Back to Journals » International Journal of Nanomedicine » Volume 13
Non-ionic thiolated cyclodextrins — the next generation
Authors Moghadam A ![]() , Ijaz M
, Ijaz M ![]() , Asim MH, Mahmood A, Jelkmann M, Matuszczak B, Bernkop-Schnürch A
, Asim MH, Mahmood A, Jelkmann M, Matuszczak B, Bernkop-Schnürch A ![]()
Received 4 October 2017
Accepted for publication 12 May 2018
Published 10 July 2018 Volume 2018:13 Pages 4003—4013
DOI https://doi.org/10.2147/IJN.S153226
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Ali Moghadam,1,2 Muhammad Ijaz,2,3 Mulazim Hussain Asim,2,4 Arshad Mahmood,2,5 Max Jelkmann,2 Barbara Matuszczak,6 Andreas Bernkop-Schnürch2
1Institute of Biotechnology, College of Agriculture, Shiraz University, Shiraz, Iran; 2Center for Chemistry and Biomedicine, Department of Pharmaceutical Technology, Institute of Pharmacy, University of Innsbruck, Innsbruck, Austria; 3Department of Pharmacy, COMSATS Institute of Information and Technology, Lahore, Pakistan; 4Department of Pharmaceutics, Faculty of Pharmacy, University of Sargodha, Sargodha, Pakistan; 5Department of Pharmacy, COMSATS Institute of Information Technology Abbottabad, Pakistan; 6Center for Chemistry and Biomedicine, Department of Pharmaceutical Chemistry, Institute of Pharmacy, University of Innsbruck, Innsbruck, Austria
Introduction: The current study was aimed at developing a novel mucoadhesive thiolated cyclodextrin (CD) without ionizable groups and an intact ring backbone for drug delivery.
Materials and methods: Thiolated beta CD (β-CD) was prepared through bromine substitution of its hydroxyl groups followed by replacement to thiol groups using thiourea. The thiolated β-CD was characterized in vitro via dissolution studies, cytotoxicity studies, mucoadhesion studies on freshly excised porcine intestinal mucosa, and inclusion complex formation with miconazole nitrate.
Results: Thiolated β-CDs namely β-CD-SH600 and β-CD-SH1200 displayed 558.66 ± 78 and 1,163.45 ± 96 µmol thiol groups per gram of polymer, respectively. Stability constant (Kc) of 190 M-1 confirmed the inclusion complex formation of miconazole nitrate with β-CD-SH. Inclusion complexes of β-CD-SH600 and β-CD-SH1200 resulted in 157- and 257-fold increased solubility of miconazole nitrate, respectively. In addition, more than 80% of thiol groups were stable even after 6 hours at pH 5. Both β-CD-SH compounds showed at least 1.3-fold improved solubility in water. In contrast to cationic thiolated CDs of the first generation, both thiomers showed no significant cytotoxicity. The mucoadhesive properties of the new thiolated CDs were 39.73- and 46.37-fold improved, respectively.
Conclusion: These results indicate that β-CD-SH might provide a new favorable tool for delivery of poorly soluble drugs providing a prolonged residence time on mucosal surfaces.
Keywords: thiolated cyclodextrin, mucosal delivery, inclusion complex
Introduction
Cyclodextrins (CDs) are considered as probably the smallest polymeric backbone for thiomers representing some of the most broadly considered mucoadhesive excipients.1,2 Because of the ability of thiol groups to form disulfide bonds with mucus glycoproteins covering mucosal membranes,3 thiomers are covalently attached to the mucus layer.4,5 These unique properties guarantee the benefits of prolonged drug residence time on specific target mucosal tissues and a consequently more efficient local therapy.6–8
The first generation of thiolated CD derivatives have been developed recently.1,2 Although they were obtained under comparatively harsh conditions via oxidative opening of the glucose rings, these derivatives showed strongly improved mucoadhesive properties. Nonetheless, their ring-opened structure is certainly disadvantageous to form stable inclusion complexes especially with hydrophobic drugs and to provide subsequently sustained drug release properties. Moreover, the method of oxidative ring opening followed by the covalent attachment of thiol-bearing amino ligands such as cysteamine via reductive amination results in cationic thiolated CDs. These cationic thiolated CDs, however, showed a significant toxic effect on cells.1,2 In addition, an unintended pH-dependent drug release and even incompatibility with anionic drugs are expected consequences.9,10
Having on one hand the strongly prolonged mucosal residence time of thiolated CDs and taking on the other hand the drawbacks of synthesis via oxidative-ring opening and reductive amination into account, it was therefore the aim of this study to generate thiolated CDs via an alternative synthetic pathway. Beta CD (β-CD) was chosen for this study as various derivatives of this type of CD have already shown great potential in drug delivery.8,11 In order to obtain non-ionic thiolated CDs without a ring opening, hydroxyl groups on the glucose subunits of β-CD are replaced by thiol groups via bromination followed by reaction with thiourea.5 The resulting non-ionic thiolated CDs are investigated regarding cytotoxicity, mucoadhesive properties, and their ability to incorporate miconazole, serving as a hydrophobic model drug.
The novelty of this work is the synthesis of non-ionic thiolated CDs that have so far not been tested in the pharmaceutical field. The hypothesis underpinning this research is that the non-ionic thiolated CDs are less toxic and inclusion complexes of higher drug payload can be formed exhibiting a prolonged drug release compared to cationic thiolated CDs.
