1158/0008-5472.CAN-05-2050
78. Bougherara H, Georgin-Lavialle S, Damaj G, et al. Relocalization of KIT D816V to cell surface after dasatinib treatment: potential clinical implications. Clin Lymphoma Myeloma Leuk. 2013;13(1):62–69. doi:10.1016/j.clml.2012.08.004
79. Verstovsek S, Tefferi A, Cortes J, et al. Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin Cancer Res. 2008;14(12):3906–3915. doi:10.1158/1078-0432.CCR-08-0366
80. Snead JL, O‘Hare T, Adrian LT, et al. Acute dasatinib exposure commits Bcr-Abl-dependent cells to apoptosis. Blood. 2009;114(16):3459–3463. doi:10.1182/blood-2007-10-113969
81. Kasamon YL, Ko CW, Subramaniam S, et al. FDA approval summary: midostaurin for the treatment of advanced systemic mastocytosis. Oncologist. 2018. doi:10.1634/theoncologist.2018-0222
82. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464. doi:10.1056/NEJMoa1614359
83. Growney JD, Clark JJ, Adelsperger J, et al. Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood. 2005;106(2):721–724. doi:10.1182/blood-2004-12-4617
84. Midostaurin (Rydapt). East Hanover (NJ): Novartis Pharmaceuticals. June 2018. (package insert). Available from: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/rydapt.pdf. Accessed April 23, 2019.
85. Baird JH, Gotlib J. Clinical validation of KIT inhibition in advanced systemic mastocytosis. Curr Hematol Malig Rep. 2018;13(5):407–416. doi:10.1007/s11899-018-0469-3
86. DeAngelo DJ, Quiery AT, Radia D, et al. Clinical activity in a phase 1 study of Blu-285, a potent, highly-selective inhibitor of KIT D816V in advanced systemic mastocytosis (AdvSM). Blood. 2017;130(Suppl 1):2.
87. Deininger MW, Gotlib J, Robinson WA, et al. Avapritinib (BLU-285), a selective KIT inhibitor, is associated with high response rate and tolerable safety profile in advanced systemic mastocytosis (AdvSM): results of a phase 1 study. EHA Annual Meeting, 2018. Abstract PF612.
88. Qiu W, Yang K, Lei M, et al. SCF/c-kit signaling is required in 12-O-tetradecanoylphorbol-13-acetate-induced migration and differentiation of hair follicle melanocytes for epidermal pigmentation. Cell Tissue Res. 2015;360(2):333–346. doi:10.1007/s00441-014-2101-8
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.