Materials and methods
Materials
β-CD (molecular weight: 1,134.98 Da), lithium bromide (LiBr), triphenylphosphine (Ph3P), N,N-dimethylacetamide (DMA), N-bromosuccinimide (NBS), thiourea, 5,5′-dithiobis(2-nitrobenzoic acid) (Ellman’s reagent), Hanks’ balanced salt solution (HBSS), resazurin (7-hydroxy-3H-phenoxazin-3-one 10-oxide) sodium salt, cysteine, sodium borohydride (NaBH4), dimethyl sulfoxide-d6 (DMSO-d6), tetramethylsilane (TMS), fluorescein diacetate (FDA), minimum essential medium Eagle (MEM), sodium chloride, miconazole nitrate, methanol, trifluoroacetic acid, hydrochloric acid (HCl), acetic acid, monopotassium phosphate, dipotassium phosphate, and Triton-X 100 were all purchased from Sigma-Aldrich, Vienna, Austria.
Cell culture medium was prepared by using MEM powder 9.66 g/L (modified with Earle’s salts; phenol red; 19 amino acids; and the non-essential amino acids L-asp, L-asn, L-glu, L-ser, L-ala, L-gly, and L-pro), L-glutamine 2 mM, sodium bicarbonate 2.2 g/L, 1% penicillin-streptomycin solution, and 10% fetal bovine serum (FBS). FBS and 100 mM phosphate-buffered saline (PBS) pH 6.8 were purchased from Gibco (Invitrogen, Lofer, Austria). Pretreated standard grade regenerated dialysis tubing of cellulose membrane (molecular weight cutoff of 500–1,000 Da) was purchased from Spectrum Lab, Göttingen, Germany. All other materials were of analytical grade and received from commercial sources.
Synthesis of thiolated β-CD
β-CD as a hydrophilic polymer was chosen for the substitution of its primary and secondary hydroxyl groups with bromine moieties that were later replaced by thiol groups. Thiolated β-CD was synthesized by replacement of hydroxyl groups with bromine that were replaced by thiol groups based on a method described previously.12 Both steps: bromination and thiolation (Figure 1) were carried out under nitrogen gas. β-CD, NBS, and Ph3P were dried in a desiccator under diminished pressure in the presence of molecular sieves 4°A. LiBr anhydrous was dried at 150°C in an oven prior to the experiment.
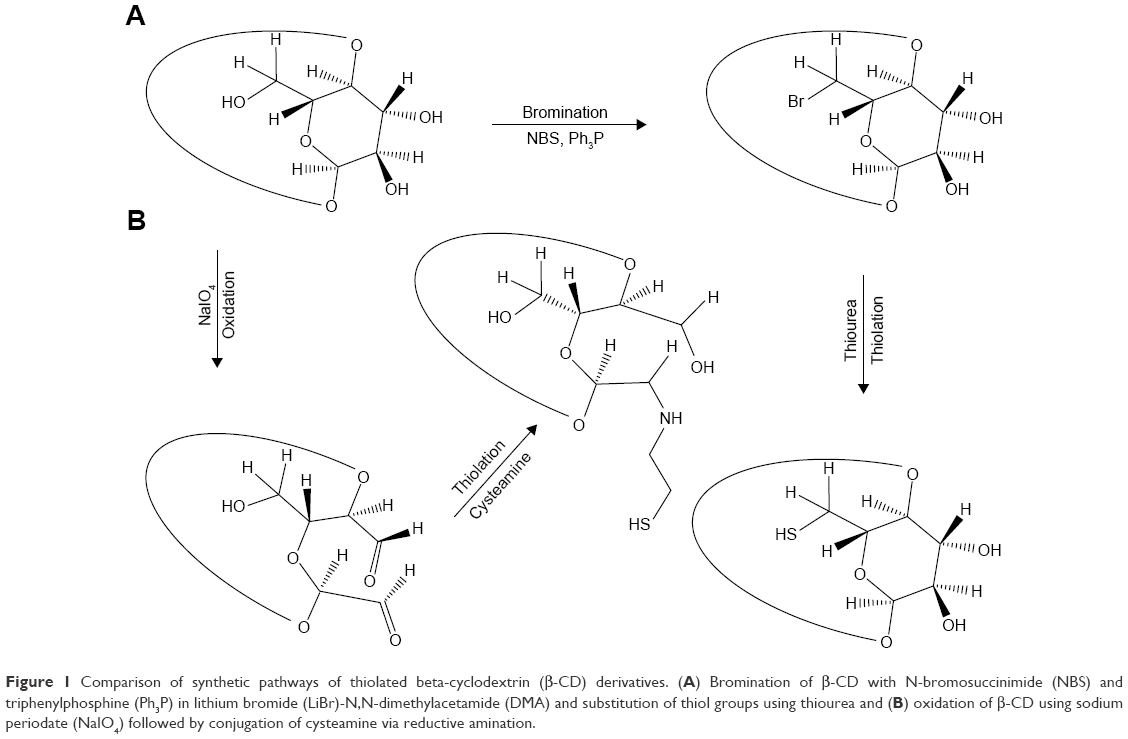 | Figure 1 Comparison of synthetic pathways of thiolated beta-cyclodextrin (β-CD) derivatives. (A) Bromination of β-CD with N-bromosuccinimide (NBS) and triphenylphosphine (Ph3P) in lithium bromide (LiBr)-N,N-dimethylacetamide (DMA) and substitution of thiol groups using thiourea and (B) oxidation of β-CD using sodium periodate (NaIO4) followed by conjugation of cysteamine via reductive amination. |
First, 1 g of dried β-CD was dissolved in 100 mL of DMA, the mixture was heated for 1 h at 90°C under stirring, and then 24 g of LiBr was added slowly. This LiBr-DMA system was established to guarantee a homogeneous bromination of β-CD.5,13 The reaction mixture was kept for 1 h at 90°C until LiBr was completely dissolved and then maintained at 70°C for 14 h until a clear solution was obtained.
NBS and Ph3P were separately dissolved in 100 mL of DMA in concentrations as listed in Table 1 and gradually added to the reaction mixture, having been cooled down in ice water.
 | Table 1 Overview on applied reaction conditions and resulting amount of immobilized thiol (-SH) and disulfide (-S-S-) groups |
The final solution was kept at 70°C for 3 h while stirring. The brominated CD was precipitated by the addition of 400 mL of acetone (1:4). The precipitate was filtered with a Whatman filter paper, washed three times with acetone, dried in an oven at 40°C, dissolved in water, dialyzed 2 days against water, and finally freeze-dried (−51°C, 0.01 mbar, Gamma LSC 1–16, Martin Christ, Osterode am Harz, Germany).
The brominated product was treated with thiourea to convert the bromide moieties to thiols. First, 1 g of bromo-β-CD was dissolved in a solution of thiourea (0.5%; m/v) in 100 mL of DMA and the mixture was stirred for 19 h at 80°C. After cooling the reaction mixture in ice water, 20 mL of 3 M NaOH solution was added slowly to release the free thiolated β-CD. After stirring for 15 min at room temperature, the mixture was neutralized with 3 M H2SO4 solution. The final product was precipitated in acetone as already described, dialyzed using cellulose membrane (molecular weight cutoff of 500–1,000 Da) against water, freeze dried, and stored at 4°C before use.
Synthesis of a cationic thiolated β-CD
For comparison, a cationic thiolated β-CD was synthesized in a two-step synthetic pathway by oxidizing diol groups of CD with NaIO4, followed by covalent coupling of cysteamine via reductive amination.1
Inclusion constant measurement
For measurement of the stability constant phase, solubility studies were carried out according to the method of Higuchi and Connors.14 Briefly, an excess amount of miconazole nitrate was mixed in 100 mM phosphate buffer pH 7.0 containing increasing amounts of β-CD-SH, dispersed by sonicator for 30 min, and then oscillated by a laboratory stirrer for 24 h at room temperature. The degree of absorption in solution was measured by ultraviolet (UV)/visible spectrophotometer at 258 nm after equilibration and converted into the concentration of miconazole nitrate by using a calibration curve. The experiments were carried out in triplicate for each sample.
The phase solubility diagram was plotted by using β-CD-SH concentration as x-coordinate and miconazole nitrate concentration as y-axis. The stability constant, Kc, was calculated from the straight-line portion of the phase solubility diagram according to the Higuchi–Connors equation:
|
|
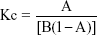
Where A is slope and B is intercept, which could be obtained from the straight line of the phase solubility diagram.
Study of thiol stability at different pH environments
β-CD-SH was hydrated in demineralized water in a final concentration of 0.25% (m/v) and the pH values were stabilized with 50 mM acetate buffer at pH 5.0, 50 mM phosphate buffer at pH 6.0, and 50 mM Tris buffer at pH 7.2, respectively. The samples and controls were incubated at 37°C under permanent shaking. At predetermined time points, aliquots of 500 μL were withdrawn and 50 μL of 1 M HCl was added in order to quench any further reaction.15 The amount of remaining thiol groups was determined by Ellman’s method as described in the following text.
Determination of the thiol content
Ellman’s reagent was used to determine free thiol groups of thiolated β-CD as described previously.16 The amount of thiol groups was assessed by using a standard curve of cysteine prepared in exactly the same way as the samples. Disulfide content was determined after reduction with NaBH4 via Ellman’s test.13
Characterization of thiolated β-CD structure
Infrared (IR) spectra were recorded on a Bruker ALPHA fourier transform (FT)-IR apparatus (Billerica, MA, USA) equipped with a platinum attenuated total reflection module. The nuclear magnetic resonance spectra were recorded in DMSO-d6 solution in 5 mm tubes at 30°C on a Varian Gemini 200 spectrometer (199.98 MHz for 1H, 50.29 MHz for 13C). The center of the solvent multiplet (DMSO-d6) was used as internal standard (chemical shifts in δ ppm), which was related to TMS with δ 2.49 ppm (1H) and 39.5 ppm (13C).
Water solubility
Solubility of unmodified and modified β-CD was determined using turbidity measurement method.17 In brief, gradually increasing amounts of test compounds from 5, 10 to 30 mg were dissolved in 1 mL of distilled water and vortexed for 5 min at room temperature. Subsequently turbidity was measured using a UV-visible spectrophotometer (UV-1240; Shimadzu, Kyoto, Japan) at a wavelength of 595 nm.
Moisture content
All the complexes were weighed (W1) and kept in a desiccator containing anhydrous calcium chloride at 37 °C for 24 h. After that, complexes were removed from the desiccators and reweighed until a constant weight (W2) was obtained. The percentage moisture loss was calculated using the following equation:18
|
|
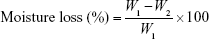
W1 is the initial weight of the complex and W2 is the final weight of the complex.
Liquid chromatography – mass spectrometry
Mass spectrometry detection was carried out using micrOTOF-Q II (Bruker) controlled by a computer running the “HyStar 3.2” acquisition software.
Source conditions were as follows: capillary voltage: 4.5 kV; end plate offset: −500 V; quadrupole ion energy: 5 eV; nebulizer: 4.4 psi; cone gas flow (nitrogen) gas: 6 L/min; dry temperature: 200 °C. Sample solutions were prepared by dissolving the complex mixtures in the mobile phase (methanol, 0.1% formic acid; 85:15%) to reach the concentration of 500 ng/mL.
Mass spectral studies were performed in positive electrospray ionization (ESI) mode in the mass range of m/z 417→415 to quantify the drug. In order to get a clear mass spectrum without any background noise, the drug was directly infused using a syringe pump into the mass spectrometer.
The stock solution of miconazole was prepared in a concentration of 1.0 mg/mL (methanol/0.1% formic acid 85:15% v/v mixture) for mass spectrometric analysis performed via direct infusion of the samples. Calibration standards were prepared by diluting the stock solution to the concentrations of 7.81, 15.62, 31.25, 62.5, 125, 250, and 500 ng/mL. The complex concentrations were prepared in the same way as calibration samples to the concentrations of 125, 250, and 500 ng/mL.
Cytotoxicity studies
Resazurin assay
Caco-2 cells were purchased from the European Collection of Authenticated Cell Cultures (Wiltshire, UK). Cells (passage number 25–33) were seeded at a density of 2.5 × 104 cells/well in a 24-well plate in a final volume of 500 μL of MEM with Earle’s balanced salts supplemented with 10% FBS, 1% penicillin-streptomycin, and 2.0 mM L-glutamine at 37 °C in a 5% CO2 environment. The medium was refreshed every other day.
Resazurin assay is performed as an oxidation-reduction indicator in cell viability assays. Resazurin (7-hydroxy-3H-phenoxazin-3-one 10-oxide) is a blue dye, itself weakly fluorescent until it is irreversibly reduced to the pink colored and highly red fluorescent resorufin by alive cells. When the cells were ~80% confluent (80% of surface of flask covered by cell monolayer after 8–10 days), they were washed twice with PBS pre-warmed at 37 °C. Test solutions including unmodified β-CD, β-CD-SH600, β-CD-SH1200 (Table 1), the cationic reference β-CD-SH (0.5% and 1%; m/v) positive control prepared in white MEM, and negative control (1% v/v Triton X-100) were added in 500 μL volume in triplicate to the cell culture. Then, the treated cells were incubated at 37°C in a 5% CO2 environment for 24 and 72 h. Then, test solutions were removed, and cells were washed twice with pre-warmed PBS. A diluted resazurin solution (2.2 μM) in 500 μL volume was added to each well and cells were incubated for 3 h. Supernatant (100 μL) was afterwards transferred to a black 96-well plate and fluorescence intensity was measured using microplate reader (M200 spectrometer; Tecan Infinite, Grödig, Austria) at a wavelength of 540 nm with background subtraction at 590 nm. Percentage of viable cells was calculated according to the following equation:
|
|

LDH assay
In parallel to the resazurin assay, lactate dehydrogenase (LDH) release assay was performed. In brief, if test samples damaged cell membrane integrity, cytoplasmic LDH is released into culture medium.6 The amount of LDH leakage is then measured using a commercial test kit (Promega, Madison, WI, USA). In detail, the cells with ~80% confluence were washed with pre-warmed HBSS before being treated with unmodified or modified samples (0.5% and 1%; m/v) as already described. HBSS was used as negative control and Triton-X 100 (1%; m/v) served as positive control. After 24 and 72 h of incubation at 37°C in 5% CO2 environment, supernatant was collected and LDH assay was performed according to the manufacturer’s instruction. Fluorescence was recorded using a microplate reader (M200 spectrometer; Tecan Infinite) with an excitation wavelength of 560 nm and an emission wavelength of 590 nm. The percentage of cell toxicity was estimated according to the following equation:
|
|
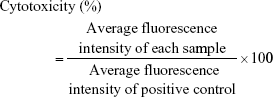
In vitro evaluation of mucoadhesive properties
For the evaluation of mucoadhesive properties, β-CDs were fluorescently labeled by incorporation of FDA. Briefly, 20 mg of CD was dissolved in 20 mL of demineralized water. pH was adjusted to 6.5 with 100 mM HCl. In all, 1 mg of FDA was dissolved in 5 mL of 96% ethanol and 1 mL of this solution was added to each CD solution. After 24 h of stirring at room temperature, the suspensions were filtered in order to eliminate unentrapped FDA and freeze dried.
Freshly excised porcine intestinal mucosa collected from a local slaughterhouse was used for the mucoadhesion test. First, the porcine small intestine was rinsed with 100 mM PBS pH 6.8. Then, it was cut into smaller pieces of 4 × 2 cm and fixed on half-cut 50 mL falcon tubes that were placed at an angle of 45° in an incubation chamber of 37°C and 100% relative humidity. Subsequently, the mucosa was continuously rinsed with the same phosphate buffer for 5 min at a flow rate of 1 mL/min. Then, 20 mg of FDA-labeled β-CD samples were separately placed on the mucosa and after 10 min, the phosphate buffer flow was restarted. Then, 30 mL of phosphate buffer flowing down the mucosa was collected at the following time points: 30, 60, 90, 120, and 180 min. In parallel, standard (100%) samples were prepared by allowing 30 mL of phosphate buffer to flow on the mucosa and then dissolving 20 mg of CDs in the collected buffer.
To quantitatively hydrolyze FDA to sodium fluorescein, 25 mL of NaOH 5 M was added to 30 mL of the collected samples and the remaining intestine on the falcon tube. Samples were incubated while shaking at 37°C for 20 min and then centrifuged at 13,400 rpm at room temperature for 5 min. Finally, 100 μL of each sample was transferred to the microplate reader (M200 spectrometer; Tecan Infinite) and fluorescence intensity was measured at an emission wavelength of 535 nm and an excitation wavelength of 485 nm.
HPLC analysis
Drug loading was measured by high-performance liquid chromatography (HPLC) using ProntoSIL C18 column (250 4.6 mm, 5 μm; Bischoff Chromatography, Leonberg, Germany) and a mobile phase containing 0.1% trifluoroacetic acid and methanol in a ratio of 15:85. Detection was performed in UV at λ = 235 nm, where the miconazole nitrate reached the maximum level of absorption, while retention time was at 7.6 min.
Measurement was conducted by integration of peak areas, and loaded drug was quantified by using a standard curve (coefficient r2 = 0.9997) that was prepared by dissolving miconazole nitrate in methanol.
Inclusion complexes and drug loading
Inclusion complexes of miconazole nitrate with unmodified and thiolated β-CDs were prepared according to a method described by Wang and Cai.15 In brief, 25 mg of miconazole nitrate dissolved in 5 mL of ethanol was added to 30 mL of 50 mM phosphate buffer pH 6.8 containing 75 mg of CDs (molar ratio; 1:1), separately. Reaction mixtures were stirred at room temperature for 24 h. Then, the mixtures were filtered using a 0.2 μm size membrane filter and freeze dried.
Dissolution study
To calculate the influence of the inclusion complex of unmodified β-CD and thiolated β-CD on dissolution of miconazole nitrate, 30 mg of each inclusion complex of pure miconazole nitrate was compressed into tablets (single puncher machine, D-7064; Paul Weber, Remshalden-Grunbach, Germany). The compressed tablets were placed in Erlenmeyer flasks containing 6 mL of 100 mM phosphate buffer pH 6.8. The flasks were placed on a rocker in an incubator at 37°C with 100% relative humidity. Aliquots of 200 μL were taken at predetermined time points and replaced by an equal volume of fresh medium. All samples were centrifuged and filtrated. The concentration of dissolved drug in these samples was determined via HPLC as already described.
Statistical analysis
The statistical difference among groups was compared by one-way analysis of variance (ANOVA) and p < 0.05 or p < 0.01 was considered statistically significant (GraphPad Prism 5.01; GraphPad Software, Inc., CA, USA). In addition, Student’s t-test was applied with a confidence interval (p < 0.05) for the analysis of two groups.
Results and discussion
Synthesis of thiolated β-CD
In order to synthesize thiolated β-CD, the hydroxyl groups on the carbohydrate backbone were replaced by thiol moieties as shown in Figure 1A. In contrast to the synthetic pathway having been used as illustrated in Figure 1B, there was no ring opening necessary. The intermediate products were odorless, composed of fine structures, and light yellow most likely due to bromine ion on the carbohydrate ring. As the primary hydroxyl groups display the highest reactivity to nucleophilic substitution compared to secondary hydroxyl groups, hydroxyl groups at the terminal site of polysaccharide chains are likely favored for this type of substitution.19,20 The final thiolated β-CD product appeared as a fibrous structured polymer, odorless, white, and being soluble at physiological pH in aqueous media. The yield in hydroxyl groups having been substituted by thiol groups in dependence on the applied concentration of NBS and Ph3P is illustrated in Table 1.
Comparing the content of immobilized thiol groups on β-CD-SH revealed almost two times higher degree of substitution (DS) when a twice as high concentration of Ph3P and NBS was applied in the first step of the reaction. This effect might be explained by the higher bromination substitutions on the backbone unit of the polymer, due to these reaction improvements, as shown in Table 1. Moreover, as increasing concentrations of thiourea had no effect on the thiolation efficiency, it can be assumed that the efficiency of the thiolation reaction is strongly dependent on the degree of bromination.5 An efficient regioselective bromination of cellulose was achieved using NBS/Ph3P in DMA/LiBr, giving a 6-bromo-6-deoxycellulose with DS = 0.9.21 Similarly, thiolation reactions of other polysaccharides in this homogeneous system led to high regioselectivity and high degrees of substitutions.5,20 To a minor extent, these thiol groups were oxidized to disulfide substructures. Generally, in the case of β-CD-SH1200, on average each CD molecule exhibited one free thiol group.
The successful immobilization of thiol groups was further confirmed by the results obtained from FT-IR spectroscopy. The FT-IR spectra of β-CD-SH and β-CD are shown in Figure S1. The infrared spectra of the thiolated derivatives differ significantly from that of the unmodified CD. In detail, in the case of β-CD-SH600 and β-CD-SH1200, there are strong bonds at 1,137 or 1,136 cm−1 as well as moderate bonds at 624 cm−1 that should be interpreted as CS stretching vibrations.6
Chemical structure of β-CD-SH was confirmed via 13C NMR spectral analysis. Comparison of the 13C NMR spectrum of β-CD-SH with that of unmodified β-CD showed numerous additional signals of aliphatic carbon atoms present in β-CD-SH indicating the introduction of the sulfhydryl ligand. Comparison of the 1H NMR spectra of unmodified β-CD with those of the modified derivatives indicates that in the case of β-CD-SH600 and β-CD-SH1200 there are additional signals at 7.4, 7.1, and 6.9 ppm that are exchangeable with D2O and may be assigned to the protons of the thiol groups (Figure S2A–D).
The 13C NMR spectra of thiolated CDs are characterized by additional signals in the aliphatic range (at 26.0, 21.9, and 20.5 for β-CD-SH600, and at 26.0 and 20.5 ppm for β-CD-SH1200) representing thiol-bearing carbon atoms (Figure S3A–D).
Phase solubility and the inclusion constant
Phase solubility studies were carried out at room temperature in buffer at pH 7 to calculate the solubility constant Kc. The phase solubility diagram of miconazole nitrate with β-CD-SH is illustrated in Figure 2 showing a linear trend. According to these results, the formation of 1:1 inclusion complexes seems likely. The stability constant Kc of the complexes of miconazole nitrate with β-CD-SH at pH 7 was calculated using Equation 1 and is summarized in Table 2.
 | Figure 2 Phase solubility study of miconazole nitrate with beta-cyclodextrin (β-CD)-SH in 100 mM phosphate buffer pH 7.0. |
 | Table 2 The inclusion constant of miconazole nitrate with β-CD-SH |
Study of thiol stability in different pH environments
Depending on the pH value of the thiomer solution, the thiol groups of the polymer conjugates were oxidized, thereby forming intermolecular as well as intramolecular disulfide bonds. These results are illustrated in Figure 3. At pH 5, the thiol groups of the β-CD-SH remained more stable towards oxidation, whereas a significant decrease in the thiol group content could be observed at pH 6 and 7.2. After 6 hours at pH 5, more than 80% thiols were stable. This observation can be explained by the decreasing H+ concentration on raising pH values resulting in turn in a higher amount of negative thiolate anions, S−, representing the active form for oxidation.
 | Figure 3 Stability of thiol groups at different pH environments at 37°C. |
Solubility
H-bond supported that self-aggregation of hydrated β-CD causes limited aqueous solubility and leads to physical instability of aqueous β-CD solutions.22 This comparatively poor aqueous solubility of β-CD is certainly limiting its potential use for various pharmaceutical applications. The solubility of β-CD, however, was improved from 1.85 g/100 mL to 2.89 g/100 mL in the case of β-CD-SH1200, corresponding to a 1.36-fold improvement. (Figure 4; Table S1).
 | Figure 4 Water solubility determined with the turbidity method at 595 nm. |
This unexpected result can be explained by the disruption of the stable, restrictive hydrogen bond system around the CD ring through insertion of disturbing thiol groups. Szente and Szejtli, for instance, could also show improved solubility of CD by introducing some methylated moieties to the ring system.23
Cytotoxicity
The potential use of CDs and their synthetic derivatives has been widely regarded as improving certain properties of the drugs, such as stability, solubility, and bioavailability.24 Safety is a primary concern when considering new excipients intended for use in pharmaceutical formulations, and so the toxicological matters together with the biological fate of CDs must be comprehensively investigated before practical use can be considered.24 The cytotoxic effect of both β-CD and β-CD-SH on Caco-2 cells was assessed by two methods, resazurin and LDH assays. Cells were treated with β-CD-SH600, β-CD-SH1200, and the cationic β-CD-SH of the first generation in two concentrations. Results confirmed that the new thiolated CDs do not show significant toxicity (p < 0.05), as cell viability was more than 92% even after treatment with the higher concentration of 1% (Figure 5A).
 | Figure 5 Cytotoxic effect of thiolated beta-cyclodextrin (β-CD) on Caco-2 cells as demonstrated by (A) resazurin and (B) lactate dehydrogenase (LDH) assays. |
In addition, no significant difference could be detected between incubation periods of 24 and 72 h for both concentrations and even with increasing degree of thiolation as shown in Figure 5A. By contrast, the cationic β-CD-SH showed a pronounced toxic effect increasing over time (Figure 5A). Results of LDH assay are shown in Figure 3. Cationic β-CD-SH exposure led to cell toxicity, whereas exposure with 0.5% and 1% β-CD-SH of the second generation showed no significant difference in comparison to the negative control, within 24 h (Figure 5B). These results are in good accordance with the resazurin results (Figure 5B).
It has been previously confirmed that thiol groups are key regulators of intracellular adhesion molecule-1 (ICAM-1) expression in the process of cell attachment.25,26 For example, L-cysteine blocks the ketone-induced and high-glucose adhesion of monocytes to endothelial cells because of depressing ICAM-1 expression through inhibition of the reactive oxygen species pathway. On the other hand, Mukherjee et al verified that extremely excessive N-acetylcysteine generates a tense condition in endothelial cells. This phenomenon results in induced expression of ICAM-1, which becomes the target of endosomal and proteolytic degradation, thus increasing functional ICAM-1 on the cell surface.25 According to these considerations, thiol groups of β-CDs will likely interact with cells. Results, however, showed that these presumptive interactions have no significant impact on cell viability. By contrast, the charge of β-CDs seems to have a pronounced effect. The comparatively high toxicity of cationic β-CD-SH can be explained by its positive charge at physiological pH due to the secondary amino groups likely ionically interacting with the anionic cell surfaces substructures, such as heparin sulfate proteoglycans, and subsequently destabilizing cell membrane integrity.25 Apart from that, it is well known that β-CDs interact with cell membrane cholesterol causing cytotoxicity. Kiss et al, for instance, confirmed that even cholesterol-solubilizing properties can be a predictive factor for β-CD cell toxicity, which depends on the structure of the CD derivative, eg, on the number and position of the new groups and the presence of ionic groups.33 Due to the addition of cholesterol to final formulations, however, this toxic effect might be compensated.11
Mucoadhesive properties
The enhancement of drug activity and selective transfer or the reduction of side effects can be achieved by inclusion complex formation.27 The CD complexation enables the development of formulations for drugs that are difficult to formulate and deliver with the existing pharmaceutical excipients, which is reflected in the increasing number of pharmaceutical products being available in the market as CD-based formulations.24 However, actual and potential use of CD derivatives with more favorable safety profiles has renewed interest in extended use of CDs for drug delivery via various routes.24 Mucosal administration is thereby among the most attractive ones.
The mucoadhesive properties of polymers are dependent on interactions between functional moieties of the mucus gel layer and the mucoadhesive polymers.28 In the case of CDs, the most likely important interactions being responsible for mucoadhesive properties – namely ionic interactions and chain entanglements – cannot take place without sacrificing their non-ionic character, thus explaining their comparatively poor mucoadhesive properties. The mucoadhesive properties of CDs and hydroxypropyl-β-CD have been reported previously by Gavini et al and Francois et al.29,30 The mucoadhesive properties of CDs can likely be strongly improved via thiolation allowing CDs to form disulfide bonds with cysteine-rich subdomains of mucus glycoproteins.
Mucoadhesion study of thiolated β-CD on porcine intestinal mucosa confirmed that this type of thiomers show strongly improved mucoadhesive properties. In detail, β-CD-SH600 and β-CD-SH1200 exhibited 39.73- and 46.37-fold higher mucoadhesion in comparison to unmodified β-CDs, respectively (Table 3). As shown in Figure 6, thiolated β-CDs were able to adhere and remain for a longer period on the intestinal mucosa and more than 52% of β-CD-SH1200 remained on the intestinal mucosa after 3 h. Application of just the fluorescence marker FDA to the mucosa showed that it is – although insoluble in aqueous media – washed off the mucosa in a considerably short time period in a similar manner as the unmodified β-CD. These data demonstrated that applying poorly soluble drugs in suspension to mucosal membranes could not provide a prolonged residence time there.
 | Table 3 Comparison of percentage of thiolated CDs remaining attached to porcine intestinal mucosa for a period of 3 h and the resulting improvement ratio with respect to unmodified β-CD |
 | Figure 6 Time course of percentage of remaining thiolated and unmodified beta-cyclodextrin (β-CD) on porcine intestinal mucosa continuously rinsed with 100 mM phosphate buffer pH 6.8 at 37°C and 100% relative humidity. |
Inclusion complex forming properties
The maximum amount of miconazole nitrate that could be incorporated in unmodified and modified CDs is shown in Table S2. Inclusion complexes prepared with β-CD-SH600 and β-CD-SH1200 increased drug solubility by 157- and 257-fold, respectively, in comparison to a just 2-fold improvement in the case of unmodified β-CD (Figure 7).
 | Figure 7 Dissolution profile of miconazole nitrate formulated in the inclusion complex with thiolated and unmodified beta-cyclodextrin (β-CD). |
Hydrophobic properties of miconazole nitrate were reduced by encapsulation, because the lipophilic groups of the drug were trapped within the lipophilic cavity of the intact ring structure of the novel thiolated CDs.31 The improved dissolution of inclusion complexes with these thiolated CDs demonstrates their advantage over the first generation of thiolated CDs providing just a 2-fold improvement in the aqueous solubility of miconazole nitrate.1
Dissolution studies
Complexation mechanisms of drug-like chemical compounds with different CDs to establish a host-guest complex in solution result in the improvement of pharmacokinetic parameters and physicochemical properties of the guest component, such as higher stability, increased aqueous solubility, decreased plasma protein binding, and cellular toxicity.32 In order to obtain data about the dissolution kinetic of unmodified and thiolated β-CD/miconazole inclusion complexes, dissolution studies were performed. Results showed a significantly different dissolution profile for all tested complexes being based on β-CD-SH1200 exhibiting much higher dissolution velocity than the others. The β-CD complexes were found to be more stable and soluble in water. Results as illustrated in Figure 7 were in good agreement with solubility data listed in Table S1 showing highest solubility for the β-CD-SH1200 complex followed by the β-CD-SH600 and the unmodified complex.
Moisture content
Moisture content influences the weight, appearance, and physical properties of the final product. An accurate moisture content determination plays therefore a key role in ensuring quality of CDs. Moisture uptake of the complexes was very low and ranged from about 0.81% ± 0.28% to 1.19% ± 0.54% for all three complexes. According to these results, moisture does not seem to have a significant impact on the properties of formed complexes.
Liquid chromatography – mass spectrometry
Liquid chromatography – mass spectrometry under optimized conditions with ESI mode provided a highly selective method for determination of miconazole. Direct infusion through a syringe pump was used to achieve the most suitable mass spectrometry conditions for miconazole. Methanol acidified with 0.1% formic acid was identified as the most appropriate eluent. The total run time for miconazole was 3.8 min. The protonated active pharmaceutical ingredient was detected as the major peak in all complexes. The spectrum of complexes showed base peak ions at mass-to-charge ratio (m/z) of 416.1 for miconazole. The calibration curve was linear for concentrations in the range of 7.81–500 ng/mL and the representative regression equation showed r2 = 0.9961.
The miconazole content was 18.25%, 25.71%, and 33.41% in β-CD-miconazole, β-CD-SH600-miconazole, and β-CD-SH1200-miconazole complexes, respectively, as shown in Table S3.
Conclusion
Thiomers are one of the most studied mucoadhesive excipients in drug delivery. In this study, a novel non-ionic β-CD-SH with intact ring structure was developed. This new generation of β-CD-SH was compared with a cationic β-CD-SH of ring-opened structure. The new generation had no toxic effect on the cell even at high concentration (1%), while the thiolated CD first generation showed a time- and concentration-dependent toxicity. The novel β-CD-SH showed comparatively high mucoadhesive properties. In contrast to ionic CDs, the new derivative formed stable inclusion complexes with miconazole nitrate and provided improved drug solubility. These favorable properties guarantee the benefits of localized delivery of poorly soluble drugs on mucosal membranes, in particular where gels and creams are irritating, such as the intraoral or ocular mucosa.
Acknowledgment
The authors would like to thank Shiraz University and University of Innsbruck for supporting this research.
Disclosure
The authors report no conflicts of interest in this work.
References
Ijaz M, Matuszczak B, Rahmat D, et al. Synthesis and characterization of thiolated β-cyclodextrin as a novel mucoadhesive excipient for intra-oral drug delivery. Carbohydr Polym. 2015;132:187–195. | ||
Ijaz M, Ahmad M, Akhtar N, Laffleur F, Bernkop-Schnürch A. Thiolated α-cyclodextrin: the invisible choice to prolong ocular drug residence time. J Pharm Sci. 2016;105(9):2848–2854. | ||
Gum JR Jr, Hicks JW, Toribara NW, Rothe EM, Lagace RE, Kim YS. The human MUC2 intestinal mucin has cysteine-rich subdomains located both upstream and downstream of its central repetitive region. J Biol Chem. 1992;267(30):21375–21383. | ||
Bernkop-Schnürch A. Thiomers: a new generation of mucoadhesive polymers. Adv Drug Deliv Rev. 2005;57(11):1569–1582. | ||
Sarti F, Staaf A, Sakloetsakun D, Bernkop-Schnürch A. Thiolated hydroxyethylcellulose: synthesis and in vitro evaluation. Eur J Pharm Biopharm. 2010;76(3):421–427. | ||
Peppas NA, Buri PA. Surface, interfacial and molecular aspects of polymer bioadhesion on soft tissues. J Control Release. 1985;2:257–275. | ||
Lehr CM. From sticky stuff to sweet receptors – achievements, limits and novel approaches to bioadhesion. Eur J Drug Metab Pharmacokinet. 1996;21(2):139–148. | ||
Shityakov S, Puskás I, Pápai K, et al. Sevoflurane-sulfobutylether-β-cyclodextrin complex: preparation, characterization, cellular toxicity, molecular modeling and blood-brain barrier transport studies. Molecules. 2015;20(6):10264–10279. | ||
Kafedjiiski K, Krauland AH, Hoffer MH, Bernkop-Schnürch A. Synthesis and in vitro evaluation of a novel thiolated chitosan. Biomaterials. 2005;26(7):819–826. | ||
Schmitz T, Grabovac V, Palmberger TF, Hoffer MH, Bernkop-Schnürch A. Synthesis and characterization of a chitosan-N-acetyl cysteine conjugate. Int J Pharm. 2008;347(1–2):79–85. | ||
Shityakov S, Sohajda T, Puskás I, Roewer N, Förster C, Broscheit JA. Ionization states, cellular toxicity and molecular modeling studies of midazolam complexed with trimethyl-β-cyclodextrin. Molecules. 2014;19(10):16861–16876. | ||
Furuhata K, Koganei K, Chang HS, Aoki N, Sakamoto M. Dissolution of cellulose in lithium bromide-organic solvent systems and homogeneous bromination of cellulose with N-bromosuccinimide-triphenylphosphine in lithium bromide-N,N-dimethylacetamide. Carbohydr Res. 1992;230(1):165–177. | ||
Iqbal J, Sarti F, Perera G, Bernkop-Schnürch A. Development and in vivo evaluation of an oral drug delivery system for paclitaxel. Biomaterials. 2011;32(1):170–175. | ||
Higuchi T, Connors KA. Phase-solubility techniques. Adv Anal Chem Instrum. 1965;4:117–212. | ||
Wang JH, Cai Z. Investigation of inclusion complex of miconazole nitrate with β-cyclodextrin. Carbohydr Polym. 2008;72(2):255–260. | ||
Hombach J, Palmberger TF, Bernkop-Schnürch A. Development and in vitro evaluation of a mucoadhesive vaginal delivery system for nystatin. J Pharm Sci. 2009;98(2):555–564. | ||
Le HT, Jeon HM, Lim CW, Kim TW. 6-Triazolyl-6-deoxy-β-cyclodextrin derivatives: synthesis, cellular toxicity, and phase-solubility study. Carbohydr Res. 2014;391:22–28. | ||
Ofokansi KC, Kenechukwu FC, Ogwu NN. Design of novel miconazole nitrate transdermal films based on Eudragit RS100 and HPMC hybrids: preparation, physical characterization, in vitro and ex vivo studies. Drug Deliv. 2015;22(8):1078–1085. | ||
Fox SC, Li B, Xu D, Edgar KJ. Regioselective esterification and etherification of cellulose: a review. Biomacromolecules. 2011;12(6):1956–1972. | ||
Suchaoin W, Bonengel S, Hussain S, Huck CW, Ma BN, Bernkop-Schnürch A. Synthesis and in vitro evaluation of thiolated carrageenan. J Pharm Sci. 2015;104(8):2523–2530. | ||
Matsui Y, Ishikawa J, Kamitakahara H, Takano T, Nakatsubo F. Facile synthesis of 6-amino-6-deoxycellulose. Carbohydr Res. 2005;340(7):1403–1406. | ||
Coleman AW, Nicolis I, Keller N, Dalbiez JP. Aggregation of cyclodextrins: An explanation of the abnormal solubility of β-cyclodextrin. J Incl Phenom. 1992;13(2):139–143. | ||
Szente L, Szejtli J. Highly soluble cyclodextrin derivatives: chemistry, properties, and trends in development. Adv Drug Deliv Rev. 1999;36(1):17–28. | ||
Irie T, Uekama K. Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. J Pharm Sci. 1997;86(2):147–162. | ||
Mukherjee TK, Mishra AK, Mukhopadhyay S, Hoidal JR. High concentration of antioxidants N-acetylcysteine and mitoquinone-Q induces intercellular adhesion molecule 1 and oxidative stress by increasing intracellular glutathione. J Immunol. 2007;178(3):1835–1844. | ||
Kanikarla-Marie P, Jain SK. L-Cysteine supplementation reduces high-glucose and ketone-induced adhesion of monocytes to endothelial cells by inhibiting ROS. Mol Cell Biochem. 2014;391(1–2):251–256. | ||
Albers E, Müller BW. Cyclodextrin derivatives in pharmaceutics. Crit Rev Ther Drug Carrier Syst. 1995;12(4):311–337. | ||
Iqbal J, Shahnaz G, Dünnhaupt S, Müller C, Hintzen F, Bernkop-Schnürch A. Preactivated thiomers as mucoadhesive polymers for drug delivery. Biomaterials. 2012;33(5):1528–1535. | ||
Francois M, Snoeckx E, Putteman P, et al. A mucoadhesive, cyclodextrin-based vaginal cream formulation of itraconazole. AAPS PharmSci. 2003;5(1):E5. | ||
Gavini E, Rassu G, Haukvik T, Lanni C, Racchi M, Giunchedi P. Mucoadhesive microspheres for nasal administration of cyclodextrins. J Drug Target. 2009;17(2):168–179. | ||
Gidwani B, Vyas A. Synthesis, characterization and application of epichlorohydrin-β-cyclodextrin polymer. Colloids Surf B Biointerfaces. 2014;114:130–137. | ||
Felton LA, Popescu C, Wiley C, Esposito EX, Lefevre P, Hopfinger AJ. Experimental and computational studies of physicochemical properties influence NSAID-cyclodextrin complexation. AAPS PharmSciTech. 2014;15(4):872–881. | ||
Kiss T, Fenyvesi F, Bácskay I, et al. Evaluation of the cytotoxicity of β-cyclodextrin derivatives: Evidence for the role of cholesterol extraction. Eur J Pharm Sci. 2010;40(4):376–380. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.